Coronin 1 Is Required for Integrin β2 Translocation in Platelets



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
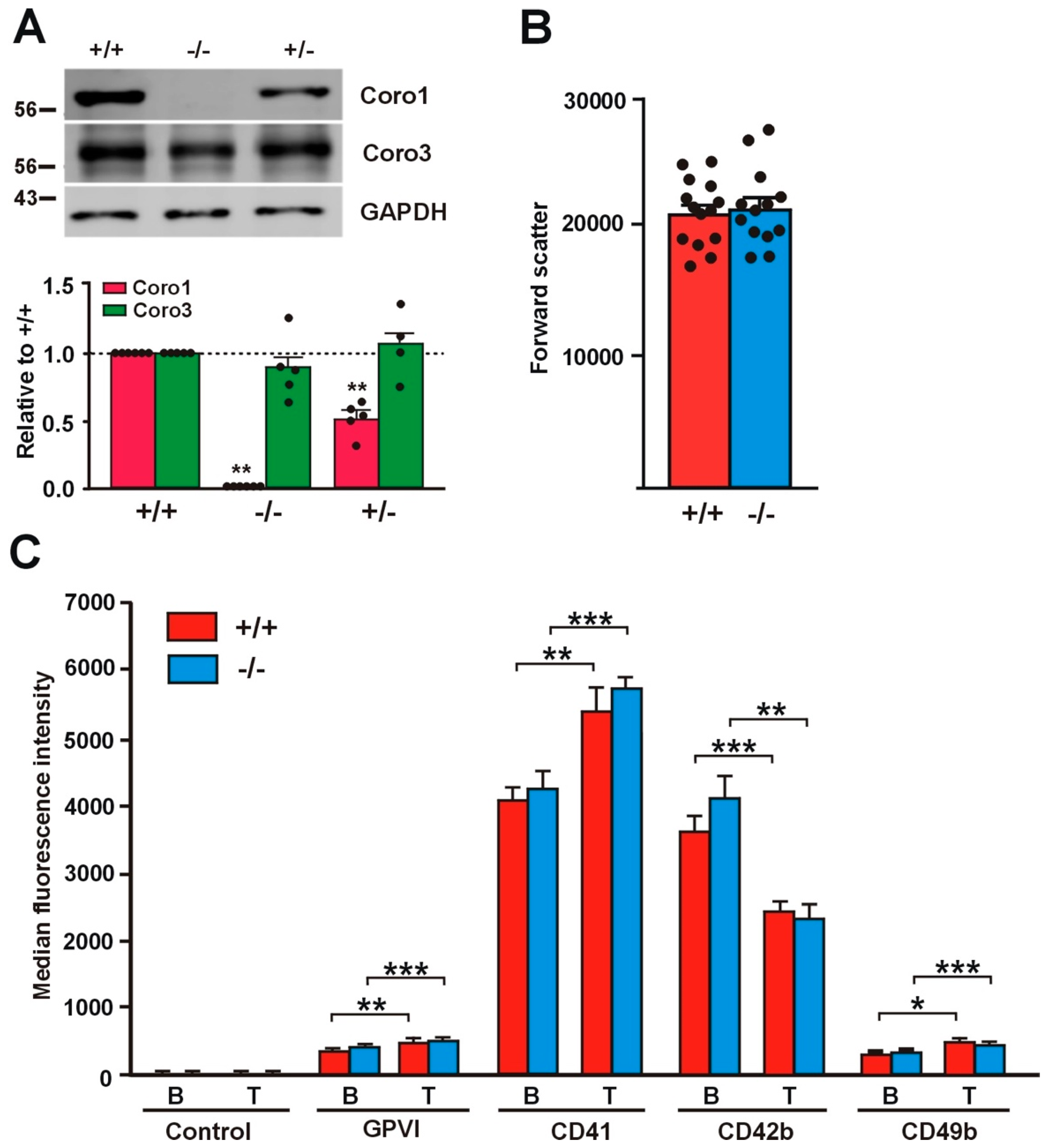
2.1. Absence of Coro1 Is Not Compensated by Increased Coro3
2.2. Receptor Expression Is Not Affected in Coro1 Deficient Platelets
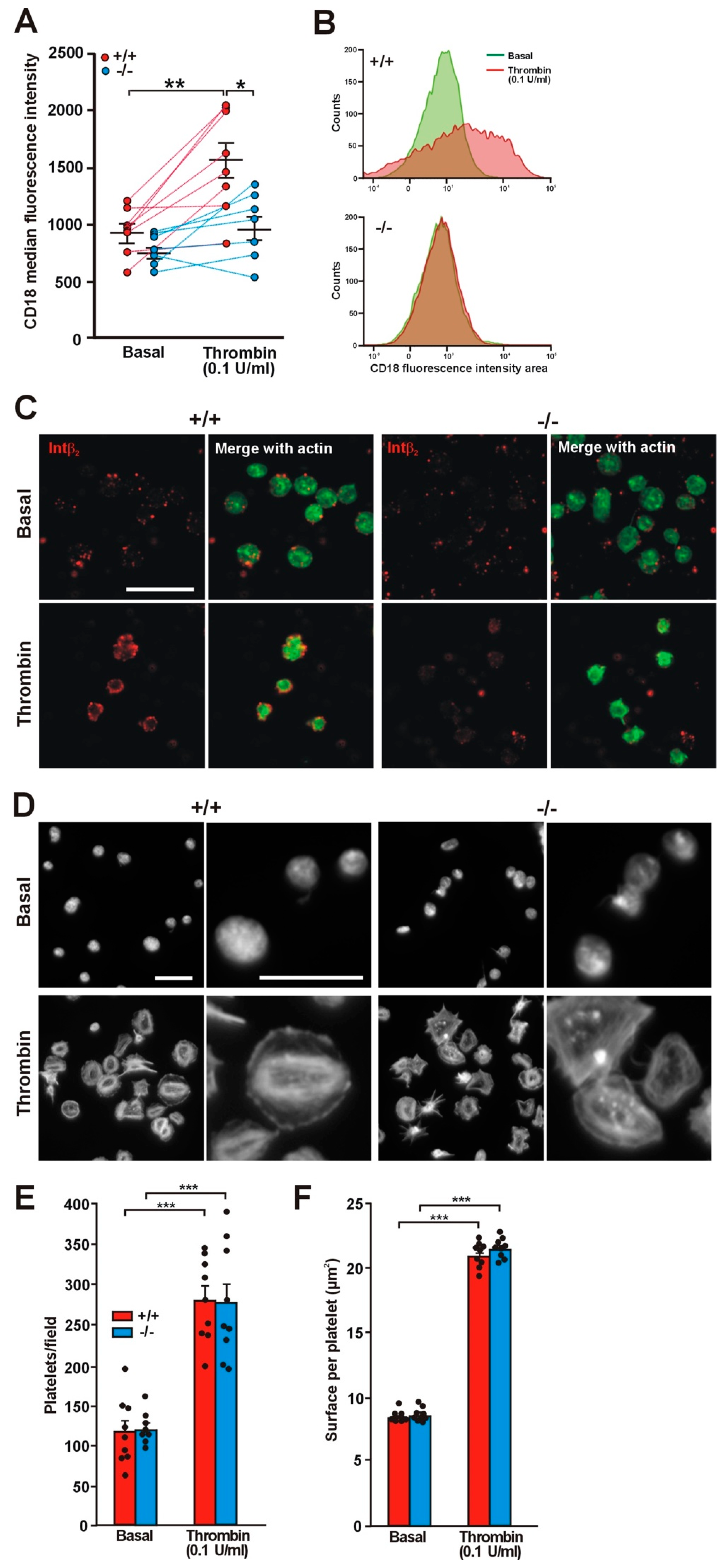
2.3. Translocation of Integrin β2 Is Impaired in the Absence of Coro1
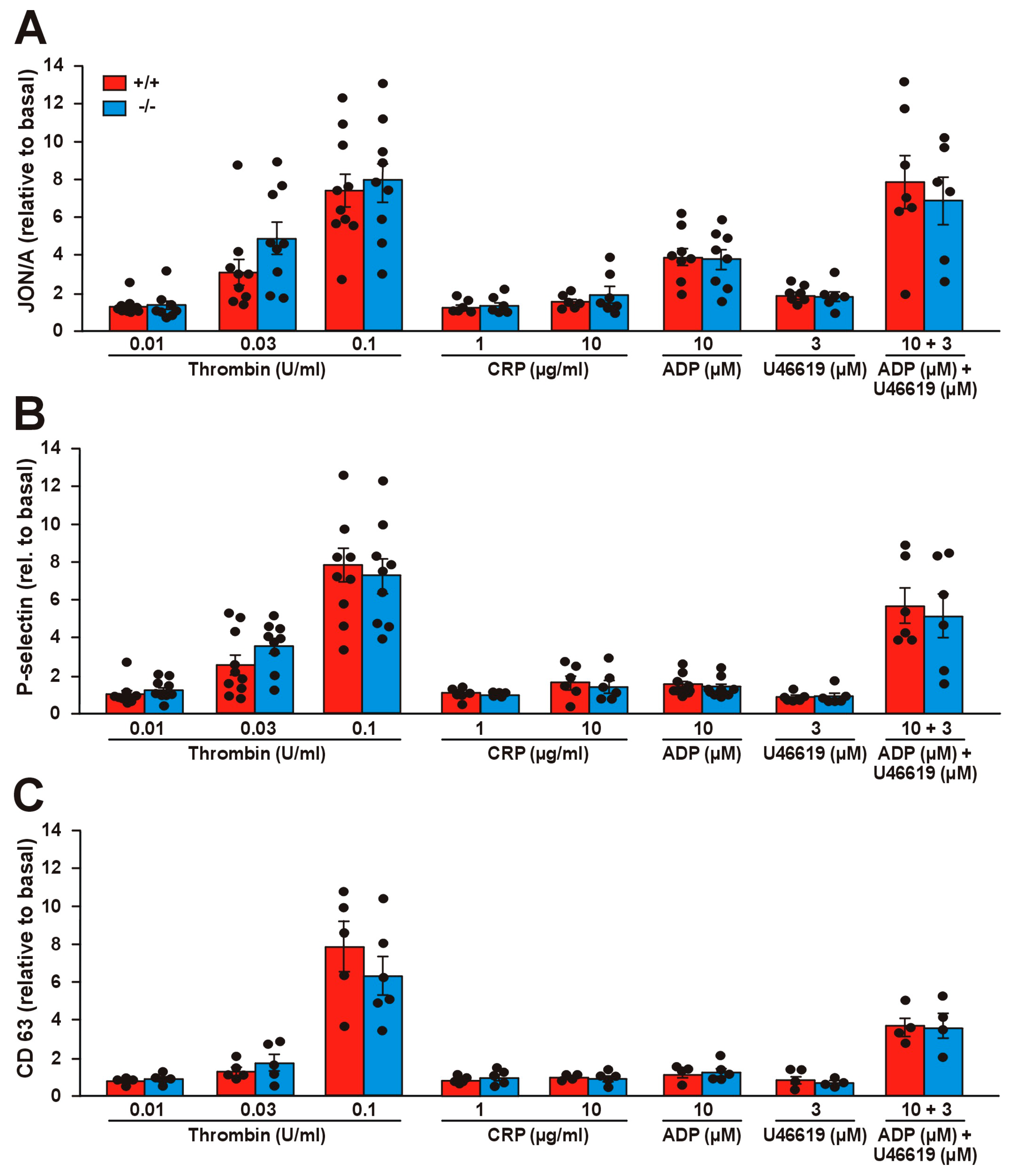
2.4. Effect of Coro1 Deficiency on Integrin αIIbβ3 Activation and Granule Secretion
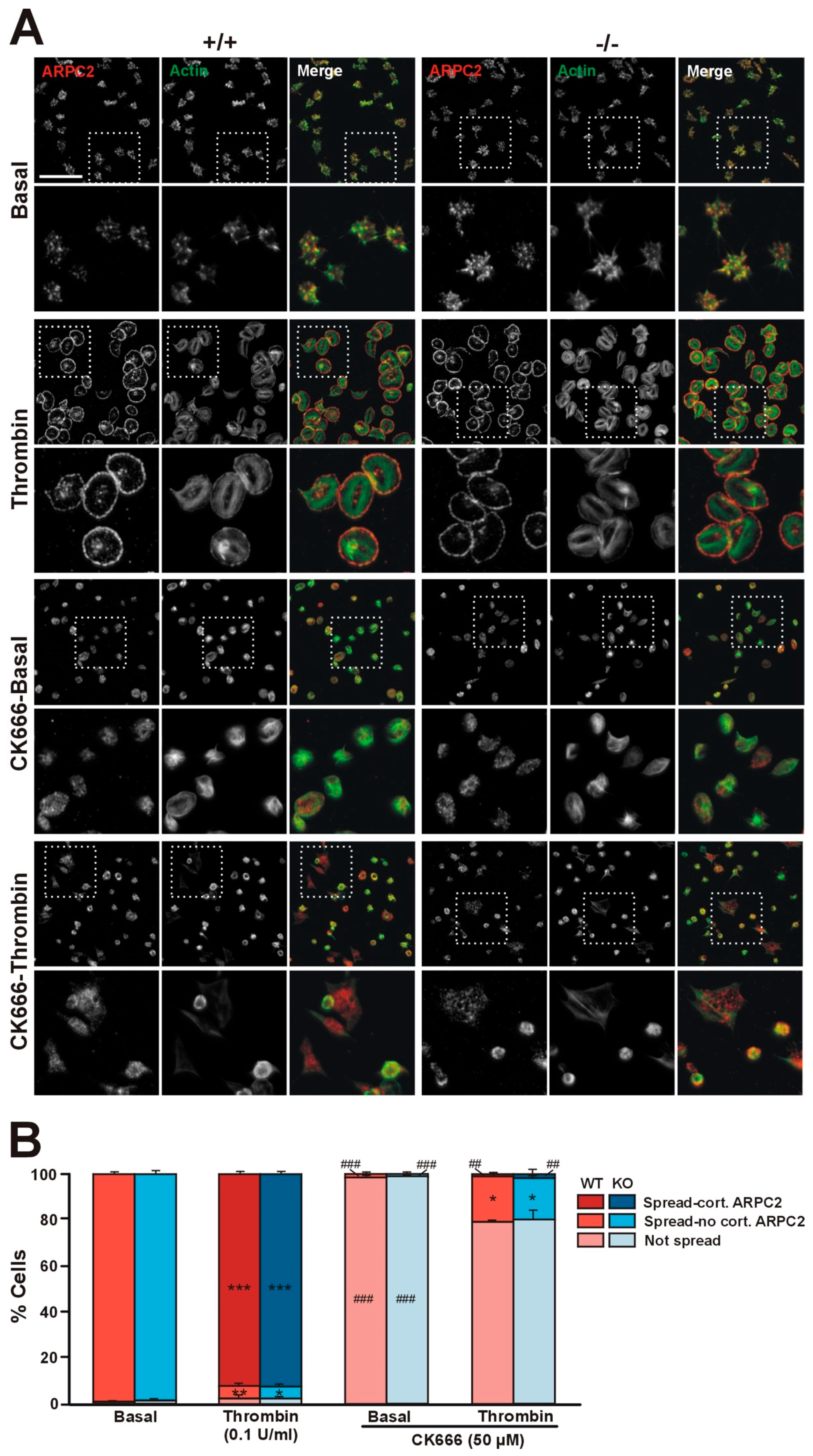
2.5. Effect of Coro1 Deficiency on Platelet Aggregation and Spreading
2.6. Coro1 Is Dispensable for Arp2/3 Complex Localization
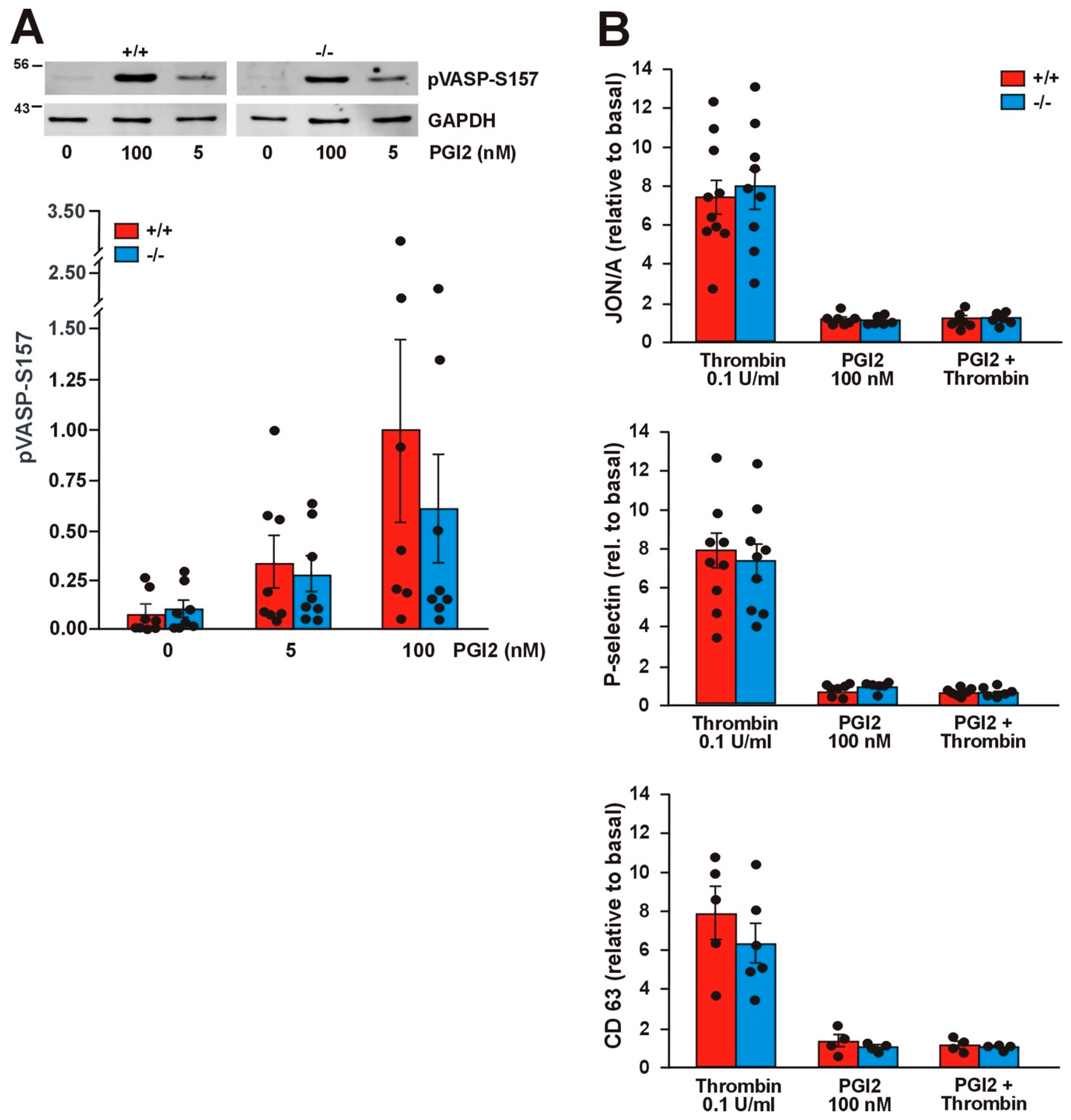
2.7. Absence of Coro1 Does Not Affect cAMP Signaling
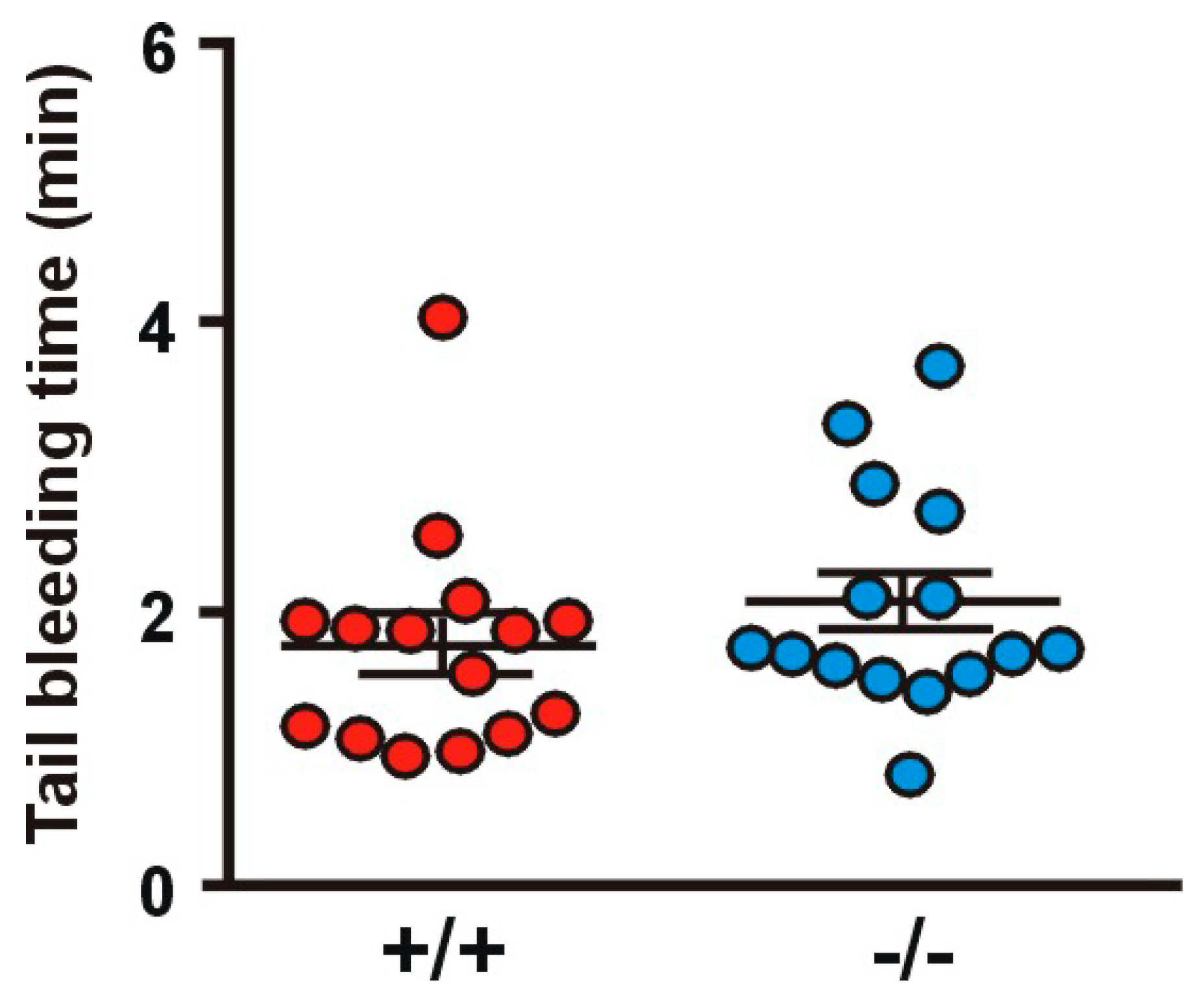
2.8. Absence of Coro1 Does Not Impair Hemostasis
3. Discussion
4. Materials and Methods
4.1. Reagents
4.2. Experimental Animals
4.3. Mouse Platelet Preparation
4.4. Western Blot
4.5. Flow Cytometry
4.6. Aggregation, Spreading, and Immunostaining
4.7. Tail Bleeding Assay
4.8. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| ACD | acid citrate dextrose |
| ADP | adenosine diphosphate |
| ARPC2 | p34-Arc, component of the Arp2/3 complex |
| BSA | bovine serum albumin |
| cAMP | cyclic adenosine monophosphate |
| CDK5 | cyclin-dependent kinase 5 |
| CDx | cluster of differentiation x |
| CRP | collagen related peptide |
| FACS | fluorescence-activated cell sorting |
| FITC | fluorescein isothiocyanate |
| GEFOGER | Gly-Phe-Hyp-Gly-Glu-Arg |
| ICAM-1 | intercellular adhesion molecule 1 |
| KO | knockout |
| LFA-1 | lymphocyte function-associated antigen 1 |
| PAGE | polyacrylamide gel electrophoresis |
