Are Leukaemic Stem Cells Restricted to a Single Cell Lineage?



{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Nature of Cancer Stems Cells and Their Progeny
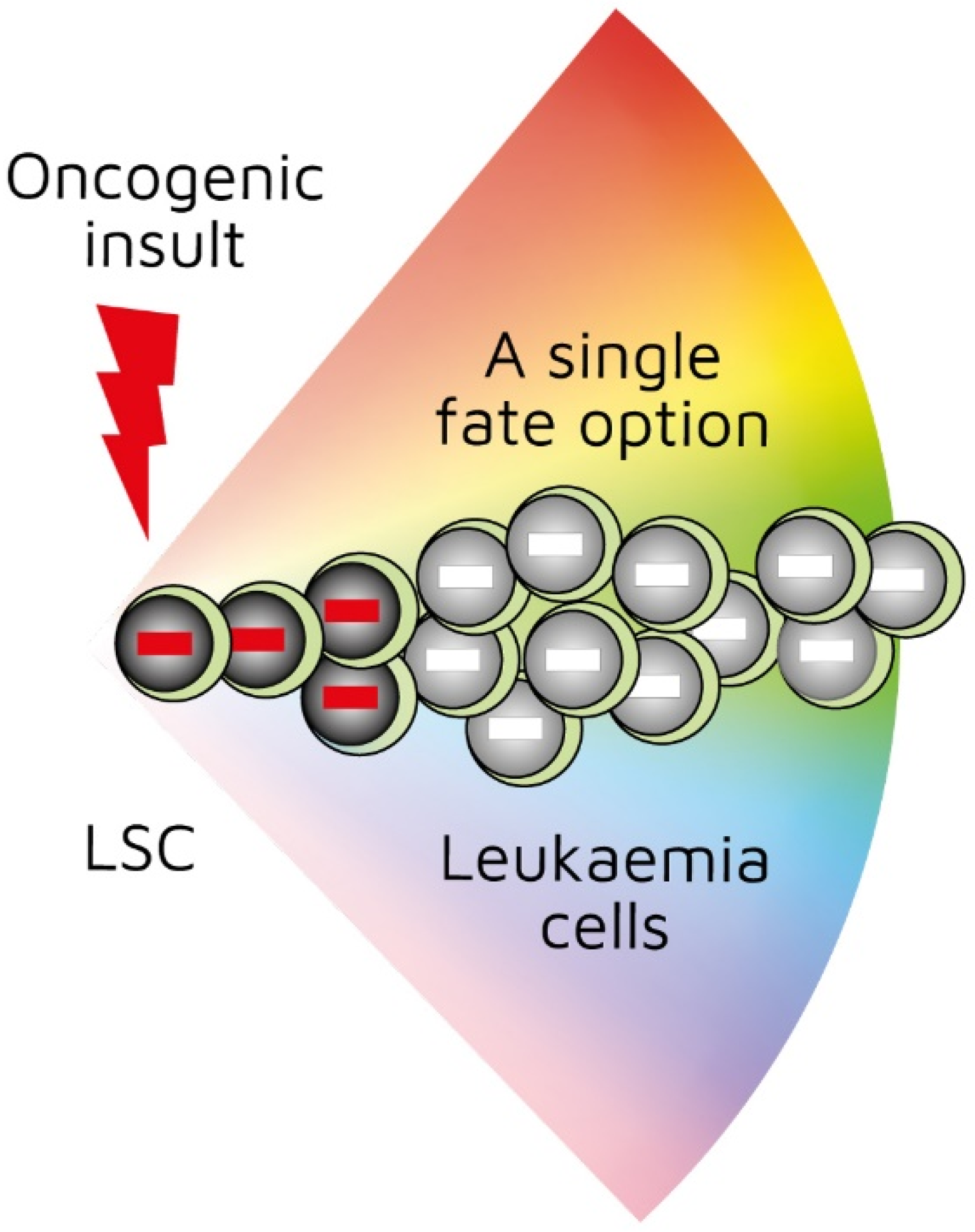
3. Leukaemic Cells Belong to One Lineage
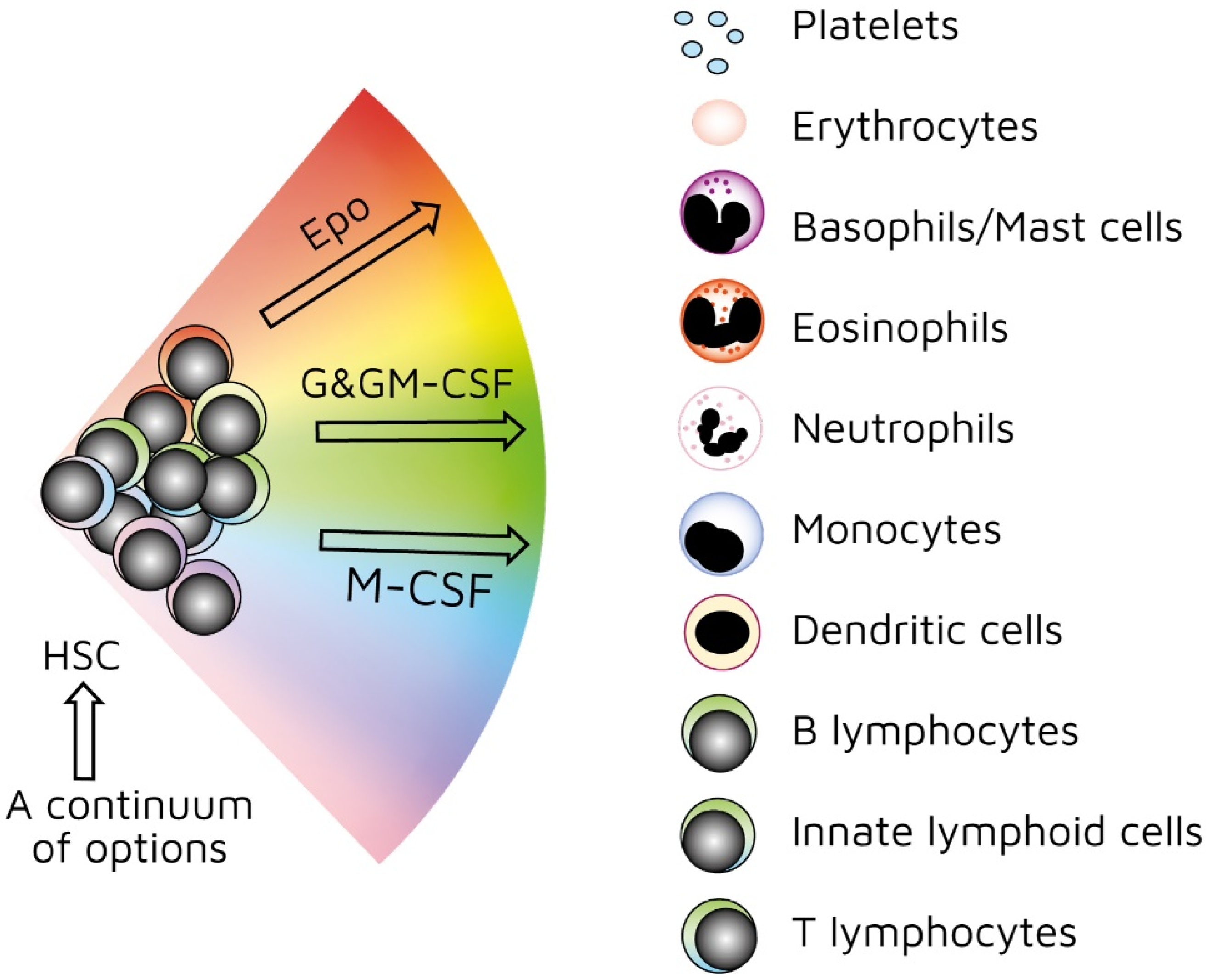
4. Do Normal Stem Cells Behave Differently?
5. Concluding Remarks
Author Contributions
Funding
Conflicts of Interest
References
- Dick, J.E. Stem cell concepts renew cancer research. Blood 2008, 112, 4793–4808. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Salmon, S.; Seligman, M. B cell neoplasia in man. Lancet 1974, 304, 1230–1233. [Google Scholar] [CrossRef]
- Wright, N.A. Boveri at 100: Cancer evolution, from preneoplasia to malignancy. J. Pathol. 2014, 234, 146–151. [Google Scholar] [CrossRef] [PubMed]
- Greaves, M.F.; Wiemels, J.; Ford, A.M. Leukaemia in twins: Lessons in natural history. Blood 2003, 102, 2321–2333. [Google Scholar] [CrossRef]
- Bonnet, D.; Dick, J.E. Human acute myeloid leukaemia is organised as a hierarchy that originates from a primitive hematopoietic cell. Nat. Med. 1997, 7, 730–737. [Google Scholar] [CrossRef]
- Quintiana, E.; Shakleton, M.; Sabel, M.S.; Fullen, D.R.; Johnson, T.M.; Morrison, S.J. Efficient tumour formation by single human carcinoma cells. Nature 2008, 456, 593–598. [Google Scholar] [CrossRef] [Green Version]
- Vicente-Duenas, C.; Perez-Caro, M.; Abullo-Jimenez, F.; Cobaleda, C.; Sanchez-Garcia, I. Stem cell driven cancer: “hands-off” regulation of cancer development. Cell Cycle 2009, 8, 1314–1318. [Google Scholar] [CrossRef] [Green Version]
- Yates, L.R.; Campbell, P.J. Evolution of the cancer genome. Nat. Rev. Genet. 2012, 13, 795–806. [Google Scholar] [CrossRef] [Green Version]
- Drayson, M.T.; Michell, R.H.; Durham, J.; Brown, G. Cell proliferation and CD11b expression are controlled independently during HL60 differentiation initiated by 1, 25α-dihydroxyvitamin D3 or all-trans-retinoic acid. Exp. Cell Res. 2001, 266, 126–134. [Google Scholar] [CrossRef]
