Epigenetic Regulation in Etiology of Type 1 Diabetes Mellitus


Abstract
:1. Introduction
1.1. HLA Class II Alleles as a Main Predisposition Genetic Factor in T1DM
1.2. Environmental Risk Factors in T1DM
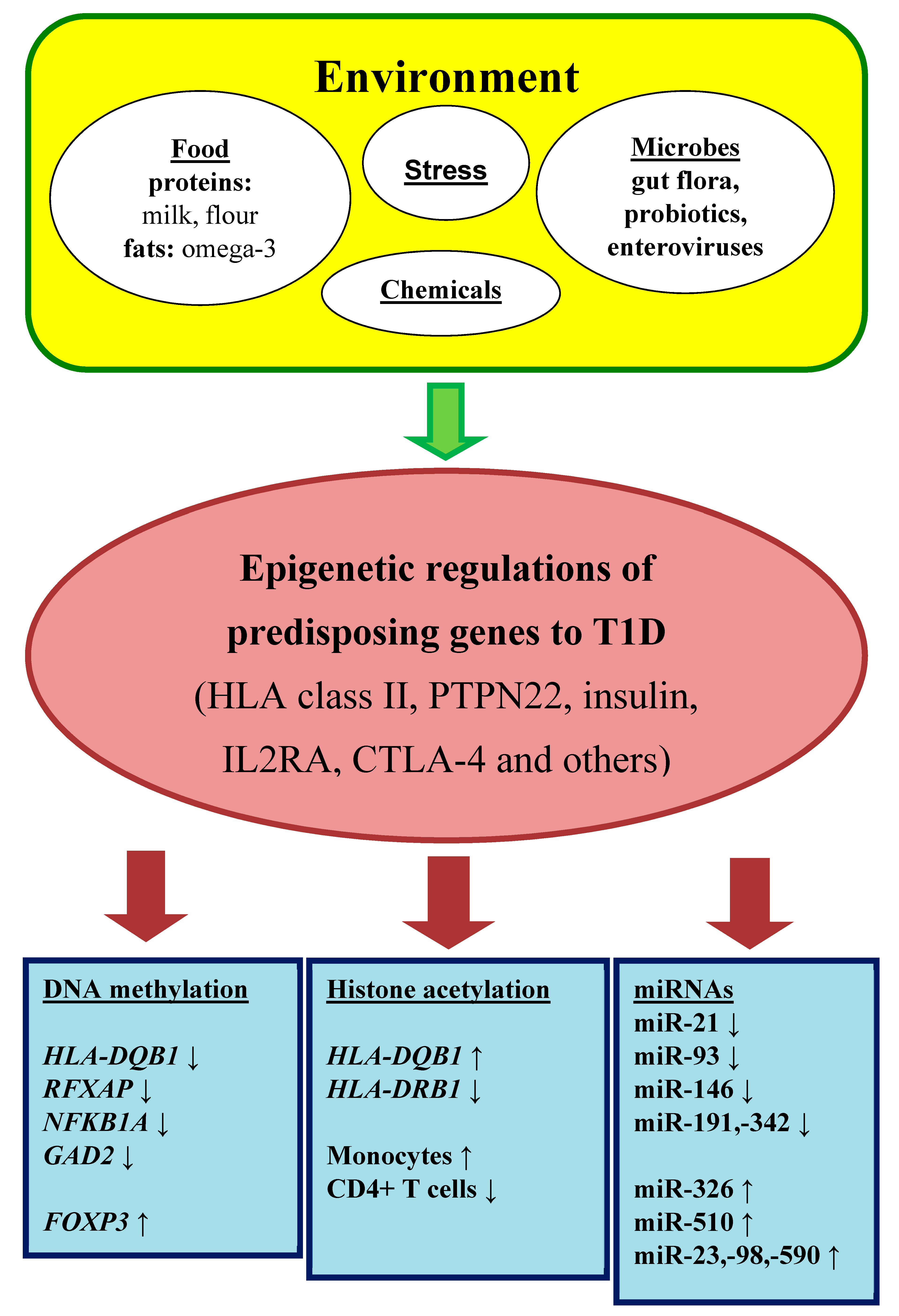
1.3. Epigenetic Regulations as a Connection between Environment and Genome
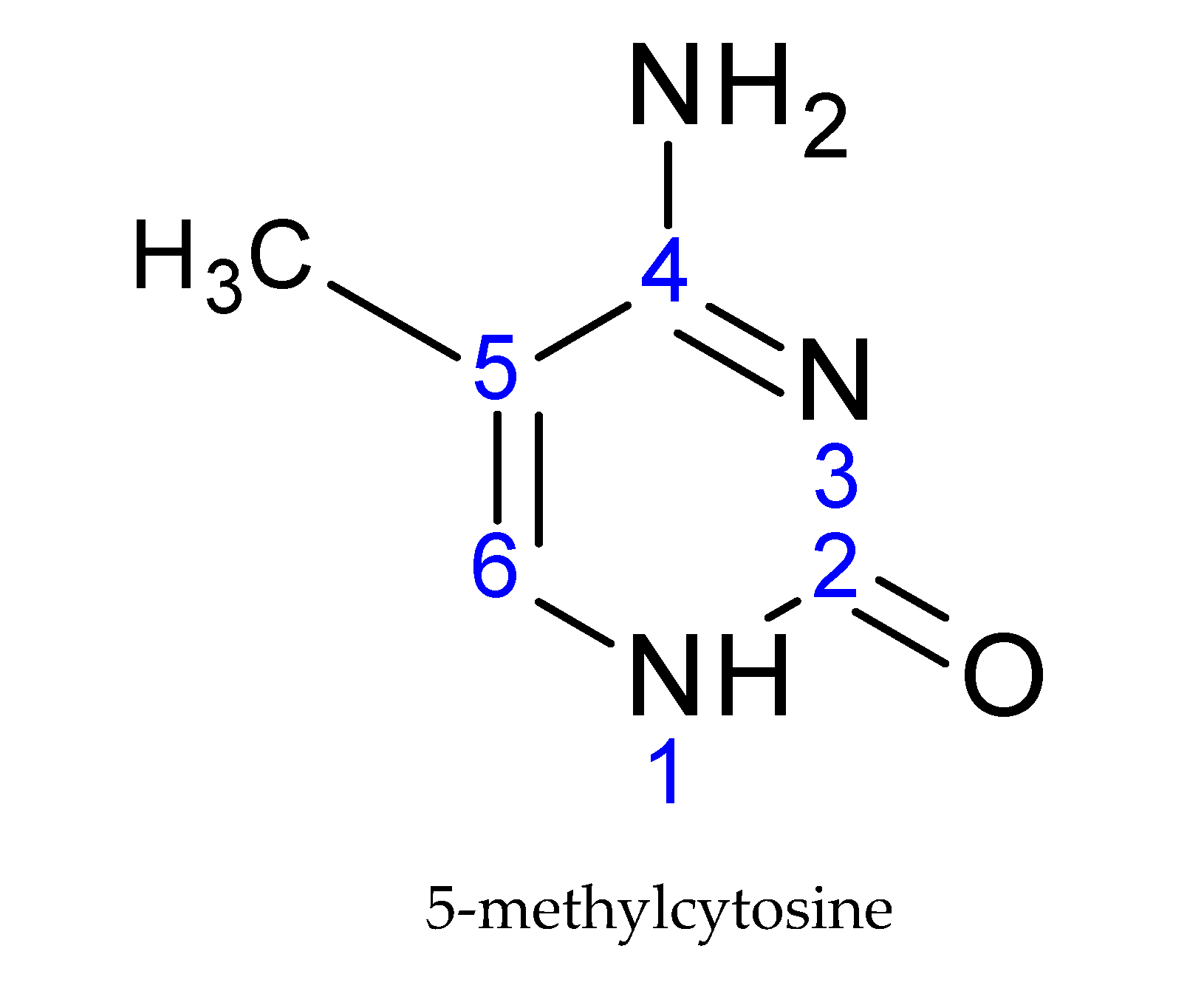
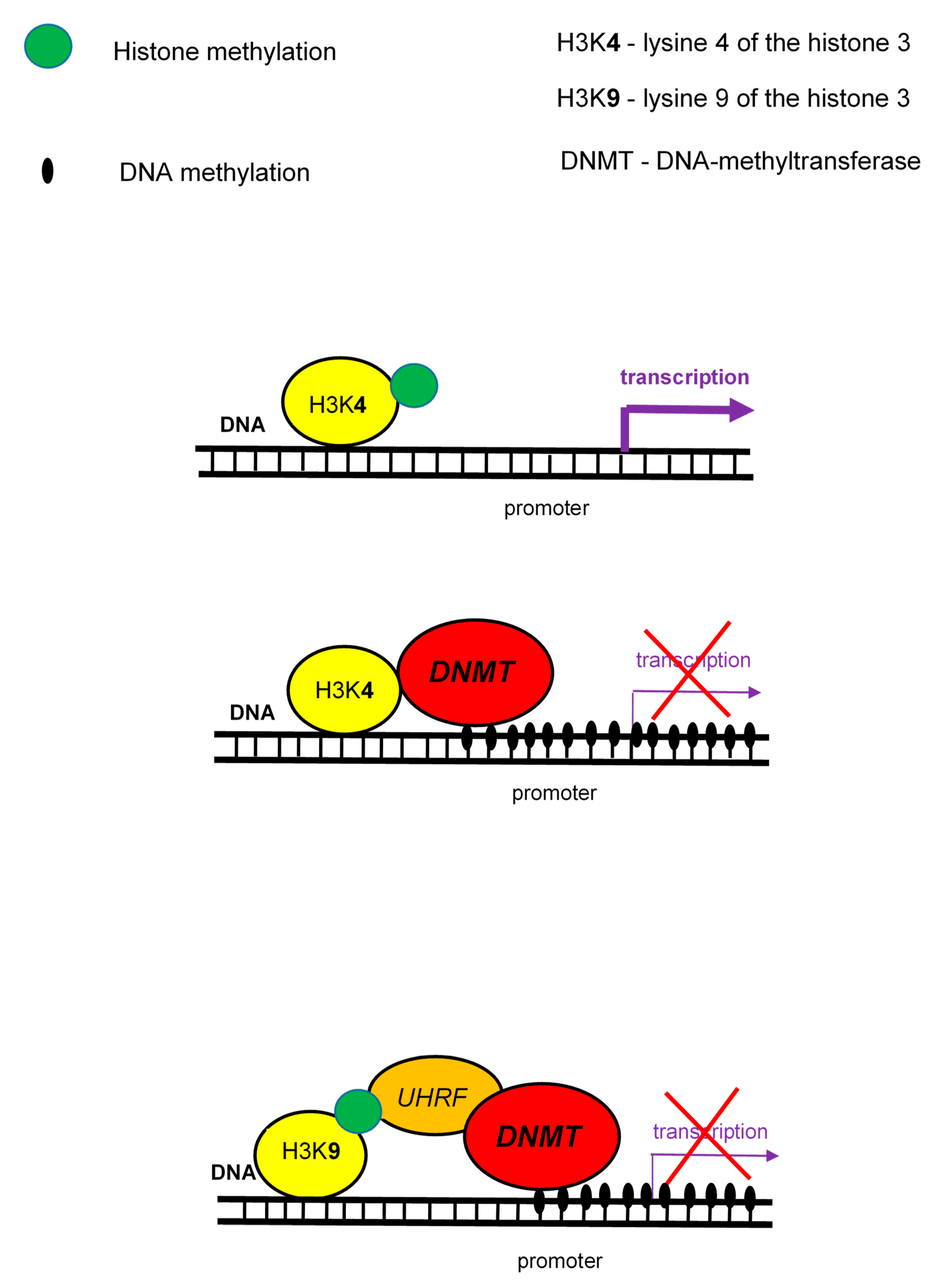
2. DNA Methylation and Its Role in T1DM
2.1. Monozygotic Twin Studies
2.2. The Decrease of Immune Tolerance is Regulated by DNA Methylation
2.3. Insulin Gene and Its Epigenetic Modifications
2.4. Interleukin 2 Receptor α-Chain Gene and Its Epigenetic Modifications
2.5. The Intestinal Microbiome and Epigenome in T1DM
3. Histone Modifications and Their Role in T1DM
3.1. The Studies of Natarajan’s Group
3.2. The Role of Innate Immunity
3.3. The Decrease of Immune Tolerance is Regulated by Histone Acetylation
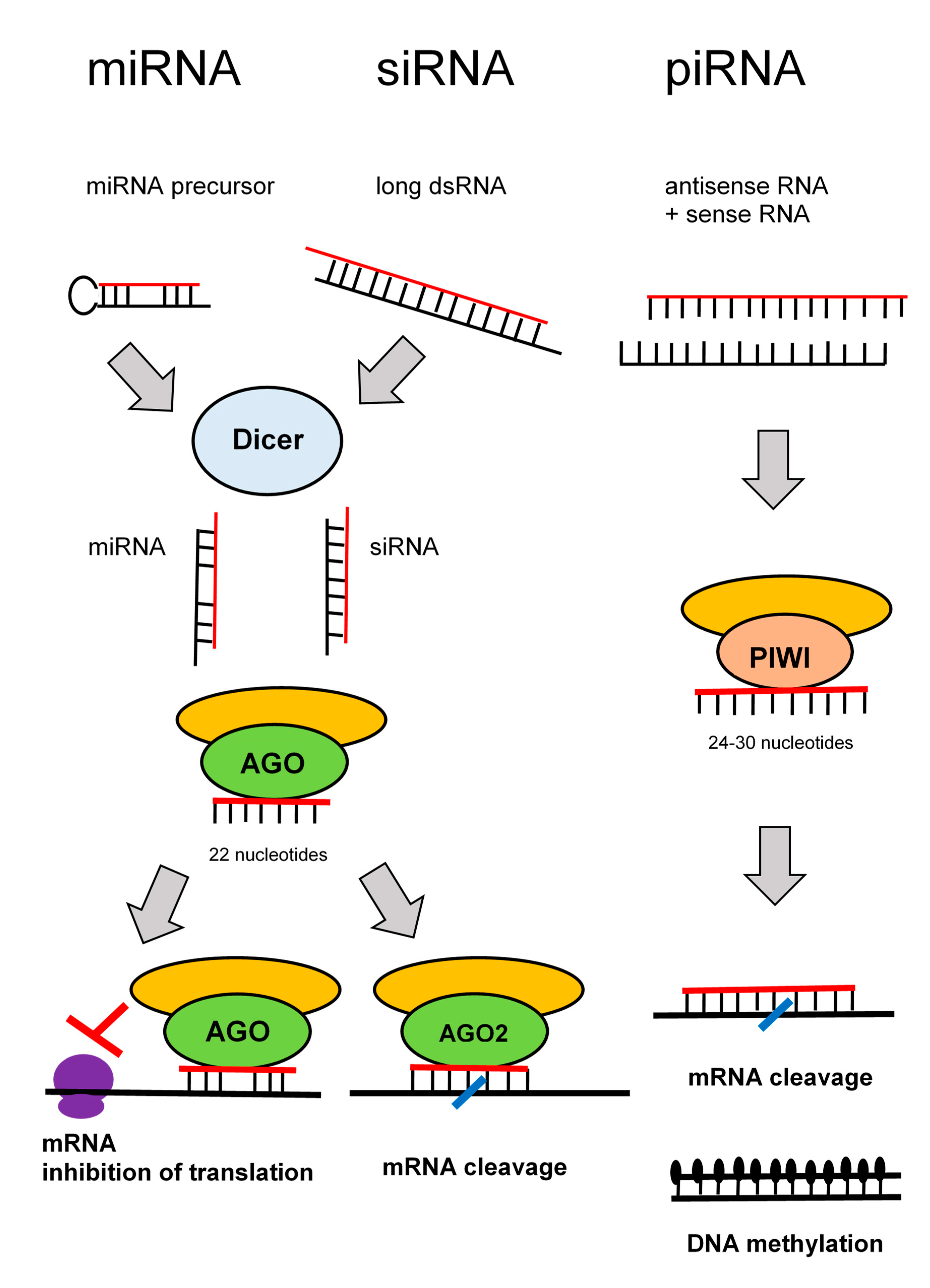
4. RNA Interference and Their Role in T1DM
4.1. The Decrease of Immune Tolerance is Regulated by miRNAs
4.2. The Detection of miRNAs in Peripheral Blood Mononuclear Cells
4.3. The Experimental Studies in Cultured Cells and Animal Models
5. Conclusions
Funding
Conflicts of Interest
Abbreviations
| APC | antigen presenting cells |
| CIITA | class II transactivator |
| COX | cyclooxygenase |
| CREBBP | cAMP-response element binding protein (CREB) binding protein |
| DNMTs | DNA-methyltransferases |
| FOXP | “forkhead box P” transcription factor |
| GAD | glutamic acid decarboxylase |
| HATs | histone-acetyltransferases |
| HDACs | histone-deacetylases |
| HLA | human leukocyte antigen |
| HPA | hypothalamic-pituitary-adrenal |
| INS | insulin |
| LADA | latent autoimmune diabetes in adults |
| MBD | methyl-CpG-binding proteins |
| mTOR | mammalian/mechanistic target of rapamycin (serine-threonine kinase) |
| MZ twins | monozygotic twins |
| NF-κB | nuclear factor kappa B |
| NFKB1A | gene for the inhibitor of NF-κB, IκBα |
| NOD mice | non-obese diabetic mice |
| NRF1 | nuclear respiratory factor 1 |
| PDCD4 | programmed cell death protein 4 |
| PTPN22 | protein tyrosin-phosphatase non-receptor-type 22 |
| RFXAP | regulatory factor X associated protein |
| RISC | RNA-induced silencing complex |
| SAM | S-adenosyl methionine donor |
| SNP | single nucleotide polymorphism |
| T1DM | type 1 diabetes mellitus |
| Tregs | regulatory T cells |
| TSA | trichostatin A |