| PBS | phosphate buffered saline |
| PDE4 | phosphodiesterase 4 |
| PFA | paraformaldehyde |
| PGI2 | prostacyclin |
| PRP | platelet-rich plasma |
| SEM | standard error of the mean |
| TNF | tumor necrosis factor |
| TRITC | tetramethylrhodamine isothiocyanate |
| VASP | vasodilator-stimulated phosphoprotein |
| WT | wild type |
References
- Tomaiuolo, M.; Brass, L.F.; Stalker, T.J. Regulation of platelet activation and coagulation and its role in vascular injury and arterial thrombosis. Interv. Cardiol. Clin. 2017, 6, 1–12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hartwig, J.H. The platelet cytoskeleton. In Platelets, 3rd ed.; Michelson, A.D., Ed.; Academic Press: London, UK, 2013; pp. 145–168. [Google Scholar]
- Smolenski, A. Novel roles of cAMP/cGMP-dependent signaling in platelets. J. Thromb. Haemost. 2012, 10, 167–176. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chan, K.T.; Creed, S.J.; Bear, J.E. Unraveling the enigma: Progress towards understanding the coronin family of actin regulators. Trends Cell Biol. 2011, 21, 481–488. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pieters, J.; Muller, P.; Jayachandran, R. On guard: Coronin proteins in innate and adaptive immunity. Nat. Rev. Immunol. 2013, 13, 23765056. [Google Scholar] [CrossRef] [PubMed]
- Riley, D.R.J.; Khalil, J.S.; Naseem, K.M.; Rivero, F. Biochemical and immunocytochemical characterization of coronins in platelets. Platelets 2019, 4, 1–12. [Google Scholar] [CrossRef] [Green Version]
- Suzuki, K.K.; Nishihata, J.; Arai, Y.; Honma, N.; Yamamoto, K.; Irimura, T.; Toyoshima, S. Molecular cloning of a novel actin-binding protein, p57, with a WD repeat and a leucine zipper motif. FEBS Lett. 1995, 364, 283–288. [Google Scholar] [CrossRef] [Green Version]