- Brown, G.; Hughes, P.J.; Michell, R.H. Cell differentiation and proliferation-simultaneous but independent? Exp. Cell Res. 2003, 291, 282–288. [Google Scholar] [CrossRef]
- Fialkow, P.J.; Denman, A.M.; Jacobson, R.J.; Lowenthal, M.N. Chronic myelocytic leukaemia: Origin of some lymphocytes from leukaemic stem cells. J. Clin. Investig. 1978, 62, 815–823. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kowal-Vern, A.; Mazzella, F.M.; Cotelingham, J.D.; Shrit, M.A.; Rector, J.T.; Schumacher, H.R. Diagnosis andcharacterisation of acute erythroleukaemia subsets by determining the percentages of myeloblasts and proerythroblasts in 69 cases. Am. J. Hematol. 2000, 65, 5–13. [Google Scholar] [CrossRef]
- Greaves, M.F. Molecular genetics, natural history and the demise of childhood leukaemia. Eur. J. Cancer 1999, 35, 173–185. [Google Scholar] [CrossRef]
- Cox, C.V.; Blair, A. A primitive cell origin for B-cell precursor ALL? Stem Cell Rev. 2005, 1, 189–196. [Google Scholar] [CrossRef]
- Grimwade, D.; Enver, T. Acute promyelocytic leukaemia: Where does it stem from? Leukemia 2004, 1, 375–384. [Google Scholar] [CrossRef] [Green Version]
- Edwards, R.H.; Wasik, M.A.; Finan, J.; Rodriguez, R.; Moore, J.; Kamoun, M.; Rennert, H.; Bird, J.; Nowell, P.C.; Salhany, K.E. Evidence for early hematopoietic progenitor cell involvement in acute promyelocytic leukemia. Am. J. Clin. Pathol. 1999, 112, 819–827. [Google Scholar] [CrossRef] [Green Version]
- Kikushige, Y.; Ishikawa, F.; Miyamoto, T.; Shima, T.; Urata, S.; Yoshimoto, G.; Mori, Y.; Iino, T.; Yamauchi, T.; Eto, T.; et al. Self-renewing hematopoietic stem cell is the primary target in pathogenesis of human chronic lymphocytic leukemia. Cancer Cell 2011, 20, 246–259. [Google Scholar] [CrossRef] [Green Version]
- Nowell, P.; Hungerford, D. A minute chromosome in human granulocytic leukaemia. Science 1960, 132, 1497. [Google Scholar]
- Yunis, J.J.; Oken, M.M.; Kaplan, M.E.; Ensrud, K.M.; Howe, R.R.; Theologides, A. Distinctive chromosomal abnormalities in histologic subtypes of non-Hodgkin’s lymphoma. N. Eng. J. Med. 1982, 307, 1231–1236. [Google Scholar] [CrossRef]
