The Terrestrial Carnivorous Plant Utricularia reniformis Sheds Light on Environmental and Life-Form Genome Plasticity

 , , , ,
, , , ,  and
and 
Abstract
:1. Introduction
2. Results
2.1. The Utricularia reniformis Genome
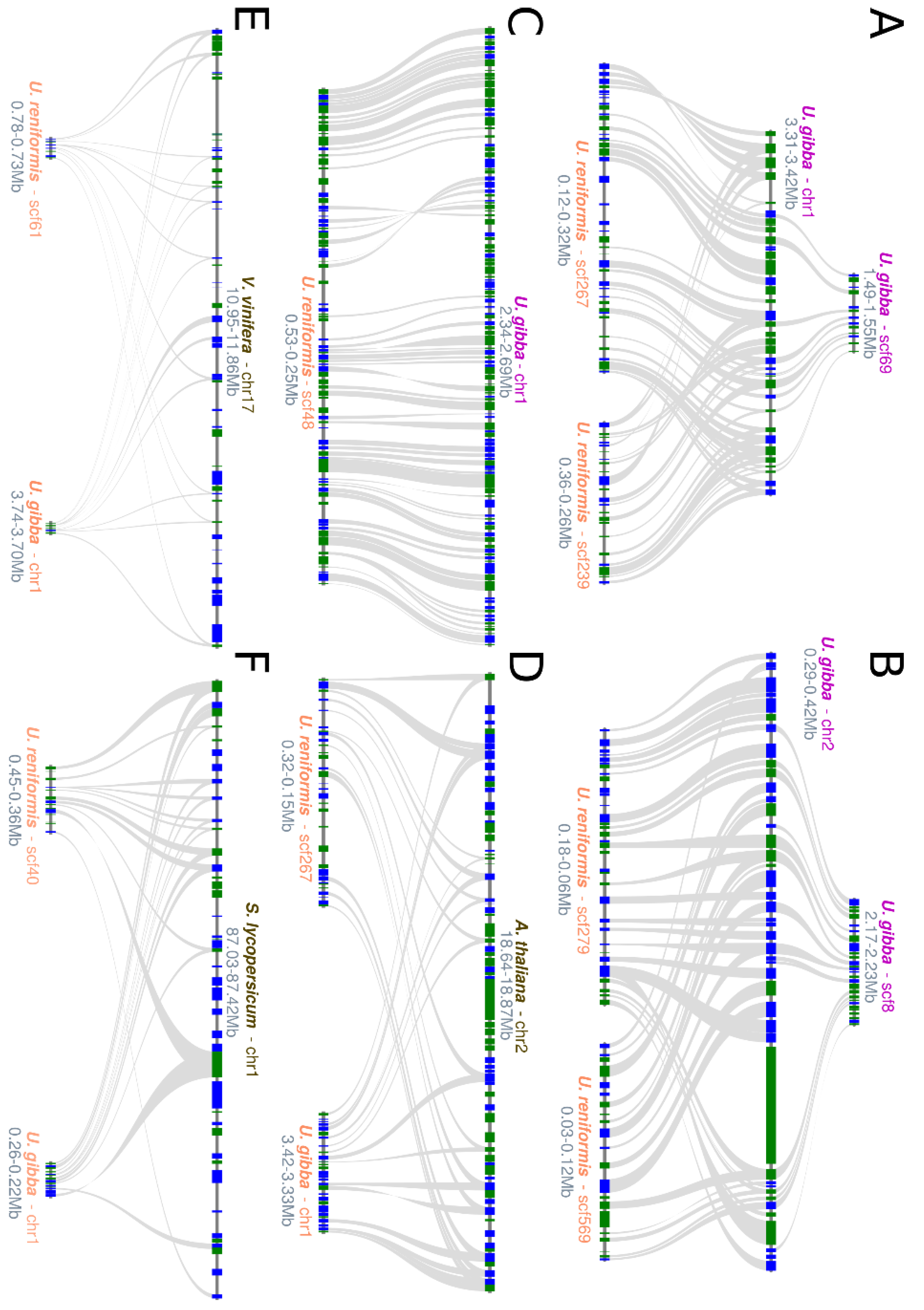
2.2. Structural Comparative Analysis
2.3. Utricularia reniformis Comparative Annotation
2.4. Utricularia reniformis Shows a Massive Expansion of LTR from the Gypsy Superfamily
2.5. Distinct Functional Enrichment Patterns Among Tandem and Dispersed Duplicate Genes
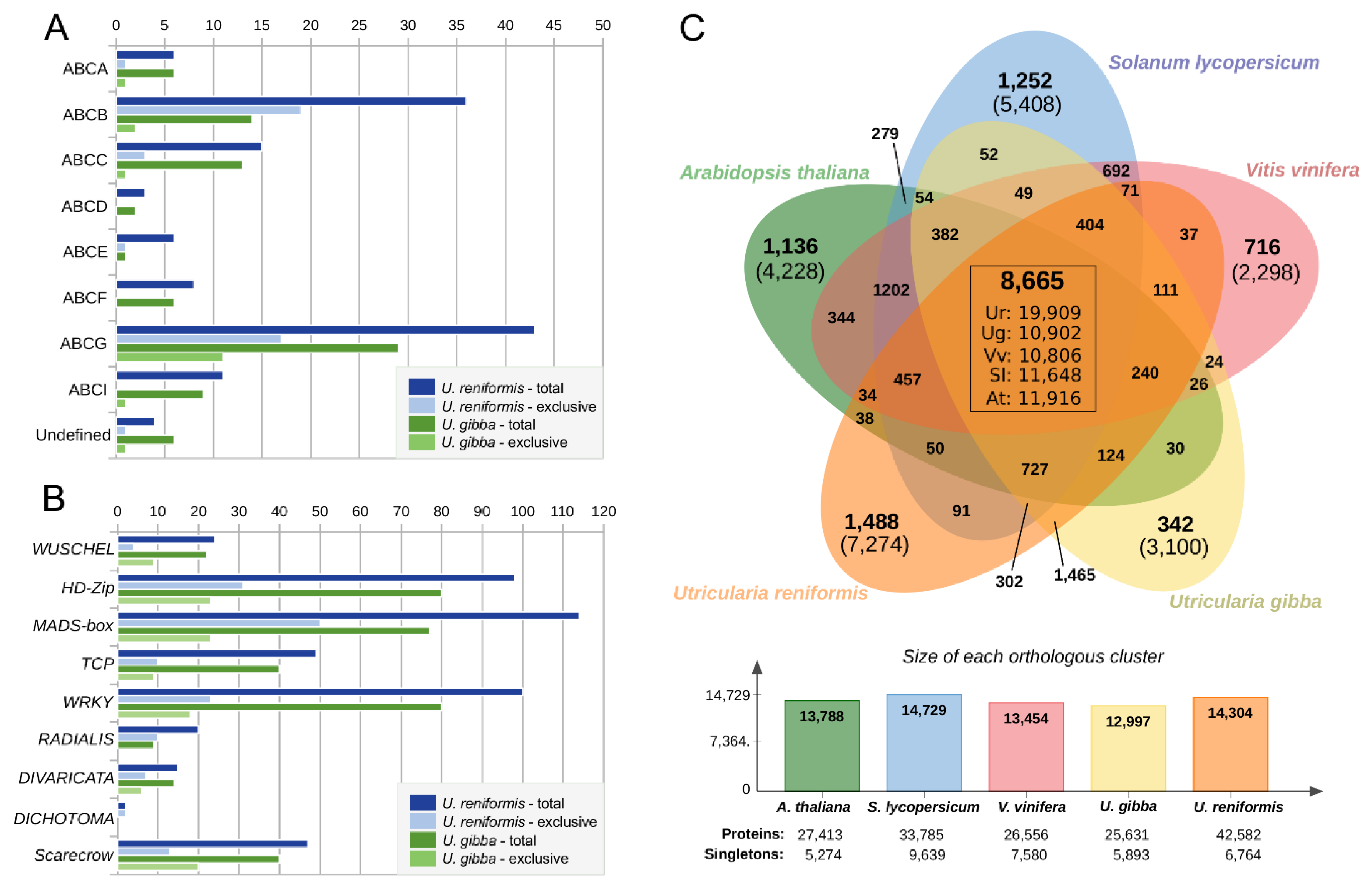
2.6. Utricularia reniformis Displays Unique Patterns of Carnivory-Associated, Land Adaptation, and Developmentally Related Genes
2.7. Comparative Annotation Among Other Angiosperms Genomes Reveals Species-Specific Genes Strictly Related to the Environment and Life-Form Adaptations
3. Discussion
3.1. The Role of WGD in Utricularia reniformis Genome Evolution
3.2. LTR-Retrotransposons are Key Agents Governing Utricularia Genome Size Changes
3.3. The Genomic Landmarks for Terrestrial Adaptation
4. Materials and Methods
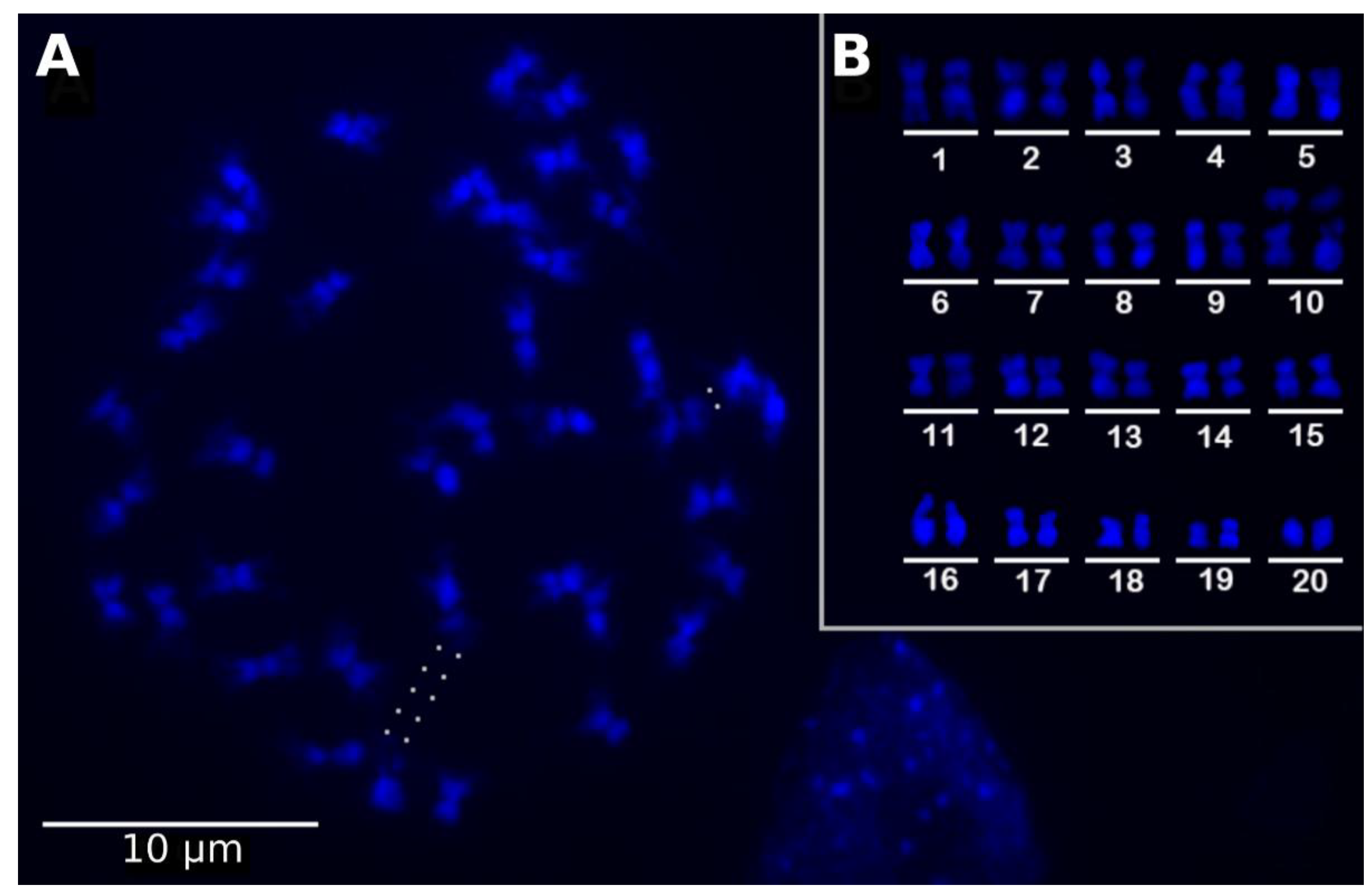
4.1. Plant Material, Genome Size Estimation, and Cytogenetic Analysis
4.2. Genome and Transcriptome Sequencing
4.3. Genome Assembly and WGD Analysis
4.4. Transposable Elements Identification, Classification, and Genome Annotation
4.5. Phylogenetic Analysis
4.6. Comparative Analysis
4.7. Availability of Supporting Data
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Taylor, P. The Genus Utricularia-A Taxonomic Monograph; The Royal Botanic Gardens, Kew: London, UK, 1989. [Google Scholar]
- Leushkin, E.V.; Sutormin, R.A.; Nabieva, E.R.; Penin, A.A.; Kondrashov, A.S.; Logacheva, M.D. The miniature genome of a carnivorous plant Genlisea aurea contains a low number of genes and short non-coding sequences. BMC Genom. 2013, 14, 476. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vu, G.T.H.; Schmutzer, T.; Bull, F.; Cao, H.X.; Fuchs, J.; Tran, T.D.; Jovtchev, G.; Pistrick, K.; Stein, N.; Pecinka, A.; et al. Comparative genome analysis reveals divergent genome size evolution in a carnivorous plant genus. Plant Genome 2015, 8, 1–14. [Google Scholar] [CrossRef] [Green Version]
- Ibarra-Laclette, E.; Lyons, E.; Hernández-Guzmán, G.; Pérez-Torres, C.A.; Carretero-Paulet, L.; Chang, T.H.; Lan, T.; Welch, A.J.; Juárez, M.J.A.; Simpson, J.; et al. Architecture and evolution of a minute plant genome. Nature 2013, 498, 94–98. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lan, T.; Renner, T.; Ibarra-Laclette, E.; Farr, K.M.; Chang, T.H.; Cervantes-Pérez, S.A.; Zheng, C.; Sankoff, D.; Tang, H.; Purbojati, R.W.; et al. Long-read sequencing uncovers the adaptive topography of a carnivorous plant genome. Proc. Natl. Acad. Sci. USA 2017, 114, E4435–E4441. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hanson, L.; Mc Mahon, K.A.; Johnson, M.A.T.; Bennett, M.D. First nuclear DNA C-values for another 25 angiosperm families. Ann. Bot. 2001, 87, 251–258. [Google Scholar] [CrossRef] [Green Version]
- Greilhuber, J.; Borsch, T.; Müller, K.; Worberg, A.; Porembski, S.; Barthlott, W. Smallest angiosperm genomes found in Lentibulariaceae, with chromosomes of bacterial size. Plant Biol. 2006, 8, 770–777. [Google Scholar] [CrossRef] [PubMed]
- Veleba, A.; Bureš, P.; Adamec, L.; Šmarda, P.; Lipnerová, I.; Horová, L. Genome size and genomic GC content evolution in the miniature genome-sized family Lentibulariaceae. New Phytol. 2014, 203, 22–28. [Google Scholar] [CrossRef]
- Kondo, K. A Comparison of Variability in Utricularia cornuta and Utricularia juncea. Am. J. Bot. 1972, 59, 23. [Google Scholar] [CrossRef]
- Rahman, D.M.O.; Adamec, L.; Kondo, K. Chromosome numbers of Utricularia bremii and Utricularia dimorphantha (Lentibulariaceae). Chromosom. Sci. 2001, 5, 105–108. [Google Scholar]
- Casper, S.J.; Stimper, R. Chromosome numbers in Pinguicula (Lentibulariaceae): Survey, atlas, and taxonomic conclusions. Plant Syst. Evol. 2009, 277, 21–60. [Google Scholar] [CrossRef]