| VNTR | variable number tandem repeat |
References
- Sharp, S.A.; Weedon, M.N.; Hagopian, W.A.; Oram, R.A. Clinical and research uses of genetic risk scores in type 1 diabetes. Curr. Opin. Genet. Dev. 2018, 50, 96–102. [Google Scholar] [CrossRef]
- Nyaga, D.M.; Vickers, M.H.; Jefferies, C.; Perry, J.K.; O’Sullivan, J.M. The genetic architecture of type 1 diabetes mellitus. Mol. Cell. Endocrinol. 2018, 477, 70–80. [Google Scholar] [CrossRef]
- Abbas, A.K.; Lichtman, A.H.; Pillai, S. Cellular and Molecular Immunology, 7th ed.; Elsevier Saunders: Philadelphia, PA, USA, 2012. [Google Scholar]
- Cerna, M. Genetics of autoimmune diabetes mellitus. Wien. Med. Wochenschr. 2008, 158, 2–12. [Google Scholar] [CrossRef]
- Rewers, M.; Ludvigsson, J. Environmental risk factors for type 1 diabetes. Lancet 2016, 387, 2340–2348. [Google Scholar] [CrossRef] [Green Version]
- Paun, A.; Yau, C.; Danska, J.S. The influence of the microbiome on type 1 diabetes. J. Immunol. 2017, 198, 590–595. [Google Scholar] [CrossRef]
- Bach, J.F. The hygiene hypothesis in autoimmunity: The role of pathogens and commensals. Nat. Rev. Immunol. 2018, 18, 105–120. [Google Scholar] [CrossRef] [PubMed]
- Sharif, K.; Watad, A.; Coplan, L.; Amital, H.; Shoenfeld, Y.; Afek, A. Psychological stress and type 1 diabetes mellitus: What is the link? Expert Rev. Clin. Immunol. 2018, 14, 1081–1088. [Google Scholar] [CrossRef] [PubMed]
- Allis, C.D.; Jenuwein, T. The molecular hallmarks of epigenetic control. Nat. Rev. Genet. 2016, 17, 487–500. [Google Scholar] [CrossRef]
- Rose, N.R.; Klose, R.J. Understanding the relationship between DNA methylation and histone lysine methylation. Biochim. Biophys. Acta 2014, 1839, 1362–1372. [Google Scholar] [CrossRef] [Green Version]
- Garcia-Gonzalez, E.; Escamilla-Del-Arenal, M.; Arzate-Mejia, R.; Recillas-Targa, F. Chromatin remodeling effects on enhancer activity. Cell. Mol. Life Sci. 2016, 73, 2897–2910. [Google Scholar] [CrossRef] [PubMed]
- Ruiz-Hernandez, A.; Kuo, C.-C.; Rentero-Garrido, P.; Tang, W.-Y.; Redon, J.; Ordovas, J.M.; Navas-Acien, A.; Tellez-Plaza, M. Environmental chemicals and DNA methylation in adults: A systematic review of the epidemiologic evidence. Clin. Epigenet. 2015, 7, 55. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gaine, M.E.; Chatterjee, S.; Abel, T. Sleep deprivation and the epigenome. Front. Neural Circuits 2018, 12, 14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, G.; Wu, Q.; Gao, Y.; Chen, M.; Yang, M. The epigenetics of aging in invertebrates. Int. J. Mol. Sci. 2019, 20, 4535. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sapienza, C.; Issa, J.P. Diet, nutrition, and cancer epigenetics. Annu. Rev. Nutr. 2016, 36, 665–681. [Google Scholar] [CrossRef] [PubMed]