- Ferrari, G.; Langen, H.; Naito, M.; Pieters, J. A coat protein on phagosomes involved in the intracellular survival of mycobacteria. Cell 1999, 97, 435–447. [Google Scholar] [CrossRef] [Green Version]
- Jayachandran, R.; Liu, X.; Bosedasgupta, S.; Müller, P.; Zhang, C.-L.; Moshous, D.; Studer, V.; Schneider, J.; Genoud, C.; Fossoud, C.; et al. Coronin 1 regulates cognition and behavior through modulation of cAMP/protein kinase A signaling. PLoS Biol. 2014, 12, e1001820. [Google Scholar] [CrossRef] [Green Version]
- Pick, R.; Begandt, D.; Stocker, T.J.; Salvermoser, M.; Thome, S.; Böttcher, R.T.; Montanez, E.; Harrison, U.; Forné, I.; Khandoga, A.G.; et al. Coronin 1A, a novel player in integrin biology, controls neutrophil trafficking in innate immunity. Blood 2017, 130, 847–858. [Google Scholar] [CrossRef]
- Ojeda, V.; Castro-Castro, A.; Bustelo, X.R. Coronin1 proteins dictate Rac1 intracellular dynamics and cytoskeletal output. Mol. Cell. Biol. 2014, 34, 3388–3406. [Google Scholar] [CrossRef] [Green Version]
- Combaluzier, B.; Mueller, P.; Massner, J.; Finke, D.; Pieters, J. Coronin 1 is essential for IgM-mediated Ca2+ mobilization in B cells but dispensable for the generation of immune responses in vivo. J. Immunol. 2009, 182, 1954–1961. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grogan, A.; Reeves, E.; Keep, N.; Wientjes, F.; Totty, N.F.; Burlingame, A.L.; Hsuan, J.J.; Segal, A.W. Cytosolic phox proteins interact with and regulate the assembly of coronin in neutrophils. J. Cell Sci. 1997, 110, 3071–3081. [Google Scholar] [PubMed]