- Fleischmann, A.; Michael, T.P.; Rivadavia, F.; Sousa, A.; Wang, W.; Temsch, E.M.; Greilhuber, J.; Müller, K.F.; Heubl, G. Evolution of genome size and chromosome number in the carnivorous plant genus Genlisea (Lentibulariaceae), with a new estimate of the minimum genome size in angiosperms. Ann. Bot. 2014, 114, 1651–1663. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ibarra-Laclette, E.; Albert, V.A.; Herrera-Estrella, A.; Herrera-Estrella, L. Is GC bias in the nuclear genome of the carnivorous plant Utricularia driven by ros-based mutation and biased gene conversion? Plant Signal. Behav. 2011, 6, 1631–1634. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ibarra-laclette, E.; Albert, V.A.; Pérez-torres, C.A.; Zamudio-Hernández, F.; de Ortega-estrada, M.J.; Herrera-Estrella, A.; Herrera-estrella, L. Transcriptomics and molecular evolutionary rate analysis of the bladderwort (Utricularia), a carnivorous plant with a minimal genome. BMC Plant Biol. 2011, 11, 101. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Albert, V.A.; Jobson, R.W.; Michael, T.P.; Taylor, D.J. The carnivorous bladderwort (Utricularia, Lentibulariaceae): A system inflates. J. Exp. Bot. 2010, 61, 5–9. [Google Scholar] [CrossRef] [Green Version]
- Carretero-Paulet, L.; Chang, T.H.; Librado, P.; Ibarra-Laclette, E.; Herrera-Estrella, L.; Rozas, J.; Albert, V.A. Genome-wide analysis of adaptive molecular evolution in the carnivorous plant Utricularia gibba. Genome Biol. Evol. 2015, 7, 444–456. [Google Scholar] [CrossRef] [Green Version]
- Grover, C.E.; Wendel, J.F. Recent Insights into Mechanisms of Genome Size Change in Plants. J. Bot. 2010, 2010, 1–8. [Google Scholar] [CrossRef] [Green Version]
- Hakes, L.; Pinney, J.W.; Lovell, S.C.; Oliver, S.G.; Robertson, D.L. All duplicates are not equal: The difference between small-scale and genome duplication. Genome Biol. 2007, 8, R209. [Google Scholar] [CrossRef]
- Pellicer, J. Genome Size Diversity and Its Impact on the Evolution of Land Plants. Genes 2018, 9, 88. [Google Scholar] [CrossRef] [Green Version]
- Schatz, M.C.; Witkowski, J.; McCombie, W.R. Current challenges in de novo plant genome sequencing and assembly. Genome Biol. 2012, 13, 243. [Google Scholar] [CrossRef]
- Moriyama, Y.; Koshiba-Takeuchi, K. Significance of whole-genome duplications on the emergence of evolutionary novelties. Brief. Funct. Genom. 2018, 17, 329–338. [Google Scholar] [CrossRef]
- Bennetzen, J.L. Transposable element contributions to plant gene and genome evolution. Plant Mol. Biol. 2000, 42, 251–269. [Google Scholar] [CrossRef] [PubMed]
- Magallón, S.; Sánchez-Reyes, L.L.; Gómez-Acevedo, S.L. Thirty clues to the exceptional diversification of flowering plants. Ann. Bot. 2018, 123, 491–503. [Google Scholar] [CrossRef] [PubMed]
- BFG-The Brazil Flora Group Growing knowledge: An overview of Seed Plant diversity in Brazil. Rodriguésia 2015, 66, 1085–1113. [CrossRef]
- Clivati, D.; Gitzendanner, M.A.; Hilsdorf, A.W.S.; Araújo, W.L.; de Miranda, V.F.O. Microsatellite markers developed for Utricularia reniformis (Lentibulariaceae). Am. J. Bot. 2012, 99, 375–378. [Google Scholar] [CrossRef] [Green Version]
- Silva, S.R.; Alvarenga, D.O.; Aranguren, Y.; Penha, H.A.; Fernandes, C.C.; Pinheiro, D.G.; Oliveira, M.T.; Michael, T.P.; Miranda, V.F.O.; Varani, A.M. The mitochondrial genome of the terrestrial carnivorous plant Utricularia reniformis (Lentibulariaceae): Structure, comparative analysis and evolutionary landmarks. PLoS ONE 2017, 12, e0180484. [Google Scholar] [CrossRef] [Green Version]
- Silva, S.R.; Diaz, Y.C.A.; Penha, H.A.; Pinheiro, D.G.; Fernandes, C.C.; Miranda, V.F.O.; Michael, T.P.; Varani, A.M. The chloroplast genome of Utricularia reniformis sheds light on the evolution of the ndh gene complex of terrestrial carnivorous plants from the Lentibulariaceae family. PLoS ONE 2016, 11, e0165176. [Google Scholar] [CrossRef]
- Cheng, F.; Wu, J.; Cai, X.; Liang, J.; Freeling, M.; Wang, X. Gene retention, fractionation and subgenome differences in polyploid plants. Nat. Plants 2018, 4, 258. [Google Scholar] [CrossRef]
- Salojärvi, J.; Smolander, O.-P.; Nieminen, K.; Rajaraman, S.; Safronov, O.; Safdari, P.; Lamminmäki, A.; Immanen, J.; Lan, T.; Tanskanen, J.; et al. Genome sequencing and population genomic analyses provide insights into the adaptive landscape of silver birch. Nat. Genet. 2017, 49, 904–912. [Google Scholar] [CrossRef]