- Picascia, A.; Grimaldi, V.; Pignalosa, O.; De Pascale, M.R.; Schiano, C.; Napoli, C. Epigenetic control of autoimmune diseases: From bench to bedside. Clin. Immunol. 2015, 157, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Taudt, A.; Colomé-Tatché, M.; Johannes, F. Genetic sources of population epigenomic variation. Nat. Rev. Genet. 2016, 17, 319–332. [Google Scholar] [CrossRef]
- Surace, A.E.A.; Hedrich, C.M. The role of epigenetics in autoimmune/inflammatory disease. Front. Immunol. 2019, 10, 1525. [Google Scholar] [CrossRef] [Green Version]
- Klose, R.J.; Bird, A.P. Genomic DNA methylation: The mark and its mediators. Trends Biochem. Sci. 2006, 31, 89–97. [Google Scholar] [CrossRef]
- Weber, M.; Hellmann, I.; Stadler, M.B.; Ramos, L.; Pääbo, S.; Rebhan, M.; Schübeler, D. Distribution, silencing potential and evolutionary impact of promoter DNA methylation in the human genome. Nat. Genet. 2007, 39, 457–466. [Google Scholar] [CrossRef]
- Shen, H.; Qiu, C.; Li, J.; Tian, Q.; Deng, H.-W. Characterization of the DNA methylome and its interindividual variation in human peripheral blood monocytes. Epigenomics 2013, 5, 255–269. [Google Scholar] [CrossRef] [Green Version]
- Majumder, P.; Boss, J.M. DNA methylation dysregulates and silences the HLA-DQ locus by altering chromatin architecture. Genes Immun. 2011, 12, 291–299. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zajacová, M. Regulation of HLA Class II Genes Expression. Ph.D. Thesis, Charles University, Third Faculty of Medicine, Vinohrady, Czech Republic, 2018. [Google Scholar]
- Xiang, Z.; Yang, Y.; Chang, C.; Lu, Q. The epigenetic mechanism for discordance of autoimmunity in monozygotic twins. J. Autoimmun. 2017, 83, 43–50. [Google Scholar] [CrossRef] [PubMed]
- Rakyan, V.K.; Beyan, H.; Down, T.A.; Hawa, M.I.; Maslau, S.; Aden, D.; Daunay, A.; Busato, F.; Mein, C.A.; Manfras, B.; et al. Identification of type 1 diabetes-associated DNA methylation variable positions that precede disease diagnosis. PLoS Genet. 2011, 7, 1002300. [Google Scholar] [CrossRef] [PubMed]
