Mediation Analysis Supports a Causal Relationship between Maternal Hyperglycemia and Placental DNA Methylation Variations at the Leptin Gene Locus and Cord Blood Leptin Levels

Abstract
:1. Introduction
2. Results
2.1. Sample Characteristics
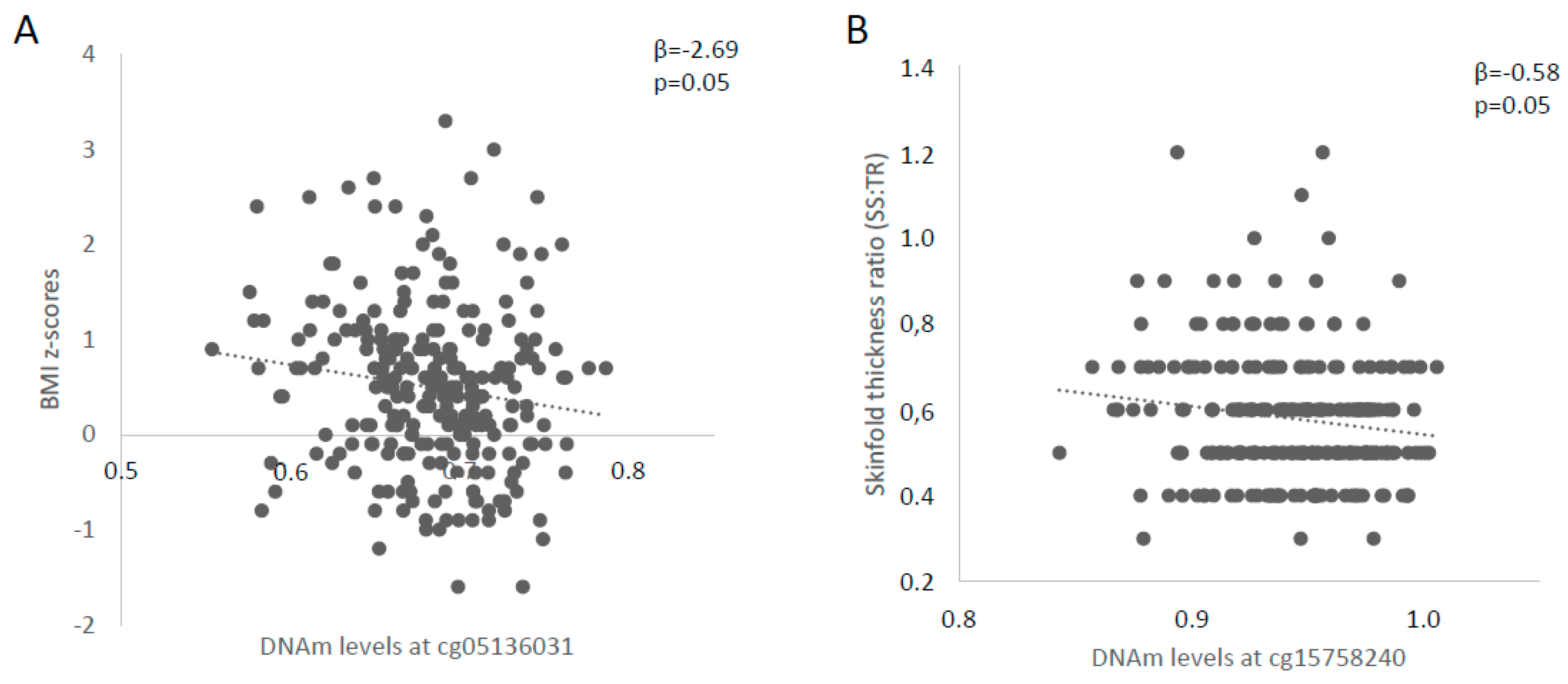
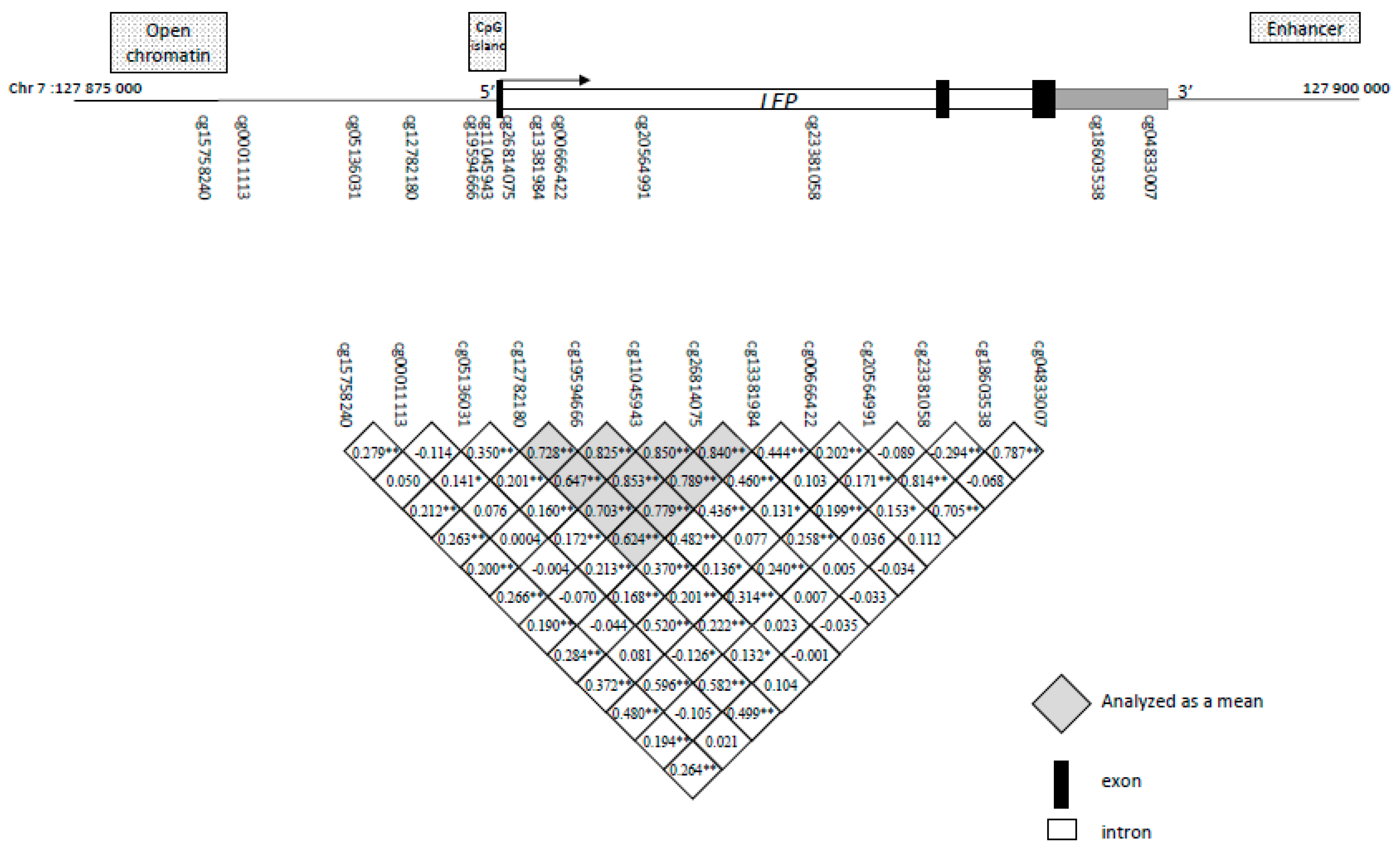
2.2. Identification of Potential Functional LEP 5′-C-phosphate-G-3′ (CpG) Sites and Association with Childhood Anthropometric Profile
2.3. Investigation of Placental DNAm Levels as Mediating Factor between Maternal Glycemia and Adiposity Markers in Offspring
3. Methods
3.1. Participants’ Selection from Gen3G Cohort
3.2. Gen3G Follow-Up During Pregnancy
3.3. Sampling and Measures at Birth
3.4. Clinical Evaluation of Children
3.5. DNA Extraction and Sample Preparation
3.6. Measurements of DNA Methylation
3.7. Statistical Analyses
3.8. Mediation Analysis
4. Discussion
Strengths and Limitations
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| BMI-z | Body mass index z-scores |
| DNAm | DNA methylation |
| CpG | 5′-C-phosphate-G-3′ |
| DNMT | DNA methyltransferase |
| DOHaD | Developmental origins of health and diseases |
| GCT | Glucose challenge test |
| GDM | Gestational diabetes mellitus |
| Gen3G | Genetics of Glucose regulation in Gestation and Growth |
| IQR | Interquartile range |
| LEP | Leptin |
| OGTT | Oral glucose tolerance test |
| RCP | Regression on correlated probes |
| RIN | RNA integrity number |
| SS | Suprascapular |
| ST | Skinfold Thickness |
| TR | Tricipital |
References
- WHO. Report of the Commission on Ending Childhood Obesity: Implementation Plan: Executive Summary; World Health Organization: Geneva, Switzerland, 2017. [Google Scholar]
- Sahoo, K.; Sahoo, B.; Choudhury, A.K.; Sofi, N.Y.; Kumar, R.; Bhadoria, A.S. Childhood obesity: Causes and consequences. J. Fam. Med. Prim. Care 2015, 4, 187–192. [Google Scholar]
- Deshmukh-Taskar, P.; Nicklas, T.A.; Morales, M.; Yang, S.J.; Zakeri, I.; Berenson, G.S. Tracking of overweight status from childhood to young adulthood: The Bogalusa Heart Study. Eur. J. Clin. Nutr. 2006, 60, 48–57. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ma, R.C.W.; Popkin, B.M. Intergenerational diabetes and obesity—A cycle to break? PLoS Med. 2017, 14, e1002415. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barker, D.J.; Osmond, C.; Kajantie, E.; Eriksson, J.G. Growth and chronic disease: Findings in the Helsinki Birth Cohort. Ann. Hum. Biol. 2009, 36, 445–458. [Google Scholar] [CrossRef]