- Rendón-Anaya, M.; Ibarra-Laclette, E.; Méndez-Bravo, A.; Lan, T.; Zheng, C.; Carretero-Paulet, L.; Perez-Torres, C.A.; Chacón-López, A.; Hernandez-Guzmán, G.; Chang, T.-H.; et al. The avocado genome informs deep angiosperm phylogeny, highlights introgressive hybridization, and reveals pathogen-influenced gene space adaptation. Proc. Natl. Acad. Sci. USA 2019, 116, 17081–17089. [Google Scholar] [CrossRef] [Green Version]
- Wheeler, G.L.; Carstens, B.C. Evaluating the adaptive evolutionary convergence of carnivorous plant taxa through functional genomics. PeerJ 2018, 6, e4322. [Google Scholar] [CrossRef] [Green Version]
- Ravee, R.; Mohd Salleh, F.-. ’Imadi; Goh, H.H. Discovery of digestive enzymes in carnivorous plants with focus on proteases. PeerJ 2018, 6, e26940. [Google Scholar] [CrossRef] [PubMed]
- Hwang, J.-U.; Song, W.-Y.; Hong, D.; Ko, D.; Yamaoka, Y.; Jang, S.; Yim, S.; Lee, E.; Khare, D.; Kim, K.; et al. Plant ABC Transporters enable many unique aspects of a terrestrial plant’s lifestyle. Mol. Plant 2016, 9, 338–355. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jiao, Y.; Leebens-Mack, J.H.; Ayyampalayam, S.; Bowers, J.E.; McKain, M.R.; McNeal, J.; Rolf, M.; Ruzicka, D.R.; Wafula, E.K.; Wickett, N.J.; et al. A genome triplication associated with early diversification of the core eudicots. Genome Biol. 2012, 13, R3. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Feldman, M.; Levy, A.A. Allopolyploidy–A shaping force in the evolution of wheat genomes. Cytogenet. Genome Res. 2005, 109, 250–258. [Google Scholar] [CrossRef]
- Alix, K.; Gérard, P.R.; Schwarzacher, T.; Heslop-Harrison, J.S.P. Polyploidy and interspecific hybridization: Partners for adaptation, speciation and evolution in plants. Ann. Bot. 2017, 120, 183–194. [Google Scholar] [CrossRef]
- Clark, J.W.; Donoghue, P.C.J. Whole-genome duplication and plant macroevolution. Trends Plant Sci. 2018, 23, 933–945. [Google Scholar] [CrossRef] [Green Version]
- Bertioli, D.J.; Jenkins, J.; Clevenger, J.; Dudchenko, O.; Gao, D.; Seijo, G.; Leal-Bertioli, S.C.M.; Ren, L.; Farmer, A.D.; Pandey, M.K.; et al. The genome sequence of segmental allotetraploid peanut Arachis hypogaea. Nat. Genet. 2019, 51, 877–884. [Google Scholar] [CrossRef] [Green Version]
- Panchy, N.; Lehti-Shiu, M.; Shiu, S.-H. Evolution of Gene Duplication in Plants. Plant Physiol. 2016, 171, 2294–2316. [Google Scholar] [CrossRef] [Green Version]
- Fedoroff, N. Transposons and genome evolution in plants. Proc. Natl. Acad. Sci. USA 2000, 97, 7002–7007. [Google Scholar] [CrossRef] [Green Version]
- Bennetzen, J.L.; Wang, H. The Contributions of Transposable Elements to the Structure, Function, and Evolution of Plant Genomes. Annu. Rev. Plant Biol. 2014, 65, 505–530. [Google Scholar] [CrossRef]
- Serrato-Capuchina, A.; Matute, D.R. The role of transposable elements in speciation. Genes 2018, 9, 254. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dubin, M.J.; Mittelsten Scheid, O.; Becker, C. Transposons: A blessing curse. Curr. Opin. Plant Biol. 2018, 42, 23–29. [Google Scholar] [CrossRef] [PubMed]
- Kim, K.M.; Caetano-Anollés, G. The proteomic complexity and rise of the primordial ancestor of diversified life. BMC Evolut. Biol. 2011, 11, 140. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Willing, E.M.; Rawat, V.; Mandáková, T.; Maumus, F.; James, G.V.; Nordström, K.J.V.; Becker, C.; Warthmann, N.; Chica, C.; Szarzynska, B.; et al. Genome expansion of Arabis alpi. Nat. Plants 2015, 1, 1–7. [Google Scholar]
- Macas, J.; Neumann, P. Ogre elements-A distinct group of plant Ty3/gypsy-like retrotransposons. Gene 2007, 390, 108–116. [Google Scholar] [CrossRef]
- Neumann, P.; Navrátilová, A.; Koblí, A.; Kejnovský, E.; Eva, H.; Hobza, R.; Widmer, A.; Dole, J. Plant centromeric retrotransposons: A structural and cytogenetic perspective. Mob. DNA 2011, 1–16. [Google Scholar] [CrossRef] [Green Version]
- Jiang, N.; Bao, Z.; Temnykh, S.; Cheng, Z.; Jiang, J.; Wing, R.A.; McCouch, S.R.; Wessier, S.R. Dasheng: A recently amplified nonautonomous long terminal repeat element that is a major component of pericentromeric regions in rice. Genetics 2002, 161, 1293–1305. [Google Scholar]