- Moriceau, S.; Kantari, C.; Mocek, J.; Davezac, N.; Gabillet, J.; Guerrera, I.C.; Brouillard, F.; Tondelier, D.; Sermet-Gaudelus, I.; Danel, C.; et al. Coronin-1 is associated with neutrophil survival and is cleaved during apoptosis: Potential implication in neutrophils from cystic fibrosis patients. J. Immunol. 2009, 182, 7254–7263. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jayachandran, R.; Sundaramurthy, V.; Combaluzier, B.; Mueller, P.; Korf, H.; Huygen, K.; Miyazaki, T.; Albrecht, I.; Massner, J.; Pieters, J. Survival of mycobacteria in macrophages is mediated by coronin 1-dependent activation of calcineurin. Cell 2007, 130, 37–50. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mueller, P.; Massner, J.; Jayachandran, R.; Combaluzier, B.; Albrecht, I.; Gatfield, J.; Blum, C.; Ceredig, R.; Rodewald, H.R.; Rolink, A.G.; et al. Regulation of T cell survival through coronin-1-mediated generation of inositol-1,4,5-trisphosphate and calcium mobilization after T cell receptor triggering. Nat. Immunol. 2008, 9, 424–431. [Google Scholar] [CrossRef] [PubMed]
- Föger, N.; Rangell, L.; Danilenko, D.M.; Chan, A.C. Requirement for coronin 1 in T lymphocyte trafficking and cellular homeostasis. Science 2006, 313, 839–842. [Google Scholar] [CrossRef] [Green Version]
- Shiow, L.R.; Roadcap, D.W.; Paris, K.; Watson, S.R.; Grigorova, I.L.; Lebet, T.; An, J.; Xu, Y.; Jenne, C.N.; Föger, N.; et al. The actin regulator coronin 1A is mutant in a thymic egress-deficient mouse strain and in a patient with severe combined immunodeficiency. Nat. Immunol. 2008, 9, 1307–1315. [Google Scholar] [CrossRef]
- Haraldsson, M.K.; Louis-Dit-Sully, C.A.; Lawson, B.R.; Gascoigne, N.R.J.; Argyrios, N.; Kono, D.H. The lupus-related Lmb3 locus contains a disease-suppressing Coronin-1A gene mutation. Immunity 2009, 28, 40–51. [Google Scholar] [CrossRef] [Green Version]