- Barker, D.J. The developmental origins of chronic adult disease. Acta Paediatr. Suppl. 2004, 93, 26–33. [Google Scholar] [CrossRef]
- Barker, D.J. The developmental origins of adult disease. J. Am. Coll. Nutr. 2004, 23, 588S–595S. [Google Scholar] [CrossRef]
- Barker, D.J. Developmental origins of adult health and disease. J. Epidemiol. Community Health 2004, 58, 114–115. [Google Scholar] [CrossRef] [Green Version]
- Barker, D.J. The developmental origins of insulin resistance. Horm. Res. 2005, 64, 2–7. [Google Scholar] [CrossRef]
- Foley, D.L.; Craig, J.M.; Morley, R.; Olsson, C.A.; Dwyer, T.; Smith, K.; Saffery, R. Prospects for epigenetic epidemiology. Am. J. Epidemiol. 2009, 169, 389–400. [Google Scholar] [CrossRef] [Green Version]
- Dolinoy, D.C.; Weidman, J.R.; Waterland, R.A.; Jirtle, R.L. Maternal genistein alters coat color and protects Avy mouse offspring from obesity by modifying the fetal epigenome. Environ. Health Perspect. 2006, 114, 567–572. [Google Scholar] [CrossRef] [Green Version]
- Garcia-Cardona, M.C.; Huang, F.; Garcia-Vivas, J.M.; Lopez-Camarillo, C.; Del Rio Navarro, B.E.; Navarro Olivos, E.; Hong-Chong, E.; Bolanos-Jimenez, F.; Marchat, L.A. DNA methylation of leptin and adiponectin promoters in children is reduced by the combined presence of obesity and insulin resistance. Int. J. Obes. 2014, 38, 1457–1465. [Google Scholar] [CrossRef] [PubMed]
- Xia, L.; Wang, C.; Lu, Y.; Fan, C.; Ding, X.; Fu, H.; Qi, K. Time-specific changes in DNA methyltransferases associated with the leptin promoter during the development of obesity. Nutr. Hosp. 2014, 30, 1248–1255. [Google Scholar] [PubMed]
- Yang, M.; Sun, J.Z.; Sun, Y.L.; You, W.; Dai, J.; Li, G.S. Association between leptin gene promoter methylation and type 2 diabetes mellitus (Article in Chinese). Zhonghua Yi Xue Yi Chuan Xue Za Zhi 2012, 29, 474–477. [Google Scholar] [PubMed]
- Maccani, M.A.; Marsit, C.J. Epigenetics in the placenta. Am. J. Reprod. Immunol. 2009, 62, 78–89. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jansson, T.; Powell, T.L. Role of the placenta in fetal programming: Underlying mechanisms and potential interventional approaches. Clin. Sci. 2007, 113, 1–13. [Google Scholar] [CrossRef] [Green Version]