- Kameyama, Y.; Toyama, M.; Ohara, M. Hybrid origins and F1 dominance in the free-floating, sterile bladderwort, Utricularia australis F. australis (Lentibulariaceae). Am. J. Bot. 2005, 92, 469–476. [Google Scholar] [CrossRef]
- Astuti, G.; Peruzzi, L. Are shoots of diagnostic value in Central European bladderworts (Utricularia L., Lentibulariaceae)? Plant Biosyst. 2018, 152, 1214–1226. [Google Scholar] [CrossRef]
- Clivati, D.; Cordeiro, G.D.; Płachno, B.J.; de Miranda, V.F.O. Reproductive biology and pollination of Utricularia reniformis A.St.-Hil. (Lentibulariaceae). Plant Biol. 2014, 16, 677–682. [Google Scholar] [CrossRef]
- Rutishauser, R.; Isler, B. Developmental genetics and morphological evolution of flowering plants, especially bladderworts (Utricularia): Fuzzy Arberian Morphology complements Classical Morphology. Ann. Bot. 2001, 88, 1173–1202. [Google Scholar] [CrossRef] [Green Version]
- Fineran, B.A. Glandular trichomes in Utricularia—A review of their structure and function. Isr. J. Bot. 1985, 34, 295–330. [Google Scholar]
- Płachno, B.J.; Adamec, L.; Kamińska, I. Relationship between trap anatomy and function in Australian carnivorous bladderworts (Utricularia) of the subgenus Polypompholyx. Aquat. Bot. 2015, 120, 290–296. [Google Scholar] [CrossRef]
- Płachno, B.J.; Stpiczyńska, M.; Krajewski, Ł.; Świątek, P.; Adamec, L.; Miranda, V.F.O. Flower palate structure of the aquatic bladderworts Utricularia bremii Heer and U. minor L. from section Utricularia (Lentibulariaceae). Protoplasma 2017, 254, 2007–2015. [Google Scholar] [CrossRef] [Green Version]
- Jobson, R.W.; Morris, E.C. Feeding ecology of a carnivorous bladderwort (Utricularia uliginosa, Lentibulariaceae). Austral Ecol. 2001, 26, 680–691. [Google Scholar] [CrossRef]
- Reifenrath, K.; Theisen, I.; Schnitzler, J.; Porembski, S.; Barthlott, W. Trap architecture in carnivorous Utricularia (Lentibulariaceae). Flora Morphol. Distrib. Funct. Ecol. Plants 2006, 201, 597–605. [Google Scholar] [CrossRef]
- Sanabria-Aranda, L.; González-Bermúdez, A.; Torres, N.N.; Guisande, C.; Manjarrés-Hernández, A.; Valoyes-Valois, V.; Díaz-Olarte, J.; Andrade-Sossa, C.; Duque, S.R. Predation by the tropical plant Utricularia foliosa. Freshw. Biol. 2006, 51, 1999–2008. [Google Scholar] [CrossRef]
- Rajasekar, C.; Rajendran, A. Prey composition of Utricularia striatula Sm. (Lentibulariaceae): Lithophytic carnivore Southern Western Ghats, India. Int. J. Fish. Aquat. Stud. 2018, 6, 382–388. [Google Scholar]
- Elhiti, M.; Stasolla, C. Structure and function of homodomain-leucine zipper (HD-Zip) proteins. Plant Signal. Behav. 2009, 4, 86. [Google Scholar] [CrossRef] [Green Version]
- Lian, G.; Ding, Z.; Wang, Q.; Zhang, D.; Xu, J. Origins and Evolution of WUSCHEL-Related Homeobox Protein Family in Plant Kingdom. Sci. World J. 2014, 2014, 1–12. [Google Scholar] [CrossRef] [Green Version]
- Messenguy, F.; Dubois, E. Role of MADS box proteins and their cofactors in combinatorial control of gene expression and cell development. Gene 2003, 316, 1–21. [Google Scholar] [CrossRef]
- Jack, T. Molecular and genetic mechanisms of floral control. Plant Cell 2004, 16, S1–S17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, J.; Ding, C.; Xu, B.; Chen, C.; Narsai, R.; Whelan, J.; Hu, Z.; Zhang, M. A Casparian strip domain-like gene, CASPL, negatively alters growth and cold tolerance. Sci. Rep. 2015, 5, 14299. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cho, M.; Cho, H.-T. The function of ABCB transporters in auxin transport. Plant Signal. Behav. 2013, 8, e22990. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Teschner, J.; Lachmann, N.; Schulze, J.; Geisler, M.; Selbach, K.; Santamaria-Araujo, J.; Balk, J.; Mendel, R.R.; Bittner, F. A Novel Role for Arabidopsis Mitochondrial ABC Transporter ATM3 in Molybdenum Cofactor Biosynthesis. Plant Cell 2010, 22, 468–480. [Google Scholar] [CrossRef] [Green Version]
- Stein, D.J.; Chamberlain, S.R.; Fineberg, N. An A-B-C Model of Habit Disorders: Hair-Pulling, Skin-Picking, and Other Stereotypic Conditions. CNS Spectr. 2006, 11, 824–827. [Google Scholar] [CrossRef] [Green Version]
- Clay, N.K.; Adio, A.M.; Denoux, C.; Jander, G.; Ausubel, F.M. Glucosinolate metabolites required for an Arabidopsis innate immune response. Science 2009, 323, 95–101. [Google Scholar] [CrossRef] [Green Version]