- Chang-Hoon, N.; Rabbits, T.H. The role of LMO2 in development and in T cell leukaemia after chromosomal translocation or retroviral insertion. Mol. Ther. 2006, 13, 15–25. [Google Scholar]
- Shurtleff, S.A.; Buijs, A.; Behm, F.G.; Rubnitz, J.E.; Raimondi, S.C.; Hancock, M.L.; Chan, G.C.; Pui, C.H.; Grosveld, G.; Downing, J.R. TEL/AML1 fusion resulting from a cryptic t(12;21) is the most common genetic lesion in pediatric ALL and defines a subgroup of patients with an excellent prognosis. Leukemia 1995, 9, 1985–1989. [Google Scholar] [PubMed]
- Pérez-Caro, M.; Cobaleda, C.; González-Herrero, I.; Vicente-Dueñas, C.; Bermejo-Rodríguez, C.; Sánchez-Beato, M.; Orfao, A.; Pintado, B.; Flores, T.; Sánchez-Martín, M.; et al. Cancer induction by restriction of oncogene expression to the stem cell compartment. EMBO J. 2009, 28, 8–20. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vicente-Dueñas, C.; Romero-Camarero, I.; González-Herrero, I.; Alonso-Escudero, E.; Abollo-Jiménez, F.; Jiang, X.; Gutierrez, N.C.; Orfao, A.; Marín, N.; Villar, L.M.; et al. A novel molecular mechanism involved in multiple myeloma development revealed by targeting MafB to haematopoietic progenitors. EMBO J. 2012, 31, 3704–3717. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Green, M.R.; Vicente-Dueñas, C.; Romero-Camarero, I.; Liu, C.L.; Dai, B.; González-Herrero, I.; García-Ramírez, I.; Alonso-Escudero, E.; Iqbal, J.; Chan, W.C.; et al. Transient expression of Bcl6 is sufficient for oncogenic function and induction of mature B-cell lymphoma. Nat. Commun. 2014, 5, 3904. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Riggi, N.; Suvà, M.L.; De Vito, C.; Provero, P.; Stehle, J.C.; Baumer, K.; Cironi, L.; Janiszewska, M.; Petricevic, T.; Suvà, D.; et al. EWS-FLI-1 modulates miRNA145 and SOX2 expression to initiate mesenchymal stem cell reprogramming toward Ewing sarcoma cancer stem cells. Genes Dev. 2010, 24, 916–932. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Garcia, C.B.; Shaffer, C.M.; Alfaro, M.P.; Smith, A.L.; Sun, J.; Zhao, Z.; Young, P.P.; VanSaun, M.N.; Eid, J.E. Reprogramming of mesenchymal stem cells by the synovial sarcoma-associated oncogene SYT-SSX2. Oncogene 2012, 31, 2323–2334. [Google Scholar] [CrossRef] [Green Version]