- Sood, R.; Zehnder, J.L.; Druzin, M.L.; Brown, P.O. Gene expression patterns in human placenta. Proc. Natl. Acad. Sci. USA 2006, 103, 5478–5483. [Google Scholar] [CrossRef] [Green Version]
- Zhou, Y.; Rui, L. Leptin signaling and leptin resistance. Front. Med. 2013, 7, 207–222. [Google Scholar] [CrossRef]
- Morris, D.L.; Cho, K.W.; Rui, L. Critical role of the Src homology 2 (SH2) domain of neuronal SH2B1 in the regulation of body weight and glucose homeostasis in mice. Endocrinology 2010, 151, 3643–3651. [Google Scholar] [CrossRef]
- Coppari, R.; Bjorbaek, C. Leptin revisited: Its mechanism of action and potential for treating diabetes. Nat. Rev. Drug Discov. 2012, 11, 692–708. [Google Scholar] [CrossRef] [Green Version]
- Allard, C.; Desgagne, V.; Patenaude, J.; Lacroix, M.; Guillemette, L.; Battista, M.C.; Doyon, M.; Menard, J.; Ardilouze, J.L.; Perron, P.; et al. Mendelian randomization supports causality between maternal hyperglycemia and epigenetic regulation of leptin gene in newborns. Epigenetics 2015, 10, 342–351. [Google Scholar] [CrossRef] [Green Version]
- Guillemette, L.; Allard, C.; Lacroix, M.; Patenaude, J.; Battista, M.C.; Doyon, M.; Moreau, J.; Menard, J.; Bouchard, L.; Ardilouze, J.L.; et al. Genetics of Glucose regulation in Gestation and Growth (Gen3G): A prospective prebirth cohort of mother-child pairs in Sherbrooke, Canada. BMJ Open 2016, 6, e010031. [Google Scholar] [CrossRef]
- Cardenas, A.; Gagne-Ouellet, V.; Allard, C.; Brisson, D.; Perron, P.; Bouchard, L.; Hivert, M.F. Placental DNA Methylation Adaptation to Maternal Glycemic Response in Pregnancy. Diabetes 2018, 67, 1673–1683. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pidsley, R.; Zotenko, E.; Peters, T.J.; Lawrence, M.G.; Risbridger, G.P.; Molloy, P.; Van Djik, S.; Muhlhausler, B.; Stirzaker, C.; Clark, S.J. Critical evaluation of the Illumina MethylationEPIC BeadChip microarray for whole-genome DNA methylation profiling. Genome Biol. 2016, 17, 208. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fortin, J.P.; Labbe, A.; Lemire, M.; Zanke, B.W.; Hudson, T.J.; Fertig, E.J.; Greenwood, C.M.; Hansen, K.D. Functional normalization of 450k methylation array data improves replication in large cancer studies. Genome Biol. 2014, 15, 503. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Johnson, W.E.; Li, C.; Rabinovic, A. Adjusting batch effects in microarray expression data using empirical Bayes methods. Biostatistics 2007, 8, 118–127. [Google Scholar] [CrossRef] [PubMed]