- Stocker, T.; Pircher, J.; Skenderi, A.; Ehrlich, A.; Eberle, C.; Megens, R.; Petzold, T.; Zhang, Z.; Walzog, B.; Müller-Taubenberger, A.; et al. The actin regulator coronin-1A modulates platelet shape change and consolidates arterial thrombosis. Thromb. Haemost. 2018, 118, 2098–2111. [Google Scholar] [CrossRef]
- Michelson, A.; Barnard, M.; Hechtman, H.; MacGregor, H.; Connolly, R.; Loscalzo, J.; Valeri, C. In vivo tracking of platelets: Circulating degranulated platelets rapidly lose surface P-selectin but continue to circulate and function. Proc. Natl. Acad. Sci. USA 1996, 93, 11877–11882. [Google Scholar] [CrossRef] [Green Version]
- Piguet, P.F.; Vesin, C.; Ryser, J.E.; Senaldi, G.; Grau, G.E.; Tacchini-Cottier, F. An effector role for platelets in systemic and local lipopolysaccharide—Induced toxicity in mice, mediated by a CD11a- and CD54-dependent interaction with endothelium. Infect. Immun. 1993, 61, 4182–4187. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guo, J.; Piguet, P.F. Stimulation of thrombocytopoiesis decreases platelet beta2 but not beta1 or beta3 integrins. Br. J. Haematol. 1998, 100, 712–719. [Google Scholar] [CrossRef] [PubMed]
- Piguet, P.F.; Vesin, C.; Rochat, A. Beta2 integrin modulates platelet caspase activation and life span in mice. Eur. J. Cell Biol. 2001, 80, 171–177. [Google Scholar] [CrossRef] [PubMed]
- Zeiler, M.; Moser, M.; Mann, M. Copy number analysis of the murine platelet proteome spanning the complete abundance range. Mol. Cell. Proteom. 2014, 13, 3435–3445. [Google Scholar] [CrossRef] [Green Version]
- Philippeaux, M.M.; Vesin, C.; Tacchini-Cottier, F.; Piguet, P.F. Activated human platelets express β2 integrin. Eur. J. Haematol. 2009, 56, 130–137. [Google Scholar] [CrossRef]
