1. Introduction
The ubiquitin/proteasome system (UPS) is responsible for the majority of intracellular eukaryotic protein turnover, and its coordinated function is essential for many cellular processes, including cell growth, differentiation, activation, and signalling [
1]. Because the UPS is a crucial regulator of cell proliferation and apoptosis, therapeutic strategies to regulate its role could potentially restore stable cellular homeostasis in some cancer patients [
2]. The first proteasome inhibitor approved by the US FDA for the treatment of multiple myeloma (MM) patients was bortezomib, a low-molecular-weight, water-soluble dipeptide boronic acid derivative that binds with high affinity to the proteasome and show slow dissociation [
3]. Bortezomib has also shown promise as possible antitumor agent in preclinical studies of tumours such as ovarian cancer, lung cancer, pancreatic cancer, prostate cancer, melanoma, glioblastoma, and most recently in MYC-driven neuroblastoma [
4,
5,
6]. Bortezomib is a reversible inhibitor of the chymotrypsin-like activity of the 26S proteasome degrading ubiquitinated proteins [
7]. This leads to the accumulation of misfolded and functional proteins in the endoplasmatic reticulum (ER) and cytosol of cells followed by ER overload, generation of excess oxygen species as well as a functional disorder of intracellular proteins, and eventually cell death [
8].
Even though inhibitory action of bortezomib is rather clearly established, the downstream mechanisms of its activity remain not fully understood [
9]; it is therefore of great interest to understand the mechanism of action of bortezomib. One of the main side effects of bortezomib treatment, occurring in up to 40% MM patients, remains peripheral neuropathy, which has been linked with neurotoxicity [
10,
11]. It has been hypothesized that specific neurotoxic action of bortezomib is through transient release of intracellular calcium stores, leading to mitochondrial calcium influx and caspase-induced apoptosis [
7]. While apoptotic transcription-associated activation in response to bortezomib has been suggested, mechanisms and new insights, particularly in the post-transcriptional gene-silencing-mediated regulation, remain largely unexplained [
12]. MicroRNAs (miRNAs, miRs), 20–22 nucleotide long non-coding RNA molecules, have recently emerged as master regulators of various genes and their role in bortezomib action is also suggested [
9,
12,
13,
14]. MiRNAs regulate gene expression at the post-transcriptional level and are involved in almost every biological process, including cell proliferation, apoptosis, as well as disease-associated processes [
15]. Differentially expressed miRNAs have been reported in a number of diseases [
16] and as a response to drug use [
17], however to date, the wide panel of mRNAs/miRNAs has not been tested in bortezomib-treated cells.
In order to better understand the underlying molecular mechanisms of bortezomib action in a neural environment, in the present study, we investigated global mRNA and miRNA expression in bortezomib-treated neuroblastoma cells and analyzed the identified miRNA–mRNA target interaction.
3. Discussion
Bortezomib action, owing to its powerful inhibiting proteasome activity, has been correlated with inhibition of anti-apoptotic proteins, p53 stabilization, cell cycle progression disruption, and other critical cellular processes [
18,