- Logue, M.W.; Smith, A.K.; Wolf, E.J.; Maniates, H.; Stone, A.; Schichman, S.A.; McGlinchey, R.E.; Milberg, W.; Miller, M.W. The correlation of methylation levels measured using Illumina 450K and EPIC BeadChips in blood samples. Epigenomics 2017, 9, 1363–1371. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rahmani, E.; Zaitlen, N.; Baran, Y.; Eng, C.; Hu, D.; Galanter, J.; Oh, S.; Burchard, E.G.; Eskin, E.; Zou, J.; et al. Sparse PCA corrects for cell type heterogeneity in epigenome-wide association studies. Nat. Methods 2016, 13, 443–445. [Google Scholar] [CrossRef] [Green Version]
- Hayes, A.F.; Rockwood, N.J. Regression-based statistical mediation and moderation analysis in clinical research: Observations, recommendations, and implementation. Behav. Res. Ther. 2017, 98, 39–57. [Google Scholar] [CrossRef]
- Preacher, K.J.; Hayes, A.F. Asymptotic and resampling strategies for assessing and comparing indirect effects in multiple mediator models. Behav. Res. Methods 2008, 40, 879–891. [Google Scholar] [CrossRef]
- Fernandez-Twinn, D.S.; Hjort, L.; Novakovic, B.; Ozanne, S.E.; Saffery, R. Intrauterine programming of obesity and type 2 diabetes. Diabetologia 2019, 62, 1789–1801. [Google Scholar] [CrossRef] [Green Version]
- Goyal, D.; Limesand, S.W.; Goyal, R. Epigenetic responses and the developmental origins of health and disease. J. Endocrinol. 2019, 242, T105–T119. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lillycrop, K.A.; Burdge, G.C. Epigenetic changes in early life and future risk of obesity. Int. J. Obes. 2011, 35, 72–83. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ling, C.; Ronn, T. Epigenetics in Human Obesity and Type 2 Diabetes. Cell Metab. 2019, 29, 1028–1044. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Calo, E.; Wysocka, J. Modification of enhancer chromatin: What, how, and why? Mol. Cell 2013, 49, 825–837. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Verdeguer, F.; Soustek, M.S.; Hatting, M.; Blattler, S.M.; McDonald, D.; Barrow, J.J.; Puigserver, P. Brown Adipose YY1 Deficiency Activates Expression of Secreted Proteins Linked to Energy Expenditure and Prevents Diet-Induced Obesity. Mol. Cell. Biol. 2015, 36, 184–196. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pan, D.Z.; Garske, K.M.; Alvarez, M.; Bhagat, Y.V.; Boocock, J.; Nikkola, E.; Miao, Z.; Raulerson, C.K.; Cantor, R.M.; Civelek, M.; et al. Integration of human adipocyte chromosomal interactions with adipose gene expression prioritizes obesity-related genes from GWAS. Nat. Commun. 2018, 9, 1512. [Google Scholar] [CrossRef]