- Zhu, X.; Subbaraman, R.; Sano, H.; Jacobs, B.; Sano, A.; Boetticher, E.; Muñoz, N.M.; Leff, A.R. A surrogate method for assessment of β2-integrin-dependent adhesion of human eosinophils to ICAM-1. J. Immunol. Methods 2000, 240, 157–164. [Google Scholar] [CrossRef]
- Liu, J.; Muñoz, N.M.; Meliton, A.Y.; Zhu, X.; Lambertino, A.T.; Xu, C.; Myo, S.; Myou, S.; Boetticher, E.; Johnson, M.; et al. β2-Integrin adhesion caused by eotaxin but not IL-5 is blocked by PDE-4 inhibition and β2-adrenoceptor activation in human eosinophils. Pulm. Pharmacol. Ther. 2004, 17, 73–79. [Google Scholar] [CrossRef]
- Jayachandran, R.; Gumienny, A.; Bolinger, B.; Ruehl, S.; Lang, M.J.; Fucile, G.; Mazumder, S.; Tchang, V.; Woischnig, A.-K.; Stiess, M.; et al. Disruption of coronin 1 signaling in T cells promotes allograft tolerance while maintaining anti-pathogen immunity. Immunity 2019, 50, 152–165. [Google Scholar] [CrossRef] [Green Version]
- Falet, H. Anatomy of the platelet cytoskeleton. In Platelets in Thrombotic and Non-Thrombotic Disorders; Graesele, P., Kleiman, N.S., Lopez, J.A., Page, C.P., Eds.; Springer: Berlin, Germany, 2017; pp. 139–156. [Google Scholar]
- Jarvis, G.E. Platelet aggregation: Turbidimetric measurements. Methods Mol. Biol. 2004, 272, 65–76. [Google Scholar]
- Jarvis, G.E. Platelet aggregation in whole blood: Impedance and particle counting methods. Methods Mol. Biol. 2004, 272, 77–87. [Google Scholar]
- Fagerholm, S.C.; Guenther, C.; Asens, M.L.; Savinko, T.; Uotila, L.M. Beta2-Integins and interacting proteins in leukocyte trafficking, immune supression, and immunodeficiency disease. Front. Immunol. 