19]. Using global mRNA and miRNA expression analysis, we demonstrate for the first time that bortezomib exposure leads to dysregulation of a number of genes and miRNAs associated with neurogenesis and apoptosis in neural cells.
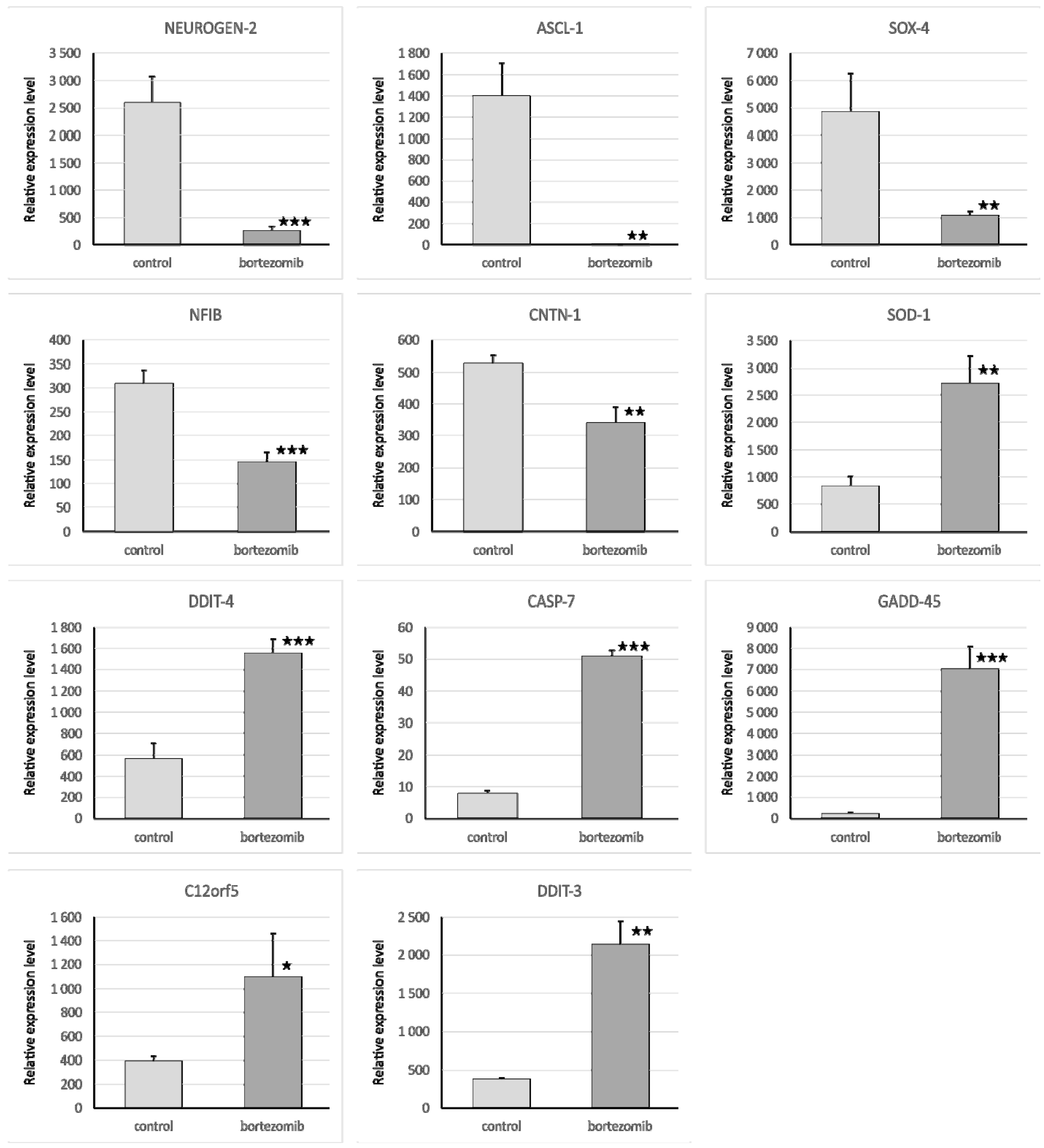
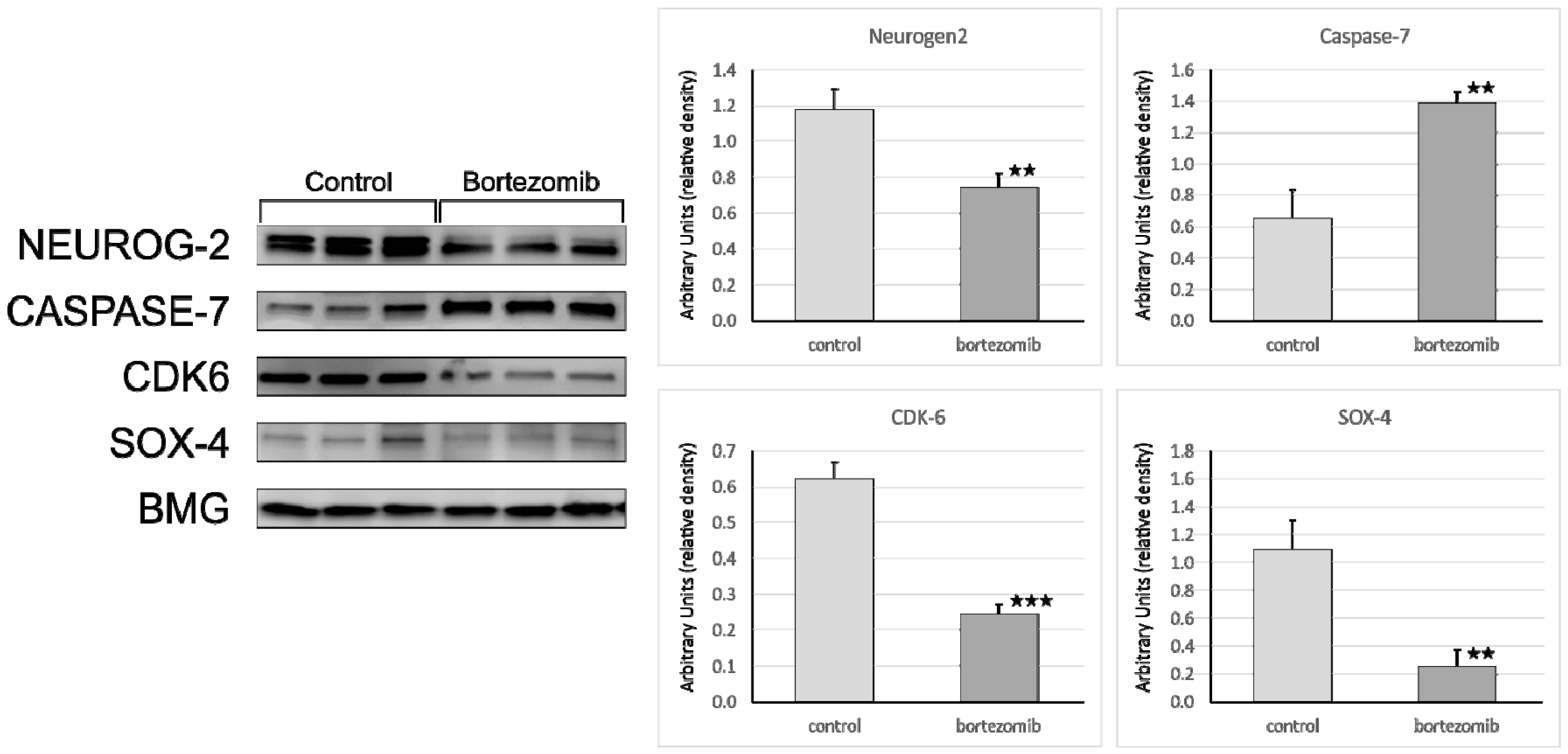
First, we looked into neurogenesis as a process that may be affected by bortezomib use. Several important genes were downregulated, including
NEUROG2, ASCL1, SYT4, however no specific correlations with miRNAs were found. The upregulation of miR-1303 could be attributed to the nature of neuroblastoma cells, as miR-1303 has been shown to promote the proliferation of SH-SY5Y cells; several studies showed that miR-1303 is upregulated in neuroblastoma cells and tissues [
20]. miR-1303 decreases doublecortin (
DCX), which plays an important role in neurogenesis and its deletion causes severe morphologic defects and delayed migration along the rostral migratory stream [
21]. Another significantly upregulated miRNA was miR-21-3p, which decreases
NCAM2, ELMO1, and
GPAM expression. Contrary to our findings, Wang et al. have reported that bortezomib decreases level of miRNA-21, which is considered as “oncomir”, since it suppresses the expression of several apoptotic and tumor suppressor genes leading to cell proliferation, migration, invasion, angiogenesis, and metastasis as well as inhibition of apoptosis in human myeloma cell lines [
14,
22]. Our results might suggest that miR-21 dysregulation contributes to neuron death, because of important roles of miR-21 targets, which it downregulates.
NCAM2 is primarily expressed in the brain, where it stimulates neurite outgrowth and facilitates dendritic and axonal compartmentalization [
23].
ELMO1 protects endothelial cells from apoptosis [
24] and it is also an important factor for glomerular protection and renal cell survival via decreasing apoptosis, especially under diabetic conditions [
25].
We also found correlations between overexpression of miRNAs that target genes essential in neuronal development and remodelling: miR-6824, -335, and -1468 all target
SOX4, which participates in Schwann cell myelination [
26], whereas miR-6836 targets
TFAP2B, which acts in the specification of GABAergic interneurons in developing cerebellum [
27]. Interestingly,
MYC-miR34a axis, in which miR-34a apoptotic actions are tightly regulated by
MYC and this can lead to drug resistance [
28], has somehow been confirmed by our study, however the role of miR-4668 (which was also upregulated and targets
MYC) in these interactions remains unclear. The overexpression of miR-3192 and consequent downregulation of
CHRNB2, which ensures communication between neurons, could be another possible neurotoxicity mechanism [
7], due to disruption of axonal transport processes. Additionally,
NCAM (fold change -5.59), which is a target for miR-21-3p, activates several signal transduction pathways or genes such as glial cell line-derived neurotrophic factor (
GDNF), fibroblast growth factor receptor (
FGFR) [
29]. This finding may support hypothesis that reduction of neurotrophins production is also involved in bortezomib action. Several of miRNAs, which showed altered expression in this study, have been described in MM patients before, including miR-21 and miR-222 [
30,
31], but a correlation between their dysregulation and bortezomib neurotoxicity is yet to be found.