- Laurila, P.P.; Soronen, J.; Kooijman, S.; Forsstrom, S.; Boon, M.R.; Surakka, I.; Kaiharju, E.; Coomans, C.P.; Van Den Berg, S.A.; Autio, A.; et al. USF1 deficiency activates brown adipose tissue and improves cardiometabolic health. Sci. Transl. Med. 2016, 8, 323ra13. [Google Scholar] [CrossRef]
- Liu, Y.; Xu, D.; Yin, C.; Wang, S.; Wang, M.; Xiao, Y. IL-10/STAT3 is reduced in childhood obesity with hypertriglyceridemia and is related to triglyceride level in diet-induced obese rats. BMC Endocr. Disord. 2018, 18, 39. [Google Scholar] [CrossRef] [Green Version]
- Tong, Q.; Dalgin, G.; Xu, H.; Ting, C.N.; Leiden, J.M.; Hotamisligil, G.S. Function of GATA transcription factors in preadipocyte-adipocyte transition. Science 2000, 290, 134–138. [Google Scholar] [CrossRef] [Green Version]
- Obermann-Borst, S.A.; Eilers, P.H.; Tobi, E.W.; de Jong, F.H.; Slagboom, P.E.; Heijmans, B.T.; Steegers-Theunissen, R.P. Duration of breastfeeding and gender are associated with methylation of the LEPTIN gene in very young children. Pediatr. Res. 2013, 74, 344–349. [Google Scholar] [CrossRef] [Green Version]
- Tobi, E.W.; Lumey, L.H.; Talens, R.P.; Kremer, D.; Putter, H.; Stein, A.D.; Slagboom, P.E.; Heijmans, B.T. DNA methylation differences after exposure to prenatal famine are common and timing- and sex-specific. Hum. Mol. Genet. 2009, 18, 4046–4053. [Google Scholar] [CrossRef] [PubMed]
- Lesseur, C.; Armstrong, D.A.; Paquette, A.G.; Li, Z.; Padbury, J.F.; Marsit, C.J. Maternal obesity and gestational diabetes are associated with placental leptin DNA methylation. Am. J. Obstet. Gynecol. 2014, 211, 654-e1. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bouchard, L.; Thibault, S.; Guay, S.P.; Santure, M.; Monpetit, A.; St-Pierre, J.; Perron, P.; Brisson, D. Leptin gene epigenetic adaptation to impaired glucose metabolism during pregnancy. Diabetes Care 2010, 33, 2436–2441. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hogg, K.; Blair, J.D.; von Dadelszen, P.; Robinson, W.P. Hypomethylation of the LEP gene in placenta and elevated maternal leptin concentration in early onset pre-eclampsia. Mol. Cell. Endocrinol. 2013, 367, 64–73. [Google Scholar] [CrossRef] [PubMed]