- Stefan, M.; Zhang, W.; Concepcion, E.; Yi, Z.; Tomer, Y. DNA methylation profiles in type 1 diabetes twins point to strong epigenetic effects on etiology. J. Autoimmun. 2014, 50, 33–37. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Elboudwarej, E.; Cole, M.; Briggs, F.B.S.; Fouts, A.; Fain, P.R.; Quach, H.; Quach, D.; Sinclair, E.; Criswell, L.A.; Lane, J.A.; et al. Hypomethylation within gene promoter regions and type 1 diabetes in discordant monozygotic twins. J. Autoimmun. 2016, 68, 23–29. [Google Scholar] [CrossRef] [Green Version]
- Paul, D.; Teschendorff, A.E.; Dang, M.A.; Lowe, R.; Hawa, M.I.; Ecker, S.; Beyan, H.; Cunningham, S.; Fouts, A.R.; Ramelius, A.; et al. Increased DNA methylation variability in type 1 diabetes across three immune effector cell types. Nat. Commun. 2016, 7, 13555. [Google Scholar] [CrossRef]
- Disanto, G.; Vcelakova, J.; Pakpoor, J.; Elangovan, R.I.; Sumnik, Z.; Ulmannova, T.; Ebers, G.C.; Ramagopalan, S.V.; Stechová, K. DNA methylation in monozygotic quadruplets affected by type 1 diabetes. Diabetologia 2013, 56, 2093–2095. [Google Scholar] [CrossRef]
- Zajacova, M.; Kotrbova-Kozak, A.; Cepek, P.; Cerna, M. Differences in promoter DNA methylation and mRNA expression of individual alleles of the HLA class II DQA1 gene. Immunol. Lett. 2015, 167, 147–154. [Google Scholar] [CrossRef]
- Cepek, P.; Zajacova, M.; Kotrbova-Kozak, A.; Silhova, E.; Cerna, M. DNA methylation and mRNA expression of HLA-DQA1 alleles in type 1 diabetes mellitus. Immunology 2016, 148, 150–159. [Google Scholar] [CrossRef] [Green Version]
- Li, Y.; Zhao, M.; Hou, C.; Liang, G.; Yang, L.; Tan, Y.; Wang, Z.; Yin, H.; Zhou, Z.; Lu, Q. Abnormal DNA methylation in CD4+ T cells from people with latent autoimmune diabetes in adults. Diabetes Res. Clin. Pract. 2011, 94, 242–248. [Google Scholar] [CrossRef]
- Redondo, M.J.; Steck, A.K.; Pugliese, A. Genetics of type 1 diabetes. Pediatr. Diabetes 2018, 19, 346–353. [Google Scholar] [CrossRef] [PubMed]
- Fradin, D.; Le Fur, S.; Mille, C.; Naoui, N.; Groves, C.; Zelenika, D.; McCarthy, M.I.; Lathrop, M.; Bougnères, P. Association of the CpG methylation pattern of the proximal insulin gene promoter with type 1 diabetes. PLoS ONE 2012, 7, 0036278. [Google Scholar] [CrossRef] [PubMed]