Bortezomib cytotoxicity is also attributed to mitochondrial-mediated dysregulation of calcium homeostasis [
7]. The mitochondrial Ca
2+ overload is one of the pro-apoptotic ways to induce the swelling of mitochondria, with perturbation or rupture of the outer membrane followed by a release of mitochondrial apoptotic factors into the cytosol [
32]. Disruption of intracellular calcium homeostasis may be an emerging mechanism involved in drug-induced neuroapoptosis [
33,
34]. Landowski et al. demonstrated that the cytotoxic action of bortezomib is mediated through a transient release of intracellular calcium store leading to mitochondrial calcium inflow and apoptosis induced by caspase-12 activation [
35,
36]. In the present study, we observed very low levels of important genes encoding calcium channels (
CACNA2D2, CACNG4) and regulating the calcium level (
ITPR1) (
Table 2), although no specific miRNA–mRNA interactions were found. The downregulation of these genes may have a significant impact on neural function as they regulate intracellular calcium release, which is fundamental to neural cells’ functions including neuronal excitability, neurite outgrowth, neurotransmitter release, mitochondrial energy production, and cell fate [
37]. These data support the hypothesis that bortezomib can induce neurotoxicity by disrupting intracellular calcium homeostasis in neural cells.
Another possibility is that bortezomib triggers oxidative stress, which contributes to proapoptotic effects and gene expression patterns reprogramming in nerves. In the present study, we observed a significant upregulation of genes engaged in apoptosis, especially related to neuron death. Among the most strongly upregulated genes were clusterin (
CLU), heme oxygenase (decycling) 1 (
HMOX1), caspase 7 (apoptosis-related cysteine peptidase,
CASP7), and DNA-damage-inducible transcript 3 (
DDIT3). Products of these genes are synthesized in response to cellular stress and appear in cells undergoing apoptosis [
38,
39,
40,
41]. Larner et al. have demonstrated that
CASP7 is significantly upregulated and activated in both neurons and astrocytes after traumatic brain injury (TBI) in rats and may play a role in the cell death in TBI in patients [
42]. An interaction between miR-124 and interleukin 6 receptor (
IL6R), which was observed in our study, could also contribute to bortezomib action, since
IL6R is a linking factor between several important pathways including NF-κB, MAPK, and JAK/STAT, which take part in a variety of cellular responses, including cell survival, inflammation, and differentiation [
43].
SQSTM1, which in our study was significantly upregulated by miR-218, is a useful indicator of autophagy—it accumulates in autophagic-defective tumor cells and decreases when autophagy is completed.
SQSTM1 upregulation observed in this study and thus enhanced autophagy might also contribute to bortezomib neurotoxicity, also by acting on ERK phosphorylation [
44]. The role of
IGF1R in bortezomib response is also suggested [
45]; in our study, miR-4423 and miR-551 significantly affected expression of this gene. One of the most prominently downregulated miRNAs in this study was miR6880-5p, responsible for downregulation of proapoptotic gene
C12orf5, which promotes neuron death (
Table 8). In case of decreased miR6880-5p,
C12orf5 expression is no longer downregulated, which was also seen in our study, as we observed upregulation of
C12orf5. In the same manner, downregulation of miR-26b-5p causes increased expression of
DDIT4, HSPD1, ADAM17, DDIT4, HSPD1, EPHA2, and
RNF216. These genes promote apoptosis, DNA damage, and neuron death. Therefore, the induction of neuronal cell apoptosis may represent a relevant step in bortezomib action.
In this study, we focused on miRNA/mRNA interactions, however there are several reports describing dysregulation of genes/miRNAs without exact interactions between them, but which are also relevant to our results. Liu et. al. have demonstrated that treatment with bortezomib significantly increased
ATF3,
c-JUN, and
CCL2 expression in DRG in rat model [
46]. Blockage of c-Jun/ATF3-CCL2 signaling pathway by siRNA has been shown to attenuate the mechanical allodynia induced by bortezomib. These findings suggested that the upregulation of
CCL2 via enhanced interaction between
c-Jun and
ATF3 contributes to bortezomib-induced mechanical allodynia [
46]. Interestingly, it has been demonstrated that the treatment with paclitaxel significantly increases the
ATF3 expression in rat DRG neurons, suggesting that
ATF3 might be involved in the chemotherapy-induced neurotoxicity [
47]. In our study, we observed increased expression of
ATF3 (fold change = 6.37) and
JUN (fold change = 2.74) in neural cells treated with bortezomib compared with control cells. However, the expression of
CCL2 was unchanged after bortezomib exposure. It may result from relatively short incubation time in setting of our study or different model of experiment.
Potential Study Limitations
Overall, our study gave some interesting results, but did not lack some drawbacks. The first limitation is only one time point and dose at which array experiment was conducted. Additionally, we did not perform functional validation assays to further test the effect of mRNA-miRNA interactions on neurogenesis, neuron death, and cellular calcium transport. We believe that different drug doses, time points, and cellular models could be of great importance to further elucidate the underlying mechanism of bortezomib action. Due to the basic and preliminary nature of this study, we should be cautious about ascribing cause and effect of bortezomib exposure on mRNA-miRNA interactions and neurotoxicity.
4. Materials and Methods
4.1. Cell Culture and Incubation
SH-SY5Y neuroblastoma cells (human, ECACC; Sigma Aldrich, St. Louis, MO, USA) were used in this study. Selected cell line is frequently used as a reliable model for studying neurotoxicity of drugs [
48,
49]. SH-SY5Y cells were incubated at a density of 4 × 10
6 cells/well in 6-well culture plates in Ham’s F-12 Nutrient Mixture (Thermo Fisher, Waltham, MA, USA) and minimum essential medium (MEM) (Sigma Aldrich, St. Louis, MO, USA) (mixed in ratio 1:1) medium supplemented with penicillin (100 U/mL), streptomycin (100 µg/mL), and L-glutamine (2 mM) without fetal bovine serum at 37 °C in saturated humidity atmosphere containing 5% CO
2. Cells were not treated (control) or treated with bortezomib 50 nM/l (Cell Signalling Technology, Danvers, MA, USA) and collected after 24 h of incubation [
50,
51]. Cells were then subjected to mRNA, miRNA, and protein isolation.