- Juniper, B.E.; Robins, R.J.; Joel, D.M. The Carnivorous Plants; Academic Press: London, UK; San Diego, CA, USA, 1989; ISBN 978-0-12-392170-3. [Google Scholar]
- Brownlee, C. Carnivorous plants: Trapping, digesting and absorbing all in one. Curr. Biol. 2013, 23, R714–R716. [Google Scholar] [CrossRef] [Green Version]
- Sirová, D.; Bárta, J.; Šimek, K.; Posch, T.; Pech, J.; Stone, J.; Borovec, J.; Adamec, L.; Vrba, J. Hunters or farmers? Microbiome characteristics help elucidate the diet composition in an aquatic carnivorous plant. Microbiome 2018, 6, 225. [Google Scholar] [CrossRef] [Green Version]
- Mette, N.; Wilbert, N.; Barthlott, W. Food composition of Aquatic Bladderworts (Utricularia, Lentibulariaceae) in Various Habiats. Beiträge Biol. Pflanz. 2000, 72, 1–13. [Google Scholar]
- Kurbatova, S.A.; Yershov, I.Y. Crustaceans and Rotifers in the Predatory Feeding of Utricularia. Inland Water Biol. 2009, 2, 271–275. [Google Scholar] [CrossRef]
- Harms, S. Prey selection in three species of the carnivorous aquatic plant Utricularia (bladderwort). Arch. Hydrobiol. 1999, 146, 449–470. [Google Scholar] [CrossRef]
- Ebihara, A.; Ishikawa, H.; Matsumoto, S.; Lin, S.-J.; Iwatsuki, K.; Takamiya, M.; Watano, Y.; Ito, M. Nuclear DNA, chloroplast DNA, and ploidy analysis clarified biological complexity of the Vandenboschia radicans complex (Hymenophyllaceae) in Japan and adjacent areas. Am. J. Bot. 2005, 92, 1535–1547. [Google Scholar] [CrossRef] [PubMed]
- Doležel, J.; Sgorbati, S.; Lucretti, S. Comparison of three DNA fluorochromes for flow cytometric estimation of nuclear DNA content in plants. Physiol. Plant. 1992, 85, 625–631. [Google Scholar] [CrossRef]
- Pellicer, J.; Leitch, I.J. The application of flow cytometry for estimating genome size and ploidy level in plants. In Molecular Plant Taxonomy. Methods in Molecular Biology (Methods and Protocols); Bresse, P., Ed.; Humana Press: Totowa, NJ, USA, 2014; pp. 279–307. ISBN 9781627037662. [Google Scholar]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef] [Green Version]
- O’Connell, J.; Schulz-Trieglaff, O.; Carlson, E.; Hims, M.M.; Gormley, N.A.; Cox, A.J. NxTrim: Optimized trimming of Illumina mate pair reads. Bioinformatics 2015, 31, 2035–2037. [Google Scholar] [CrossRef] [Green Version]
- Grabherr, M.G.; Haas, B.J.; Yassour, M.; Levin, J.Z.; Thompson, D.A.; Amit, I.; Adiconis, X.; Fan, L.; Raychowdhury, R.; Zeng, Q.; et al. Full-length transcriptome assembly from RNA-Seq data without a reference genome. Nat. Biotechnol. 2011, 29, 644–652. [Google Scholar] [CrossRef] [Green Version]
- Langmead, B.; Salzberg, S.L. Fast gapped-read alignment with Bowtie 2. Nat. Methods 2012, 9, 357–359. [Google Scholar] [CrossRef] [Green Version]
- Zimin, A.V.; Marçais, G.; Puiu, D.; Roberts, M.; Salzberg, S.L.; Yorke, J.A. The MaSuRCA genome assembler. Bioinformatics 2013, 29, 2669–2677. [Google Scholar] [CrossRef] [Green Version]
- Boetzer, M.; Henkel, C.V.; Jansen, H.J.; Butler, D.; Pirovano, W. Scaffolding pre-assembled contigs using SSPACE. Bioinformatics 2011, 27, 578–579. [Google Scholar] [CrossRef] [Green Version]
- Luo, R.; Liu, B.; Xie, Y.; Li, Z.; Huang, W.; Yuan, J.; He, G.; Chen, Y.; Pan, Q.; Liu, Y.; et al. SOAPdenovo2: An empirically improved memory-efficient short-read de novo assembler. Gigascience 2012, 1, 18. [Google Scholar] [CrossRef] [PubMed]
- Hunt, M.; Kikuchi, T.; Sanders, M.; Newbold, C.; Berriman, M.; Otto, T.D. REAPR: A universal tool for genome assembly evaluation. Genome Biol. 2013, 14, R47. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Parra, G.; Bradnam, K.; Korf, I. CEGMA: A pipeline to accurately annotate core genes in eukaryotic genomes. Bioinformatics 2007, 23, 1061–1067. [Google Scholar] [CrossRef] [PubMed]
- Simão, F.A.; Waterhouse, R.M.; Ioannidis, P.; Kriventseva, E.V.; Zdobnov, E.M. BUSCO: Assessing genome assembly and annotation completeness with single-copy orthologs. Bioinformatics 2015, 31, 3210–3212. [Google Scholar] [CrossRef] [Green Version]
- Kriventseva, E.V.; Kuznetsov, D.; Tegenfeldt, F.; Manni, M.; Dias, R.; Simão, F.A.; Zdobnov, E.M. OrthoDB v10: Sampling the diversity of animal, plant, fungal, protist, bacterial and viral genomes for evolutionary and functional annotations of orthologs. Nucleic Acids Res. 2019, 47, D807–D811. [Google Scholar] [CrossRef] [Green Version]