- Rui, J.; Deng, S.; Lebastchi, J.; Clark, P.L.; Usmani-Brown, S.; Herold, K.C. Methylation of insulin DNA in response to proinflammatory cytokines during the progression of autoimmune diabetes in NOD mice. Diabetologia 2016, 59, 1021–1029. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Husseiny, M.I.; Kuroda, A.; Kaye, A.N.; Nair, I.; Kandeel, F.; Ferreri, K. Development of a quantitative methylation-specific polymerase chain reaction method for monitoring beta cell death in type 1 diabetes. PLoS ONE 2012, 7, 0047942. [Google Scholar] [CrossRef] [PubMed]
- Akirav, E.M.; Lebastchi, J.; Galvan, E.M.; Henegariu, O.; Akirav, M.; Ablamunits, V.; Lizardi, P.M.; Herold, K.C. Detection of beta cell death in diabetes using differentially methylated circulating DNA. Proc. Natl. Acad. Sci. USA 2011, 108, 19018–19023. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fisher, M.M.; Watkins, R.A.; Blum, J.; Evans-Molina, C.; Chalasani, N.; DiMeglio, L.A.; Mather, K.J.; Tersey, S.A.; Mirmira, R.G. Elevations in circulating methylated and unmethylated preproinsulin DNA in new-onset type 1 diabetes. Diabetes 2015, 64, 3867–3872. [Google Scholar] [CrossRef] [Green Version]
- Olsen, J.A.; Kenna, L.A.; Spelios, M.G.; Hessner, M.J.; Akirav, E.M. Circulating differentially methylated amylin DNA as a biomarker of β-cell loss in type 1 diabetes. PLoS ONE 2016, 11, 0152662. [Google Scholar] [CrossRef] [Green Version]
- Belot, M.P.; Fradin, D.; Mai, N.; Le Fur, S.; Zélénika, D.; Kerr-Conte, J.; Pattou, F.; Lucas, B.; Bougnères, P. CpG methylation changes within the IL2RA promoter in type 1 diabetes of childhood onset. PLoS ONE 2013, 8, e68093. [Google Scholar] [CrossRef]
- Chen, B.; Sun, L.; Zhang, X. Integration of microbiome and epigenome to decipher the pathogenesis of autoimmune diseases. J. Autoimmun. 2017, 83, 31–42. [Google Scholar] [CrossRef]
- Knip, M.; Siljander, H. The role of the intestinal microbiota in type 1 diabetes mellitus. Nat. Rev. Endocrinol. 2016, 12, 154–167. [Google Scholar] [CrossRef]
- Davis-Richardson, A.G.; Triplett, E.W. A model for the role of gut bacteria in the development of autoimmunity for type 1 diabetes. Diabetologia 2015, 58, 1386–1393. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gowher, H.; Jeltsch, A. Mammalian DNA methyltransferases: New discoveries and open questions. Biochem. Soc. Trans. 2018, 46, 1191–1202. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, K.; Luo, Y. Histone acetylation and the regulation of major histocompatibility class II gene expression. Adv. Protein Chem. Struct. Biol. 2017, 106, 71–111. [Google Scholar] [PubMed]