2019, 10, 1–10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cerletti, C.; de Gaetano, G.; Lorenzet, R. Platelet-leukocyte interactions: Multiple links between inflammation, blood coagulation and vascular risk. Mediterr. J. Hematol. Infect. Dis. 2010, 2, e2010023. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Bosedasgupta, S.; Jayachandran, R.; Studer, V.; Rühl, S.; Stiess, M.; Pieters, J. Activation of the cAMP/protein kinase A signalling pathway by coronin 1 is regulated by cyclin-dependent kinase 5 activity. FEBS Lett. 2016, 590, 279–287. [Google Scholar] [CrossRef] [PubMed]
- Vinet, A.F.; Fiedler, T.; Studer, V.; Froquet, R.; Dardel, A.; Cosson, P.; Pieters, J. Initiation of multicellular differentiation in Dictyostelium discoideum is regulated by coronin A. Mol. Biol. Cell 2014, 25, 688–701. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Zhong, K.; Yin, Z.; Hu, J.; Wang, W.; Li, L.; Zhang, H.; Zheng, X.; Wang, P.; Zhang, Z. The seven transmembrane domain protein MoRgs7 functions in surface perception and undergoes coronin MoCrn1-dependent endocytosis in complex with Gα subunit MoMagA to promote cAMP signaling and appressorium formation in Magnaporthe oryzae. PLoS Pathog. 2019, 15, e1007382. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rondina, M.T.; Weyrich, A.S. Targeting phosphodiesterases in anti-platelet therapy. Handb. Exp. Pharmacol. 2012, 210, 225–238. [Google Scholar]
- Burkhart, J.M.; Vaudel, M.; Gambaryan, S.; Radau, S.; Walter, U.; Martens, L.; Geiger, J.; Sickmann, A.; Zahedi, R.P. The first comprehensive and quantitative analysis of human platelet protein composition allows the comparative analysis of structural and functional pathways. Blood 2012, 120, e73–e82. [Google Scholar] [CrossRef] [Green Version]