- Lesseur, C.; Armstrong, D.A.; Paquette, A.G.; Koestler, D.C.; Padbury, J.F.; Marsit, C.J. Tissue-specific Leptin promoter DNA methylation is associated with maternal and infant perinatal factors. Mol. Cell. Endocrinol. 2013, 381, 160–167. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chrzanowska, M.; Suder, A.; Kruszelnicki, P. Tracking and risk of abdominal obesity in the adolescence period in children aged 7–15. The Cracow Longitudinal Growth Study. Am. J. Hum. Biol. 2012, 24, 62–67. [Google Scholar] [CrossRef] [PubMed]
- Buck, C.O.; Eliot, M.N.; Kelsey, K.T.; Chen, A.; Kalkwarf, H.; Lanphear, B.P.; Braun, J.M. Neonatal Adipocytokines and Longitudinal Patterns of Childhood Growth. Obesity 2019, 27, 1323–1330. [Google Scholar] [CrossRef]
- Yeung, E.H.; Sundaram, R.; Xie, Y.; Lawrence, D.A. Newborn adipokines and early childhood growth. Pediatr. Obes. 2018, 13, 505–513. [Google Scholar] [CrossRef]
- Chessler, S.D.; Fujimoto, W.Y.; Shofer, J.B.; Boyko, E.J.; Weigle, D.S. Increased plasma leptin levels are associated with fat accumulation in Japanese Americans. Diabetes 1998, 47, 239–243. [Google Scholar] [CrossRef]
- Chu, N.F.; Spiegelman, D.; Yu, J.; Rifai, N.; Hotamisligil, G.S.; Rimm, E.B. Plasma leptin concentrations and four-year weight gain among US men. Int. J. Obes. 2001, 25, 346–353. [Google Scholar] [CrossRef] [Green Version]
- Lissner, L.; Karlsson, C.; Lindroos, A.K.; Sjostrom, L.; Carlsson, B.; Carlsson, L.; Bengtsson, C. Birth weight, adulthood BMI, and subsequent weight gain in relation to leptin levels in Swedish women. Obes. Res. 1999, 7, 150–154. [Google Scholar] [CrossRef] [PubMed]
- Ahmed, M.L.; Ong, K.K.; Morrell, D.J.; Cox, L.; Drayer, N.; Perry, L.; Preece, M.A.; Dunger, D.B. Longitudinal study of leptin concentrations during puberty: Sex differences and relationship to changes in body composition. J. Clin. Endocrinol. Metab. 1999, 84, 899–905. [Google Scholar] [CrossRef] [PubMed]
- Byrnes, S.E.; Baur, L.A.; Bermingham, M.; Brock, K.; Steinbeck, K. Leptin and total cholesterol are predictors of weight gain in pre-pubertal children. Int. J. Obes. 1999, 23, 146–150. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fleisch, A.F.; Agarwal, N.; Roberts, M.D.; Han, J.C.; Theim, K.R.; Vexler, A.; Troendle, J.; Yanovski, S.Z.; Yanovski, J.A. Influence of serum leptin on weight and body fat growth in children at high risk for adult obesity. J. Clin. Endocrinol. Metab. 