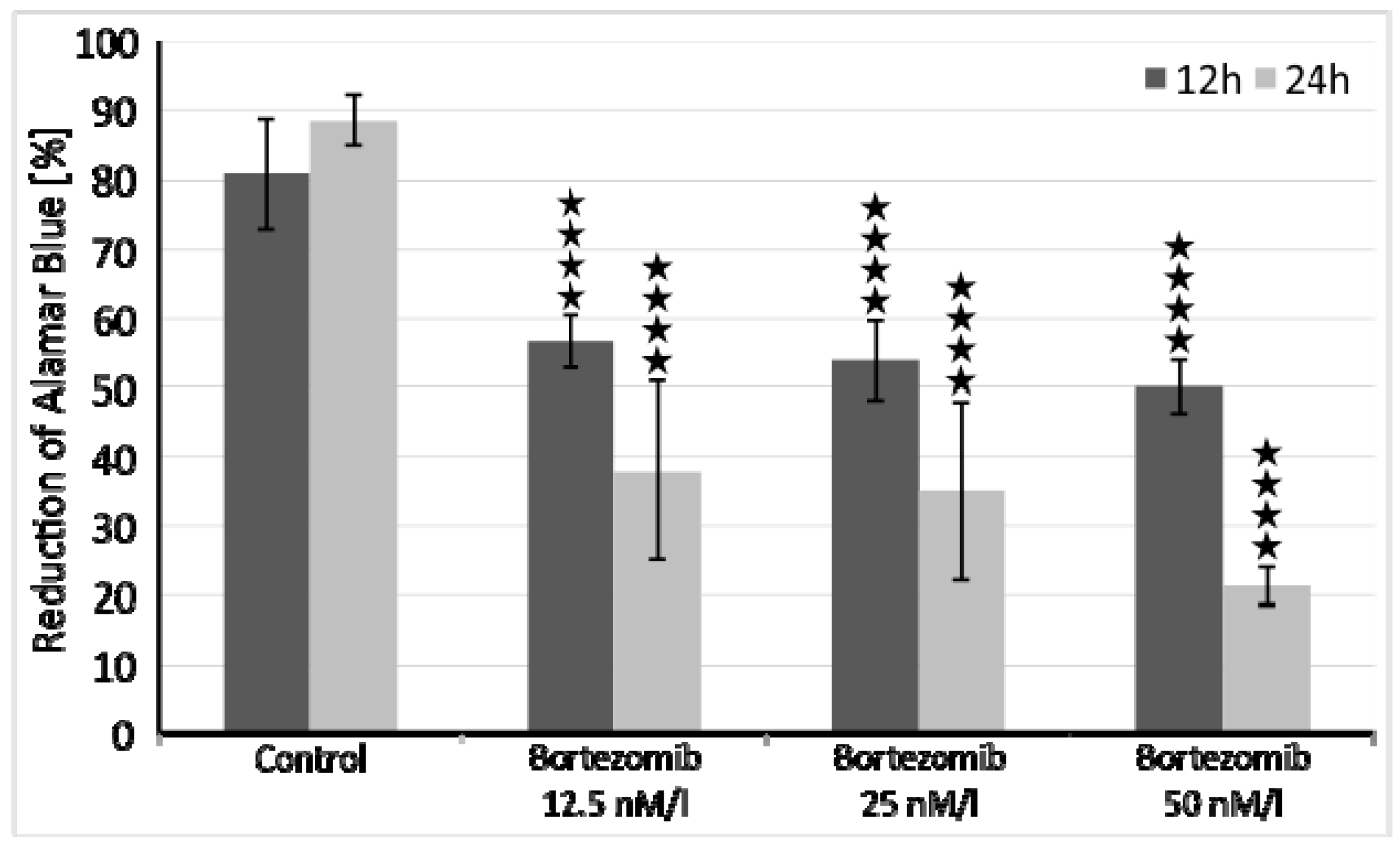
4.2. Cells Viability
The optimal dose of bortezomib for this experiment was based on the already published data [
50,
51]. However, we performed additional cell viability assays to assess cytotoxicity of tested drug. SH-SY5Y cells were incubated at a density of 1x10
4 cells/well in two 96-well culture plates (medium as in 2.1), first plate for 12 and second plate for 24 hours at 37 °C in saturated humidity atmosphere containing 5% CO
2. Each 8-well row was treated with increasing concentrations of bortezomib (12.5 nM/l, 25 nM/l, 50 nM/l) and cell viability was measured with the Alamar Blue mitochondrial dye conversion assay (Thermo Fisher, Waltham, MA, USA) [
50].
4.3. RNA and Protein Extraction
RNA and protein were isolated from three separate cell incubations for each group. Total RNA was isolated from SH-SY5Y cells (1 × 106) using the mirVana™ miRNA Isolation Kit (Thermo Fisher, Waltham, MA, USA) following the manufacturer’s instructions. The kit allows the isolation of RNA and miRNA simultaneously. Concentration and quality of the obtained RNA were assessed by Epoch spectrophotometer (Biotek, Winooski, VT, USA). SH-SY5Y cells (3 × 106) exposed to the dose of bortezomib and control cells were lysed for 10 min on ice in M-Per lysing buffer (Pierce, Rockford, IL, USA) containing protease and phosphatase inhibitors (Sigma Aldrich, St. Louis, MO, USA) (10 µg/ml leupeptin, 10 µg/mL aprotinin, 1 µg/mL pepstatin A, 1 mM sodium fluoride, and 2 mM Na3VO4). The measurement of total protein was performed using the Bradford method.
4.4. Affymetrix GeneChip Microarray and Data Analysis
Total RNA isolated from three separate cell incubations were pooled to generate one sample for subsequent experimental procedures. A sense-strand cDNA generated from the total RNA using an Ambion WT Expression Kit (Thermo Fisher Scientific, Waltham, MA, USA) was fragmented and labeled using the GeneChip WT Terminal Labeling Kit (Affymetrix, Santa Clara, CA, USA) and next hybridized onto an Affymetrix Human Gene 2.1 ST Array Strip. The hybridization and subsequent fluidics and scanning steps were carried out with an Affymetrix GeneAtlas System, with designated software (Affymetrix, Santa Clara, CA, USA). All analyses were performed using BioConductor software based on the statistical R programming language. The robust multiarray average (RMA) normalization algorithm implemented in the “Affy” library was used for normalization, background correction, and calculation of the expression levels of all of the examined genes [
52].