- Vurture, G.W.; Sedlazeck, F.J.; Nattestad, M.; Underwood, C.J.; Fang, H.; Gurtowski, J.; Schatz, M.C. GenomeScope: Fast reference-free genome profiling from short reads. Bioinformatics 2017, 33, 2202–2204. [Google Scholar] [CrossRef] [Green Version]
- Mapleson, D.; Garcia Accinelli, G.; Kettleborough, G.; Wright, J.; Clavijo, B.J. KAT: A K-mer analysis toolkit to quality control NGS datasets and genome assemblies. Bioinformatics 2017, 33, 574–576. [Google Scholar] [CrossRef]
- Gnerre, S.; MacCallum, I.; Przybylski, D.; Ribeiro, F.J.; Burton, J.N.; Walker, B.J.; Sharpe, T.; Hall, G.; Shea, T.P.; Sykes, S.; et al. High-quality draft assemblies of mammalian genomes from massively parallel sequence data. Proc. Natl. Acad. Sci. USA 2011, 108, 1513–1518. [Google Scholar] [CrossRef] [Green Version]
- Haug-Baltzell, A.; Stephens, S.A.; Davey, S.; Scheidegger, C.E.; Lyons, E. SynMap2 and SynMap3D: Web-based whole-genome synteny browsers. Bioinformatics 2017, 33, 2197–2198. [Google Scholar] [CrossRef]
- Wang, Y.; Tang, H.; Debarry, J.D.; Tan, X.; Li, J.; Wang, X.; Lee, T.; Jin, H.; Marler, B.; Guo, H.; et al. MCScanX: A toolkit for detection and evolutionary analysis of gene synteny and collinearity. Nucleic Acids Res. 2012, 40, e49. [Google Scholar] [CrossRef] [Green Version]
- Cabanettes, F.; Klopp, C. D-GENIES: Dot plot large genomes in an interactive, efficient and simple way. PeerJ 2018, 6, e4958. [Google Scholar] [CrossRef] [PubMed]
- Flutre, T.; Duprat, E.; Feuillet, C.; Quesneville, H. Considering Transposable Element Diversification in De Novo Annotation Approaches. PLoS ONE 2011, 6, e16526. [Google Scholar] [CrossRef] [PubMed]
- Neumann, P.; Novák, P.; Ho, N. Systematic survey of plant LTR- retrotransposons elucidates phylogenetic relationships of their polyprotein domains and provides a reference for element classification. Mob. DNA 2019, 10, 1–17. [Google Scholar] [CrossRef] [PubMed]
- Hoff, K.; Lomsadze, A.; Borodovsky, M.; Stanke, M. Whole-Genome Annotation with BRAKER. In Gene Prediction. Methods in Molecular Biology; Kollmar, M., Ed.; Humana: New York, NY, USA, 2019; pp. 65–95. [Google Scholar]
- Haas, B.J.; Delcher, A.L.; Mount, S.M.; Wortman, J.R.; Smith, R.K.S., Jr.; Hannick, L.I.; Maiti, R.; Ronning, C.M.; Rusch, D.B.; Town, C.D.; et al. Improving the Arabidopsis genome annotation using maximal transcript alignment assemblies. Nucleic Acids Res. 2003, 31, 5654–5666. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Haas, B.J.; Salzberg, S.L.; Zhu, W.; Pertea, M.; Allen, J.E.; Orvis, J.; White, O.; Buell, C.R.; Wortman, J.R. Automated eukaryotic gene structure annotation using EVidenceModeler and the program to assemble spliced alignments. Genome Biol. 2008, 9, R7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Conesa, A.; Götz, S. Blast2GO: A comprehensive suite for functional analysis in plant genomics. Int. J. Plant Genom. 2008, 2008, 619832. [Google Scholar] [CrossRef]
- Jones, P.; Binns, D.; Chang, H.-Y.; Fraser, M.; Li, W.; McAnulla, C.; McWilliam, H.; Maslen, J.; Mitchell, A.; Nuka, G.; et al. InterProScan 5: Genome-scale protein function classification. Bioinformatics 2014, 30, 1236–1240. [Google Scholar] [CrossRef] [Green Version]
- Huerta-Cepas, J.; Forslund, K.; Coelho, L.P.; Szklarczyk, D.; Jensen, L.J.; von Mering, C.; Bork, P. Fast genome-wide functional annotation through orthology massignment by eggNOG-Mapper. Mol. Biol. Evol. 2017, 34, 2115–2122. [Google Scholar] [CrossRef] [Green Version]
- Pundir, S.; Magrane, M.; Martin, M.J.; O’Donovan, C. Searching and navigating UniProt databases. Curr. Protoc. Bioinform. 2015, 50, 1–27. [Google Scholar] [CrossRef] [Green Version]
- Stamatakis, A. RAxML version 8: A tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics 2014, 30, 1312–1313. [Google Scholar] [CrossRef]