2007, 92, 948–954. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Johnson, M.S.; Huang, T.T.; Figueroa-Colon, R.; Dwyer, J.H.; Goran, M.I. Influence of leptin on changes in body fat during growth in African American and white children. Obes. Res. 2001, 9, 593–598. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Savoye, M.; Dziura, J.; Castle, J.; DiPietro, L.; Tamborlane, W.V.; Caprio, S. Importance of plasma leptin in predicting future weight gain in obese children: A two-and-a-half-year longitudinal study. Int. J. Obes. 2002, 26, 942–946. [Google Scholar] [CrossRef] [Green Version]
- Li, L.J.; Rifas-Shiman, S.L.; Aris, I.M.; Mantzoros, C.; Hivert, M.F.; Oken, E. Leptin trajectories from birth to mid-childhood and cardio-metabolic health in early adolescence. Metabolism 2019, 91, 30–38. [Google Scholar] [CrossRef]
- Christensen, J.L.; Wright, D.E.; Wagers, A.J.; Weissman, I.L. Circulation and chemotaxis of fetal hematopoietic stem cells. PLoS Biol. 2004, 2, E75. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| Participant Clinical Data n = 262 | Mean ± SD |
|---|---|
| Maternal characteristics | |
| 1st trimester of pregnancy | |
| Age (years) | 28.6 ± 4.2 |
| BMI (kg/m2) | 25.5 ± 5.5 |
| Smoking during pregnancy | |
| Yes | 7.4% |
| No | 91.8% |
| Glucose 1 h post 50 g GCT (mmol/l) | 5.0 ± 2.2 |
| 2nd trimester of pregnancy | |
| Fasting Glucose (mmol/L) | 4.2 ± 0.3 |
| Glucose 2 h post 75 g OGTT (mmol/L) | 5.9 ± 1.4 |
| Child characteristics | |
| At birth | |
| Gestational age at birth (weeks) | 39.6 ± 1.0 |
| Sex | |
| Boys | 55% |
| Girls | 45% |
| Birthweight (kg) | 3.4 ± 0.4 |
| Cord blood leptin levels (ng/mL) | 15,028 ± 12,791 |
| At 3-years-old | |
| Age (months) | 40.5 ± 3.0 |
| Weight (kg) | 15.2 ± 1.7 |
| BMI (kg/m2) | 16.1 ± 1.2 |
| BMI z-scores | 0.5 ± 0.9 |
| Sum of skinfolds thicknesses (mm)α | 17.5 ± 3.6 |
| Ratio of skinfold thicknesses (SS:TR)α | 0.6 ± 0.1 |
| CpG Sites from the MethylationEPIC BeadChip | Cord Blood Leptin Levels |
|---|---|
| cg15758240 | r = −0.289 p < 0.001 |
| cg00011113 | r = −0.141 p = 0.03 |
| cg05136031 | r = 0.221 p < 0.001 |
| Mean for cg12782180, cg19594666, cg11045943, cg26814075, cg13381984 | r = −0.125 p = 0.05 |
| cg00666422 | r = −0.129 p = 0.04 |
| cg20564991 | r = −0.061 p = 0.34 |
| cg23381058 | r = −0.198 p = 0.002 |
| cg18603538 | r = 0.006 p = 0.93 |
| cg04833007 | r = −0.045 p = 0.48 |