- Miao, F.; Smith, D.D.; Zhang, L.; Min, A.; Feng, W.; Natarajan, R. Lymphocytes from patients with type 1 diabetes display a distinct profile of chromatin histone H3 lysine 9 dimethylation. Diabetes 2008, 57, 3190–3198. [Google Scholar] [CrossRef] [Green Version]
- Miao, F.; Chen, Z.; Zhang, L.; Liu, Z.; Wu, X.; Yuan, Y.C.; Natarajan, R. Profiles of epigenetic histone post-translational modifications at type 1 diabetes susceptible genes. J. Biol. Chem. 2012, 287, 16335–16345. [Google Scholar] [CrossRef] [Green Version]
- Miao, F.; Gonzalo, I.G.; Lanting, L.; Natarajan, R. In vivo chromatin remodeling events leading to inflammatory gene transcription under diabetic conditions. J. Biol. Chem. 2004, 279, 18091–18097. [Google Scholar] [CrossRef] [Green Version]
- Miao, F.; Wu, X.; Zhang, L.; Yuan, Y.C.; Riggs, A.D.; Natarajan, R. Genome-wide analysis of histone lysine methylation variations caused by diabetic conditions in human monocytes. J. Biol. Chem. 2007, 282, 13854–13863. [Google Scholar] [CrossRef] [Green Version]
- Chen, S.S.; Jenkins, A.J.; Majewski, H. Elevated plasma prostaglandins and acetylated histone in monocytes in type 1 diabetes patients. Diabet. Med. 2009, 26, 182–186. [Google Scholar] [CrossRef]
- Liu, X.Y.; Xu, J.F. Reduced histone H3 acetylation in CD4+ T lymphocytes: Potential mechanism of latent autoimmune diabetes in adults. Dis. Markers 2015, 2015, 285125. [Google Scholar] [CrossRef] [Green Version]
- Holoch, D.; Moazed, D. RNA-mediated epigenetic regulation of gene expression. Nat. Rev. Genet. 2015, 16, 71–84. [Google Scholar] [CrossRef]
- Svoboda, P. Renaissance of mammalian endogenous RNAi. FEBS Lett. 2014, 588, 2550–2556. [Google Scholar] [CrossRef] [Green Version]
- Hezova, R.; Slaby, O.; Faltejskova, P.; Mikulkova, Z.; Buresova, I.; Raja, M.; Hodek, J.; Ovesna, J.; Michalek, J. MicroRNA-342, microRNA-191 and microRNA-510 are differentially expressed in T regulatory cells of type 1 diabetic patients. Cell. Immunol. 2010, 260, 70–74. [Google Scholar] [CrossRef] [PubMed]
- de Jong, V.M.; van der Slik, A.R.; Laban, S.; van ‘t Slot, R.; Koeleman, B.P.C.; Zaldumbide, A.; Roep, B.O. Survival of autoreactive T lymphocytes by microRNA-mediated regulation of apoptosis through TRAIL and Fas in type 1 diabetes. Genes Immun. 2016, 17, 342–348. [Google Scholar] [CrossRef]