- Ronquist, F.; Teslenko, M.; Van Der Mark, P.; Ayres, D.L.; Darling, A.; Höhna, S.; Larget, B.; Liu, L.; Suchard, M.A.; Huelsenbeck, J.P. Mrbayes 3.2: Efficient bayesian phylogenetic inference and model choice across a large model space. Syst. Biol. 2012, 61, 539–542. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Darriba, D.; Taboada, G.L.; Doallo, R.; Posada, D. JModelTest 2: More models, new heuristics and parallel computing. Nat. Methods 2012, 9, 772. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, L.; Dong, Z.; Fang, L.; Luo, Y.; Wei, Z.; Guo, H.; Zhang, G.; Gu, Y.Q.; Coleman-Derr, D.; Xia, Q.; et al. OrthoVenn2: A web server for whole-genome comparison and annotation of orthologous clusters across multiple species. Nucleic Acids Res. 2019, 47, W52–W58. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goodstein, D.M.; Shu, S.; Howson, R.; Neupane, R.; Hayes, R.D.; Fazo, J.; Mitros, T.; Dirks, W.; Hellsten, U.; Putnam, N.; et al. Phytozome: A comparative platform for green plant genomics. Nucleic Acids Res. 2012, 40, 1178–1186. [Google Scholar] [CrossRef]
- Klopfenstein, D.V.; Zhang, L.; Pedersen, B.S.; Ramírez, F.; Warwick Vesztrocy, A.; Naldi, A.; Mungall, C.J.; Yunes, J.M.; Botvinnik, O.; Weigel, M.; et al. GOATOOLS: A Python library for Gene Ontology analyses. Sci. Rep. 2018, 8, 10872. [Google Scholar] [CrossRef]
- Consortium, T.U. UniProt: A worldwide hub of protein knowledge. Nucleic Acids Res. 2018, 47, D506–D515. [Google Scholar] [CrossRef] [Green Version]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Technology | Library | Read Length | Raw Reads | Trimmed Reads | |
|---|---|---|---|---|---|
| DNAseq | Illumina HiScan and MiSeq | Paired-end (~350 bp) | 100 bp | 285,403,944 | 187,288,003 |
| Paired-end (~450 bp) | 300 bp | 49,185,074 | 35,963,241 | ||
| Mate-paired (3–9 Kb) | 100 bp | 177,044,066 | 60,011,867 | ||
| RNAseq | Ion Proton | Single-end - Leaves - Stolon - Utricules (preys) | ~200 bp | 46,622,745 41,894,450 112,414,083 | 40,853,284 34,716,966 97,821,388 |
| U. reniformis Genome | ||
|---|---|---|
| Total size of scaffolds (bp) | 304,550,249 | |
| Number of scaffolds | 1830 | |
| Number of contigs | 5452 | |
| Useful amount of scaffold sequences (≥25-Kb) | 297,419,257 | |
| % of assembled genome that is useful | 93.8% | |
| Longest scaffold (bp) | 1,862,935 | |
| Longest contig (bp) | 926,419 | |
| Scaffolds longer than 1-Kb | 1830 (100%) | |
| Scaffolds longer than 100-Kb | 688 (37.6%) | |
| Scaffolds longer than 1 Mb | 47 (2.6%) | |
| NG50 scaffold length (bp) | 466,988 | |
| LG50 scaffold count | 196 | |
| N50 contig length (bp) | 161,226 | |
| Percentage of assembly in scaffolded contigs | 91 | |
| Gaps number | 3677 | |
| Unknown bases (Ns) (bp) | 5,790,542 | |
| Average gap size (bp)/Longest gap (bp) | 1575 | 10,802 |
| U. reniformis | U. gibba | |
|---|---|---|
| Total number of identified genes | 42,582 | 25,509 |
| - Singletons | 3083 | 5120 |
| - Dispersed duplicates | 26,546 | 8831 |
| - Proximal duplicates | 1679 | 683 |
| - Tandem duplicates | 2994 | 999 |
| - Segmental duplicates | 8280 | 9876 |
| Annotation status | ||
| - Annotated genes † | 35,899 | 21,283 |
| - Genes with GOs | 27,751 | 17,760 |
| - Unknown and Hypothetical genes | 6683 | 4348 |
| Total gene length (bp) | 79,419,967 | 51,346,373 |
| Total exon length (bp) | 44,971,966 | 28,891,859 |
| Total intron length (bp) | 29,871,202 | 16,875,684 |
| Longest gene (bp) | 84,570 | 61,898 |
| Longest exon (bp) | 7309 | 6002 |
| Longest intron (bp) | 78,774 | 53,378 |
| Longest CDS (bp) | 15,201 | 15,801 |
| Mean gene length (bp) | 1872 | 2016 |
| Mean exons per gene | 5 | 5 |
| % of genome covered by genes | 26 | 51 |
| % of genome covered by CDS | 15 | 38 |
| % of genome covered by TEs-like regions | 56 | 32 |
| Gene Ontology Term | GO Code | U. renif n# Genes | U. gibba n# Genes |
|---|---|---|---|
| amidase activity | GO:0004040 | 13 | 10 |
| actin filament | GO:0005884 | 20 | 11 |
| alpha-galactosidase activity | GO:0004557 | 15 | 13 |
| alternative oxidase activity | GO:0009916 | 6 | 4 |
| ammonium transmembrane transport | GO:0008519; 0072488 | 18 | 16 |
| aspartic-type endopeptidase activity | GO:0004190 | 289 | 94 |
| ATP:ADP antiporter activity | GO:0005471 | 9 | 9 |
| ATPase activity | GO:0016887 | 1831 | 792 |
| beta-galactosidase activity | GO:0004565 | 720 | 125 |
| catalase activity | GO:0004096 | 17 | 11 |
| cellulase activity | GO:0008810 | 62 | 34 |
| chitinase activity | GO:0004568 | 21 | 13 |
| cinnamyl-alcohol dehydrogenase activity | GO:0045551 | 34 | 15 |