4.5. Affymetrix GeneChip miRNA Microarray and Data Analysis
Total RNA-enrichment in miRNAs isolated from three separate cell incubations were pooled to generate one sample for subsequent experimental procedures. The procedure was started with a poly (A) tailing reaction followed by ligation of the biotinylated signal molecule to the target RNA. The next step was sample hybridization onto an Affymetrix miRNA 4.1 Array Strip (Affymetrix, Santa Clara, CA, USA). The last step was streptavidin-PE addition and array scanning with Affymetrix GeneAtlas system (Affymetrix, Santa Clara, CA, USA). Analysis of microarray data was performed using BioConductor, as described earlier in 2.3. [
52]. Normalized data were combined with “pd.mirna.4.1” description file, containing, among others, names, types, and sequences of miRNAs. Differential expression was determined by applying the linear models for microarray data implemented in the “limma” library [
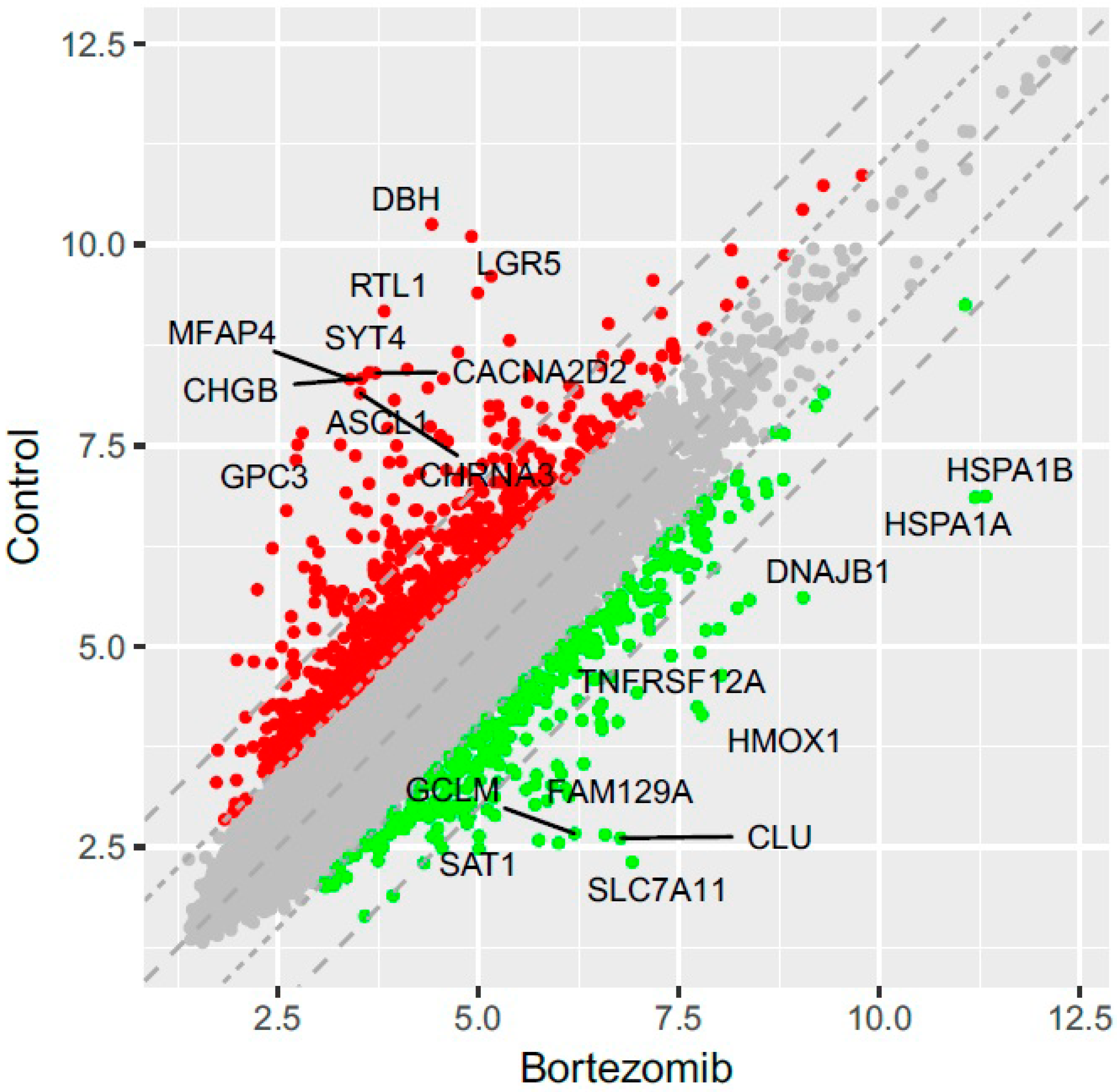
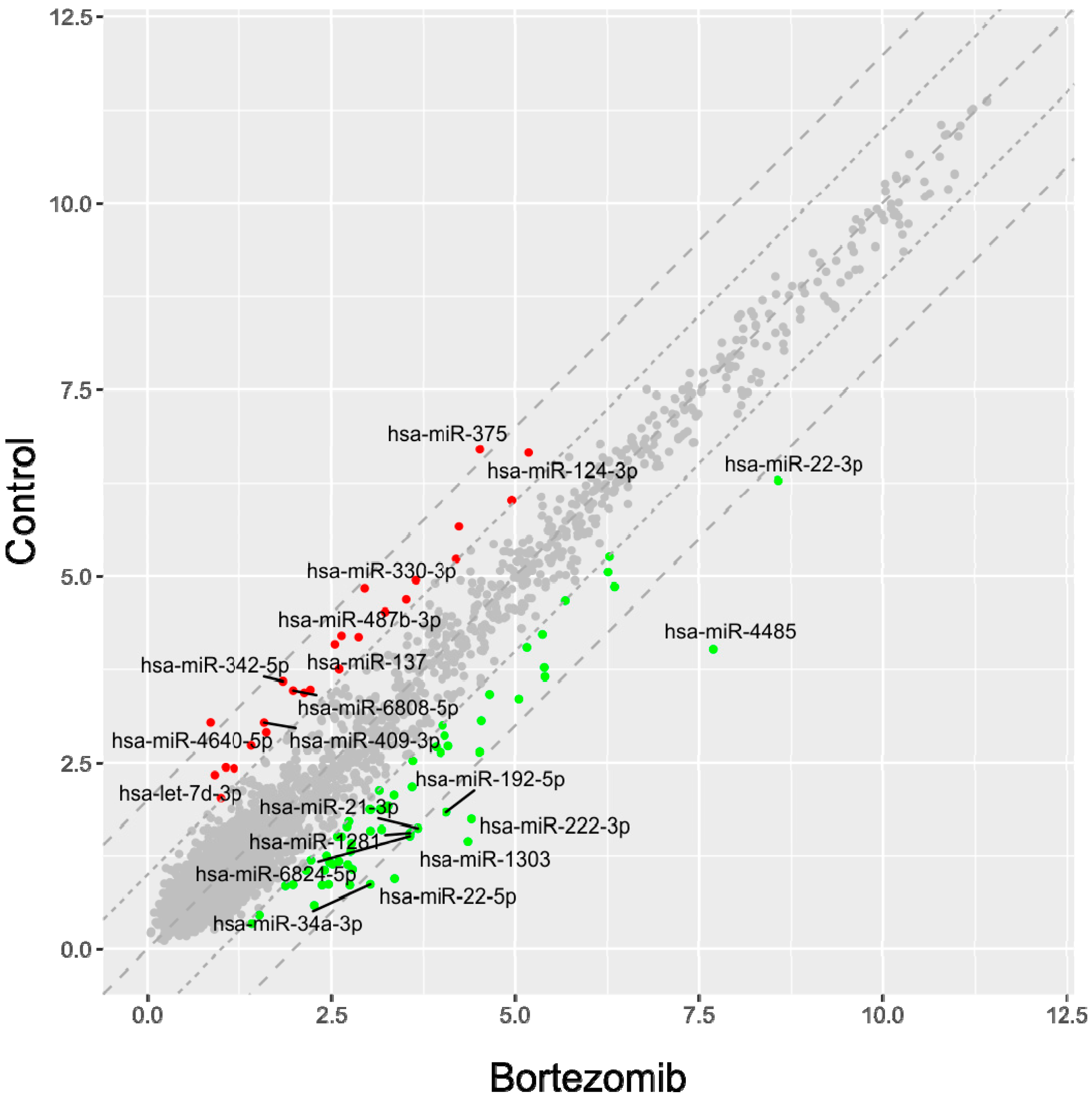
53]. Normalized miRNA expression datasets were visualized on scatterplots with relation to determined cut-off criteria (fold change > abs.2).