- Sebastiani, G.; Grieco, F.A.; Spagnuolo, I.; Galleri, L.; Cataldo, D.; Dotta, F. Increased expression of microRNA miR-326 in type 1 diabetic patients with ongoing islet autoimmunity. Diabetes Metab. Res. Rev. 2011, 27, 862–866. [Google Scholar] [CrossRef]
- Salas-Perez, F.; Codner, E.; Valencia, E.; Pizarro, C.; Carrasco, E.; Perez-Bravo, F. MicroRNAs miR-21a and miR-93 are down regulated in peripheral blood mononuclear cells (PBMCs) from patients with type 1 diabetes. Immunobiology 2013, 218, 733–737. [Google Scholar] [CrossRef]
- Wang, S.; Wan, X.; Ruan, Q. The microRNA-21 in autoimmune diseases. Int. J. Mol. Sci. 2016, 17, 864. [Google Scholar] [CrossRef] [Green Version]
- Yang, M.; Ye, L.; Wang, B.; Gao, J.; Liu, R.; Hong, J.; Wang, W.; Gu, W.; Ning, G. Decreased miR-146 expression in peripheral blood mononuclear cells is correlated with ongoing islet autoimmunity in type 1 diabetes patients. J. Diabetes 2015, 7, 158–165. [Google Scholar] [CrossRef] [PubMed]
- Zheng, Y.; Wang, Z.; Zhou, Z. miRNAs: Novel regulators of autoimmunity-mediated pancreatic β-cell destruction in type 1 diabetes. Cell. Mol. Immunol. 2017, 14, 488–496. [Google Scholar] [CrossRef] [Green Version]
- Garo, L.P.; Murugaiyan, G. Contribution of microRNAs to autoimmune diseases. Cell. Mol. Life. Sci. 2016, 73, 2041–2051. [Google Scholar] [CrossRef]
- Ruan, Q.; Wang, T.; Kameswaran, V.; Wei, Q.; Johnson, D.S.; Matschinsky, F.; Shi, W.; Chen, Y.H. The microRNA-21-PDCD4 axis prevents type 1 diabetes by blocking pancreatic β cell death. Proc. Natl. Acad. Sci. USA 2011, 108, 12030–12035. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jerram, S.T.; Dang, M.N.; Leslie, R.D. The role of epigenetics in type 1 diabetes. Curr. Diabetes Rep. 2017, 17, 89. [Google Scholar] [CrossRef]
- Zullo, A.; Sommese, L.; Nicoletti, G.; Donatelli, F.; Mancini, F.P.; Napoli, C. Epigenetics and type 1 diabetes: Mechanisms and translational applications. Transl. Res. 2017, 185, 85–93. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Publication | Specific Target | Type of Cells | Results |
|---|---|---|---|
| (1st Author + Year) | |||
| Rakyan, 2011 [25] | genome-wide profile | CD14+ monocytes | ↓ HLA-DQB1, RFXAP |
| (MZ twins) | NFKB1, AGAD2 | ||
| Stefan, 2014 [26] | genome-wide profile | B cell lines | ↑ HLA-DOB + HLA-DQA2 |
| (MZ twins) | |||
| Elboudwarej, 2016 [27] | genome-wide profile | peripheral blood | global hypomethylation |
| (MZ twins) | |||
| Paul, 2016 [28] | genome-wide profile | CD4+ T cells | long time stable variabilities |