| cyclic-nucleotide phosphodiesterase activity | GO:0004112 | 1 | 1 |
| cysteine-type peptidase activity | GO:0008234 | 384 | 185 |
| nuclease activity | GO:0004518 | 1955 | 1094 |
| fructose-bisphosphate aldolase activity | GO:0004332 | 12 | 10 |
| glutathione transferase activity | GO:0004364 | 54 | 21 |
| glutathione peroxidase activity | GO:0004602 | 14 | 9 |
| hydrolase activity, acting on ester bonds | GO:0016788 | 2767 | 1259 |
| heat shock protein activity | GO:0042026; 0006986; 0034620 | 129 | 72 |
| lipase activity | GO:0016298 | 231 | 140 |
| lipid transport | GO:0006869 | 170 | 113 |
| myosin heavy chain kinase activity | GO:0016905 | 36 | 23 |
| peroxidase activity | GO:0004601 | 192 | 140 |
| peptidase activity | GO:0008233 | 2650 | 1269 |
| phosphatase activity | GO:0016791 | 287 | 186 |
| phospholipase activity | GO:0004620 | 80 | 43 |
| polygalacturonase activity | GO:0004650 | 109 | 50 |
| polygalacturonase inhibitor activity | GO:0090353 | 5 | 1 |
| protein homodimerization activity | GO:0042803 | 809 | 525 |
| ribonuclease activity | GO:0004540 | 390 | 154 |
| serine-type carboxypeptidase activity | GO:0004185 | 82 | 38 |
| superoxide dismutase activity | GO:0004784 | 17 | 10 |
| symplast | GO:0055044 | 1060 | 679 |
| urease activity | GO:0009039 | 2 | 1 |
| water channel activity | GO:0015250 | 46 | 26 |
| Gene Ontology Term | GO Code | U. renif n# Genes | U. gibba n# Genes |
|---|---|---|---|
| developmental process involved in reproduction | GO:0003006 | 1965 | 1237 |
| reproduction | GO:0000003 | 2546 | 1600 |
| reproductive process | GO:0022414 | 2223 | 1388 |
| multicellular organism development | GO:0007275 | 4749 | 2773 |
| embryo development | GO:0009790 | 744 | 465 |
| post-embryonic development | GO:0009791 | 2447 | 1578 |
| flower development | GO:0009908 | 915 | 601 |
| developmental maturation | GO:0021700 | 264 | 171 |
| developmental process | GO:0032502 | 4126 | 2704 |
| reproductive structure development | GO:0048608 | 1685 | 1065 |
| anatomical structure development | GO:0048856 | 4046 | 2639 |
| cellular developmental process | GO:0048869 | 1352 | 880 |
| reproductive shoot system development | GO:0090567 | 655 | 437 |
| pollination | GO:0009856 | 343 | 210 |
| tropism | GO:0009606 | 215 | 129 |
| circadian rhythm | GO:0007623 | 261 | 161 |
| response to stress | GO:0006950 | 4513 | 2869 |
| response to radiation and light stimulus | GO:0009314, GO:0009416 | 1012 | 711 |
| response to external stimulus | GO:0009605 | 2108 | 1352 |
| response to biotic stimulus | GO:0009607 | 1526 | 981 |
| response to abiotic stimulus | GO:0009628 | 3161 | 2041 |
| response to endogenous stimulus | GO:0009719 | 2665 | 1693 |
| response to chemical | GO:0042221 | 4590 | 2984 |
| response to stimulus | GO:0050896 | 6579 | 4277 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Silva, S.R.; Moraes, A.P.; Penha, H.A.; Julião, M.H.M.; Domingues, D.S.; Michael, T.P.; Miranda, V.F.O.; Varani, A.M. The Terrestrial Carnivorous Plant Utricularia reniformis Sheds Light on Environmental and Life-Form Genome Plasticity. Int. J. Mol. Sci. 2020, 21, 3. https://doi.org/10.3390/ijms21010003
Silva SR, Moraes AP, Penha HA, Julião MHM, Domingues DS, Michael TP, Miranda VFO, Varani AM. The Terrestrial Carnivorous Plant Utricularia reniformis Sheds Light on Environmental and Life-Form Genome Plasticity. International Journal of Molecular Sciences. 2020; 21(1):3. https://doi.org/10.3390/ijms21010003
Chicago/Turabian StyleSilva, Saura R., Ana Paula Moraes, Helen A. Penha, Maria H. M. Julião, Douglas S. Domingues, Todd P. Michael, Vitor F. O. Miranda, and Alessandro M. Varani. 2020. "The Terrestrial Carnivorous Plant Utricularia reniformis Sheds Light on Environmental and Life-Form Genome Plasticity" International Journal of Molecular Sciences 21, no. 1: 3. https://doi.org/10.3390/ijms21010003
APA StyleSilva, S. R., Moraes, A. P., Penha, H. A., Julião, M. H. M., Domingues, D. S., Michael, T. P., Miranda, V. F. O., & Varani, A. M. (2020). The Terrestrial Carnivorous Plant Utricularia reniformis Sheds Light on Environmental and Life-Form Genome Plasticity. International Journal of Molecular Sciences, 21(1), 3. https://doi.org/10.3390/ijms21010003