- Martín-Lorenzo, A.; Auer, F.; Chan, L.N.; García-Ramírez, I.; González-Herrero, I.; Rodríguez-Hernández, G.; Bartenhagen, C.; Dugas, M.; Gombert, M.; Ginzel, S.; et al. Loss of Pax5 exploits Sca1-BCR-ABLp190 susceptibility to confer the metabolic shift essential for pB-ALL. Cancer Res. 2018, 78, 2669–2679. [Google Scholar] [CrossRef] [Green Version]
- García-Ramírez, I.; Bhatia, S.; Rodríguez-Hernández, G.; González-Herrero, I.; Walter, C.; De Tena-Dávila, S.G.; Parvin, S.; Haas, O.; Woessmann, W.; Stanulla, M.; et al. Lmo2 expression defines tumor cell identity during T-cell leukemogenesis. EMBO J. 2018, 37, e98783. [Google Scholar] [CrossRef]
- Vicente-Dueñas, C.; González-Herrero, I.; Sehgal, L.; García-Ramírez, I.; Rodríguez-Hernández, G.; Pintado, B.; Blanco, O.; Criado, F.J.G.; Cenador, M.B.G.; Green, M.R.; et al. Dnm1 links BCR-ABLp210 to epigenetic tumour stem cell priming in myeloid leukaemia. Leukaemia 2019, 33, 249–278. [Google Scholar] [CrossRef]
- Vicente-Dueñas, C.; Hauer, J.; Cobaleda, C.; Borkhardt, A.; Sánchez-García, I. Epigenetic priming in cancer initiation. Trends Cancer 2018, 4, 408–417. [Google Scholar] [CrossRef]
- Sánchez-Danés, A.; Blanpain, C. Deciphering the cells of origin of squamous cell carcinomas. Nat. Rev. Cancer 2018, 18, 549–561. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.J. Mixed-phenotype acute leukemia and beyond. Blood Res. 2016, 51, 215–216. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abdelhaleem, M. Frequent but nonrandom expression of myeloid markers on de novo childhood acute lymphoblastic leukemia. Exp. Mol. Pathol. 2007, 83, 138–141. [Google Scholar] [CrossRef] [PubMed]