- Thomas, S.G.; Poulter, N.S.; Bem, D.; Finney, B.; Machesky, L.M.; Watson, S.P. The actin binding proteins cortactin and HS1 are dispensable for platelet actin nodule and megakaryocyte podosome formation. Platelets 2017, 28, 372–379. [Google Scholar] [CrossRef] [Green Version]
- Zuidscherwoude, M.; Green, H.L.H.; Thomas, S.G. Formin proteins in megakaryocytes and platelets: Regulation of actin and microtubule dynamics. Platelets 2019, 30, 23–30. [Google Scholar] [CrossRef] [Green Version]
- Spoerl, Z.; Stumpf, M.; Noegel, A.A.; Hasse, A. Oligomerization, F-actin interaction, and membrane association of the ubiquitous mammalian coronin 3 are mediated by its carboxyl terminus. J. Biol. Chem. 2002, 277, 48858–48867. [Google Scholar] [CrossRef] [Green Version]








© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Riley, D.R.J.; Khalil, J.S.; Pieters, J.; Naseem, K.M.; Rivero, F. Coronin 1 Is Required for Integrin β2 Translocation in Platelets. Int. J. Mol. Sci. 2020, 21, 356. https://doi.org/10.3390/ijms21010356
Riley DRJ, Khalil JS, Pieters J, Naseem KM, Rivero F. Coronin 1 Is Required for Integrin β2 Translocation in Platelets. International Journal of Molecular Sciences. 2020; 21(1):356. https://doi.org/10.3390/ijms21010356
Chicago/Turabian StyleRiley, David R. J., Jawad S. Khalil, Jean Pieters, Khalid M. Naseem, and Francisco Rivero. 2020. "Coronin 1 Is Required for Integrin β2 Translocation in Platelets" International Journal of Molecular Sciences 21, no. 1: 356. https://doi.org/10.3390/ijms21010356
APA StyleRiley, D. R. J., Khalil, J. S., Pieters, J., Naseem, K. M., & Rivero, F. (2020). Coronin 1 Is Required for Integrin β2 Translocation in Platelets. International Journal of Molecular Sciences, 21(1), 356. https://doi.org/10.3390/ijms21010356