The list of experimentally validated miRNA target genes was downloaded from miRTarBase—a database of mRNA–target interactions. Only targets for differentially expressed miRNA were subtracted from the whole human miRNA-target dataset. Target gene list from each of comparisons was subjected to functional annotation and clusterization using DAVID (Database for Annotation, Visualization, and Integrated Discovery) [
54]. Target symbols of differentially expressed miRNA were uploaded to DAVID by the “RDAVIDWebService” BioConductor library [
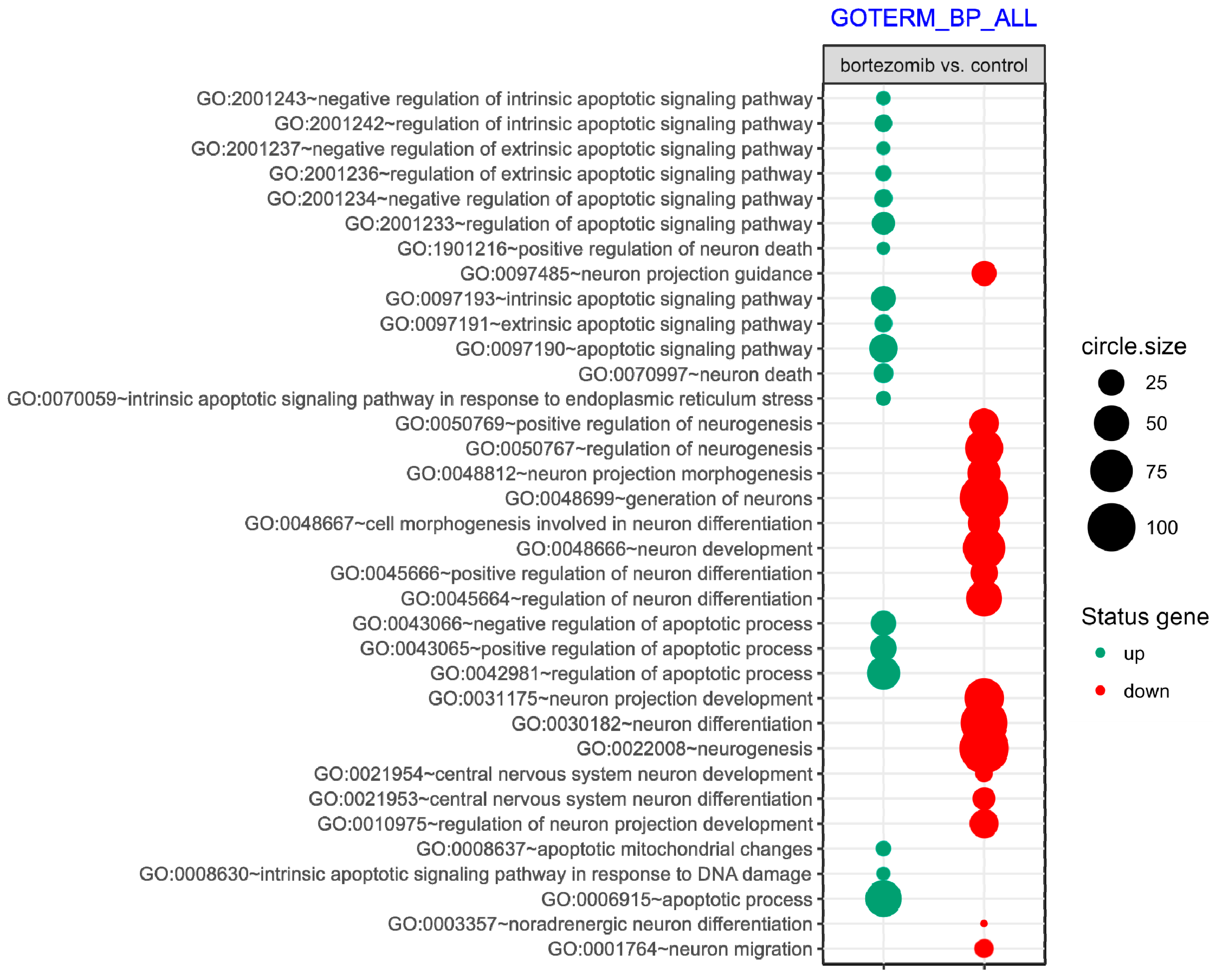
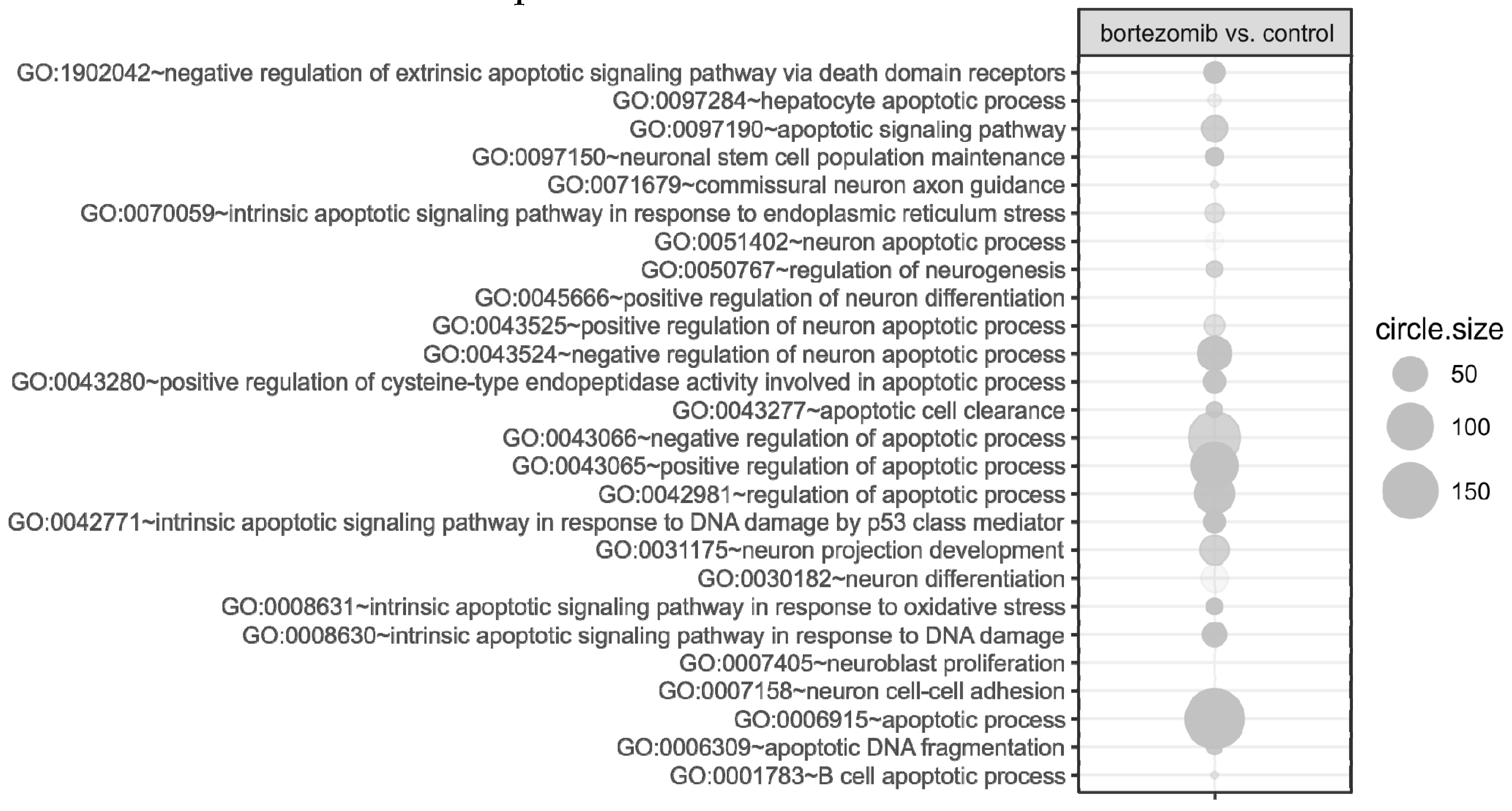
55], where targets were assigned to relevant gene ontology (GO) terms. GO ontological groups participating in the apoptosis and neurogenesis processes were visualized using bubble plot.
miRNA–target genes interaction network was performed using “networkD3” library. Taking into consideration that miRNA mostly act as repressors of target genes transcription, only target genes that negatively correlated with miRNA were visualized (if the expression of miRNA was increased, the expression of its targets should be decreased and vice versa). With such an assumption, an interaction network was performed for all of differentially expressed miRNA and target genes, as well as for miRNA and targets participating in apoptosis and neurogenesis.
4.6. DAVID
DAVID Bioinformatics Resources (Database for Annotation, Visualization, and Integrated Discovery) at
http://david.abcc.ncifcrf.gov is functional annotation and enrichment analysis. This database is an integrated tool biological knowledgebase and tools to systematically extract substantial biological terms associated with a given gene list. Functional annotation charts generated by DAVID with overrepresented gene annotations are shown as bubble plots from the BACA BioConductor package (
https://cran.r-project.org/web/packages/BACA/BACA.pdf). Bubble plots were generated with the following criteria:
p value < 0.5, adjusted method = Benjamini, and minimal number of genes per group = 5. Groups of genes fulfilling the mentioned criteria are presented in a graph in which the bubble size indicates the number of genes represented in the corresponding annotation and the condition of these genes in terms of their down- and upregulation [
54,
55].