| (MZ twins) | CD19+ B cells | in regulatory regions | |
| CD14+ monocytes | |||
| Disanto, 2013 [29] | genome-wide profile | CD14+ monocytes | association with severity |
| (MZ quadruplet) | CD4+ T cells | of disease | |
| Čepek, 2016 [31] | HLA-DQA1 gene | peripheral blood | no differences between |
| CD14+ monocytes | patients versus healthies | ||
| Li, 2011 [32] | genome-wide profile | CD4+ T cells | ↑ FOXP3 |
| Fradin, 2012 [34] | INS gene promoter | leucocytes | 4 CpG variabilities (3↓ + 1↑) |
| Belot, 2013 [40] | IL2RA gene promoter | peripheral blood | 2 CpG variabilities (both ↑) |
| Publication | Specific Target | Type of Cells | Results |
|---|---|---|---|
| (1st Author + Year) | |||
| Miao, 2008 [46] | genome-wide + H3K9me2 | monocytes | no differences |
| lymphocytes | ↑ H3K9me2 | ||
| Miao, 2012 [47] | T1DM susceptible loci | monocytes | differences in H3K9Ac: |
| + H3K9Ac, H4K16Ac, | ↑ HLA-DQB1, ↓ HLA-DRB1 | ||
| H3K4, H3K9, H3K27 me3 | lymphocytes | no differences | |
| Chen, 2009 [50] | genome-wide H4 acetylation | monocytes | ↑ H4 acetylation |
| Liu, 2015 [51] | genome-wide H3 acetylation | CD4+ T cells | ↓ H3 acetylation |
| Publication | Specific Target | Type of Cells | Results |
|---|---|---|---|
| (1st Author + Year) | |||
| Hezova, 2010 [54] | genome-wide | Tregs | ↑ miR-510 |
| ↓ miR-342 + miR-191 | |||
| de Jong, 2016 [55] | genome-target | CD8+ T cells | ↑ miR-98, miR-23b, miR-590 |
| Sebastiani, 2011 [56] | miR-326 | lymphocytes | ↑ miR-326 |
| Salas-Pérez, 2013 [57] | miR-21a + miR-93 | mononuclear cells | ↓ miR-21a + miR-93 |
| Yang, 2015 [59] | genome-wide | mononuclear cells | ↓ miR-146 |
| differences in 26 miRNAs |
© 2019 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cerna, M. Epigenetic Regulation in Etiology of Type 1 Diabetes Mellitus. Int. J. Mol. Sci. 2020, 21, 36. https://doi.org/10.3390/ijms21010036
Cerna M. Epigenetic Regulation in Etiology of Type 1 Diabetes Mellitus. International Journal of Molecular Sciences. 2020; 21(1):36. https://doi.org/10.3390/ijms21010036
Chicago/Turabian StyleCerna, Marie. 2020. "Epigenetic Regulation in Etiology of Type 1 Diabetes Mellitus" International Journal of Molecular Sciences 21, no. 1: 36. https://doi.org/10.3390/ijms21010036
APA StyleCerna, M. (2020). Epigenetic Regulation in Etiology of Type 1 Diabetes Mellitus. International Journal of Molecular Sciences, 21(1), 36. https://doi.org/10.3390/ijms21010036