| CpG Sites | BMI z-Scores | Sum of Skinfold Thickness | Ratio of Skinfold Thickness |
|---|---|---|---|
| cg15758240 | β = −0.391 p = 0.82 | β = 9.076 p = 0.21 | β = −0.581 p = 0.05 |
| cg05136031 | β = −2.687 p = 0.05 | β = −1.441 p = 0.80 | β = −0.083 p = 0.72 |
| cg23341058 | β = 0.019 p = 0.62 | β = 3.407 p = 0.35 | β = 0.015 p = 0.92 |
| CpG Sites | Maternal Glucose 1 h Post 50 g GCT at 1st Trimester of Pregnancy | Maternal Fasting Glucose at 2nd Trimester of Pregnancy | Maternal Glucose 2 h Post 75 g OGTT at 2nd Trimester of Pregnancy |
|---|---|---|---|
| cg15758240 | β < 0.001 p = 0.74 | β = −0.013 p = 0.04 | β < 0.001 p = 0.21 |
| cg05136031 | β < 0.001 p = 0.72 | Β < 0.001 p = 0.55 | Β < 0.001 p = 0.94 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gagné-Ouellet, V.; Breton, E.; Thibeault, K.; Fortin, C.-A.; Cardenas, A.; Guérin, R.; Perron, P.; Hivert, M.-F.; Bouchard, L. Mediation Analysis Supports a Causal Relationship between Maternal Hyperglycemia and Placental DNA Methylation Variations at the Leptin Gene Locus and Cord Blood Leptin Levels. Int. J. Mol. Sci. 2020, 21, 329. https://doi.org/10.3390/ijms21010329
Gagné-Ouellet V, Breton E, Thibeault K, Fortin C-A, Cardenas A, Guérin R, Perron P, Hivert M-F, Bouchard L. Mediation Analysis Supports a Causal Relationship between Maternal Hyperglycemia and Placental DNA Methylation Variations at the Leptin Gene Locus and Cord Blood Leptin Levels. International Journal of Molecular Sciences. 2020; 21(1):329. https://doi.org/10.3390/ijms21010329
Chicago/Turabian StyleGagné-Ouellet, Valérie, Edith Breton, Kathrine Thibeault, Carol-Ann Fortin, Andres Cardenas, Renée Guérin, Patrice Perron, Marie-France Hivert, and Luigi Bouchard. 2020. "Mediation Analysis Supports a Causal Relationship between Maternal Hyperglycemia and Placental DNA Methylation Variations at the Leptin Gene Locus and Cord Blood Leptin Levels" International Journal of Molecular Sciences 21, no. 1: 329. https://doi.org/10.3390/ijms21010329
APA StyleGagné-Ouellet, V., Breton, E., Thibeault, K., Fortin, C. -A., Cardenas, A., Guérin, R., Perron, P., Hivert, M. -F., & Bouchard, L. (2020). Mediation Analysis Supports a Causal Relationship between Maternal Hyperglycemia and Placental DNA Methylation Variations at the Leptin Gene Locus and Cord Blood Leptin Levels. International Journal of Molecular Sciences, 21(1), 329. https://doi.org/10.3390/ijms21010329