4.7. qRT-PCR for Validation of Expression of Selected mRNA/miRNA
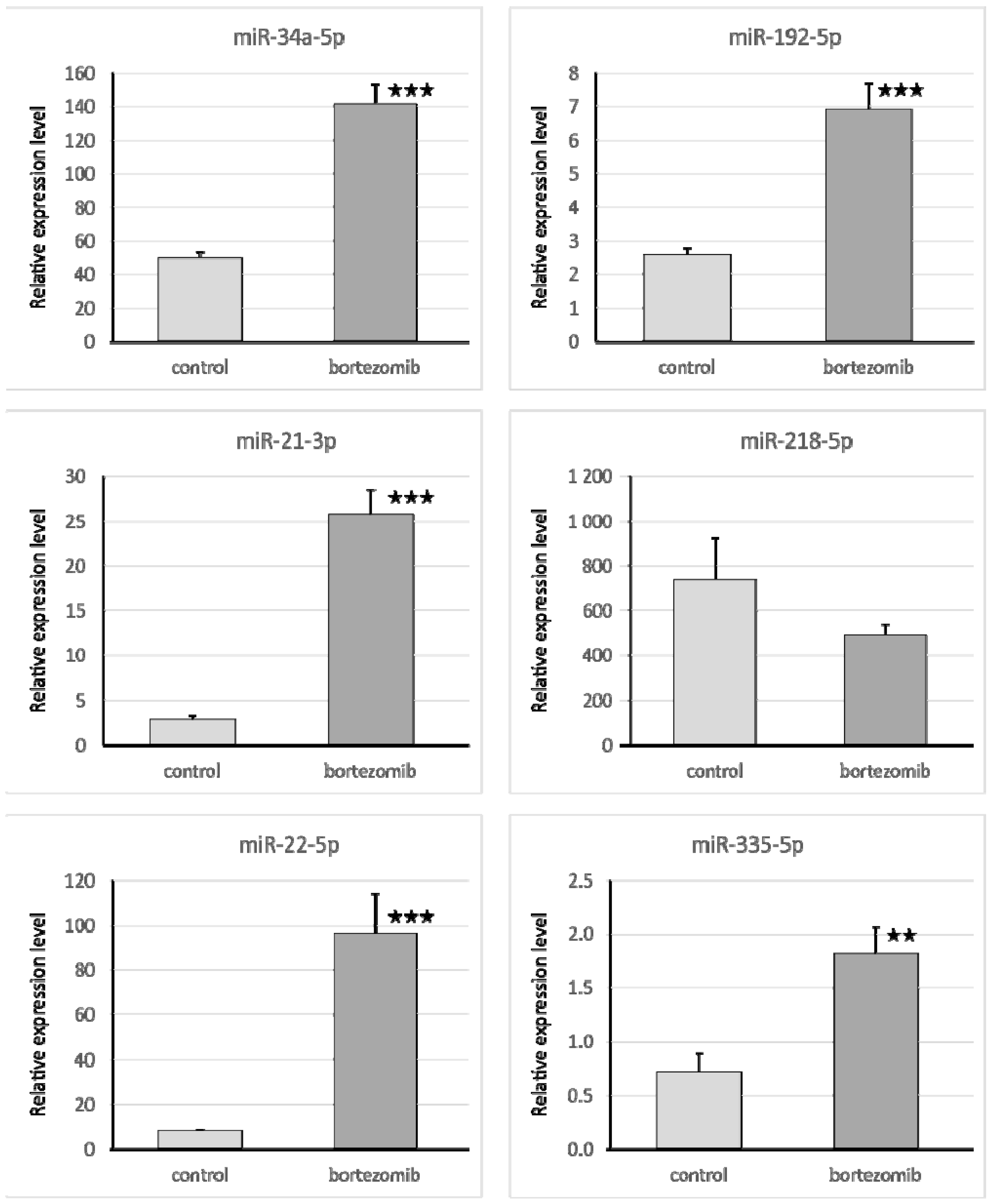
The qRT-PCR reaction was used to evaluate the expression of genes and miRNA. We selected significantly dysregulated (fold change > 2.0) mRNA/miRNA for validation of array experiments. Reaction was performed on a Bio-Rad CFX96 Real-Time PCR Detection System (Bio-Rad Inc., Hercules, CA, USA). All primers were purchased from Laboratory of DNA Sequencing and Oligonucleotide Synthesis, Institute of Biochemistry and Biophysics, Polish Academy of Sciences, Warsaw, Poland. The qRT-PCR program consisted of 10-min initial denaturation followed by a 3-step amplification program: Denaturation at 95 °C for 15 s, annealing at 56 °C for 5 s, and extension at 72 °C for 10 s. Specificity of reaction products was checked by determination of melting points (0.1 °C/s transition rate). The relative gene expression was quantified using the comparative Ct method (2ΔCt, where ΔCt = [Ct of target genes] − [Ct of endogenous control gene]).
4.7.1. Validation of Gene Expression
Isolated mRNA (0.1 µg) was reverse transcribed using the First Strand cDNA Synthesis Kit (Thermo Fisher Scientific, Waltham, MA, USA). The reaction mixture for qRT-PCR consisted of 7.5 μL of SYBR Green PCR Master Mix; 1.5 μL cDNA template; 4.2 μL Nuclease-Free Water; and 1.8 μL specific primers (0.9 μL forward primer and 0.9 μL reverse primer). The primers were designed by BLAST, and BMG was used as an endogenous control gene.
4.7.2. Validation of Selected miRNA Expression
Isolated miRNA (0.1 µg) was reverse transcribed using qScript microRNA cDNA Synthesis Kit (Quanta Biosciences, Beverly, MA, USA), which contains all necessary components for qRT-PCR. The reaction mixture for qRT-PCR contained: 5 μL PerfeCTa SYBR Green SuperMix; 0.2 μL microRNA specific primer; 0.2 μL PerfeCTa Universal PCR Primer, 1 μL microRNA cDNA; 4.6 μL Nuclease-Free Water. The primers were designed by miRPrimer, and miR93 was used as an endogenous control gene.
4.8. Western Blot
The protein samples were applied (20 µg/well) and separated on a 4%–20% sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE, miniPROTEAN II electrophoresis system, Bio-Rad) and next transferred to a 0.2 µm polyvinylidene fluoride (PVDF) membrane (Bio-Rad Inc., Hercules, CA, USA). After 2 h blocking in 3% BSA at RT conditions, the membrane was probed with a specific primary antibody as follows: Anti-Neurogenin2 (at 1:1000 dilution) (Abcam, Cambridge, UK; cat. number ab109172); anti-Cdk6 (at 1:750 dilution) (Abcam, Cambridge, UK; cat. number ab124821); anti-SOX4 (at 1:100 dilution) (Abcam, Cambridge, UK; cat. number ab80261); anti-Caspase-7 (at 1:750 dilution) (Abcam, Cambridge, UK; cat. number ab69540) and incubated overnight at 4 °C. To visualize the bands secondary antibody (specific to the primary antibody used in the previous step) conjugated with horseradish peroxidase (HRP) were used. Chemiluminescence detection was performed using the ECL Advance Detection Kit (Amersham Life Sciences, Buckinghamshire, UK), and the bands were visualized with a UVP camera (Gel DOC-It Imaging system, Bio-Rad). The ImageJ software ver. 1.8.0 (NIH, WI, USA) was used to determine the background-subtracted density of the bands. The relative protein expression levels of Neurog2, SOX4, Caspase7, and CDK6 were quantified in comparison to those of BMG.
4.9. Statistical Methods
The arithmetical means and standard deviations were calculated using MS Excel 2016. Comparisons of parameters between two groups were made using unpaired Student’s t-test and p-value < 0.05 was considered statistically significant. ANOVA with post-hoc Tukey’s test was used to calculate statistical significance in cell viability tests, p-value < 0.05 was considered significant.

 ,
, 









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}