- Iijima, K.; Sugita, K.; Inukai, T.; Goi, K.; Tezuka, T.; Uno, K.; Sato, H.; Kagami, K.; Nakazawa, S. Expression of thrombopoietin receptor and its functional role in human B-precursor leukemia cells with 11q23 translocation or Philadelphia chromosome. Leukemia 2000, 14, 1598–1605. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pérez-Caro, M.; Gutierrez-Cianca, N.; González-Herrero, I.; López-Hernández, I.; Flores, T.; Orfao, A.; Sánchez-Martín, M.; Gutiérrez-Adán, A.; Pintado, B.; Sánchez-García, I. Sustained leukaemic phenotype after inactivation of BCR-ABLp190 in mice. Oncogene 2007, 26, 1702–1713. [Google Scholar] [CrossRef]
- Kalluri, R.; Weinberg, R.A. The basics of epithelial-mesenchymal transition. J. Clin. Investig. 2009, 119, 1420–1428. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cunningham, J.J.; Ulbright, T.M.; Pera, M.F.; Looijenga, L.H. Lessons from human teratomas to guide development of safe stem cell therapies. Nat. Biotechnol. 2012, 30, 849–857. [Google Scholar] [CrossRef]
- Shinjo, K.; Takeshita, A.; Higuchi, M.; Ohnishi, K.; Ohno, R. Erythropoietin receptor expression on human bone marrow erythroid precursor cells by a newly-devised quantitative flow-cytometric assay. Br. J. Haematol. 1997, 96, 551–558. [Google Scholar] [CrossRef]
- Kondo, M.; Scherer, D.C.; Miyamoto, T.; King, A.G.; Akashi, K.; Sugamura, K.; Weissman, I.L. Cell-fate conversion of lymphoid-committed progenitors by instructive actions of cytokines. Nature 2000, 407, 383–386. [Google Scholar] [CrossRef]
- Mossadegh-Keller, N.; Sarrazin, S.; Kandalla, P.K.; Espinosa, L.; Stanley, E.R.; Nutt, S.L.; Moore, J.; Sieweke, M.H. M-CSF instructs myeloid lineage fate in single haematopoietic stem cells. Nature 2013, 497, 239–243. [Google Scholar] [CrossRef]
- Mooney, C.J.; Cunningham, A.; Tsapogas, P.; Toellner, K.M.; Brown, G. Selective expression of flt3 within the mouse hematopoietic stem cell compartment. Int. J. Mol. Sci. 2017, 18, 1037. [Google Scholar] [CrossRef] [PubMed]
- Liu, F.; Poursine-Laurent, J.; Link, D.C. Expression of the G-CSF receptor on hematopoietic progenitor cells is not required for their mobilisation by G-CSF. Blood 2000, 95, 3025–3031. [Google Scholar] [CrossRef] [PubMed]
- Balciunaite, G.; Ceredig, R.; Massa, S.; Rolink, A.G. A b220 + cd117 + cd19-hematopoietic progenitor with potent lymphoid and myeloid developmental potential. Eur. J. Immunol. 2005, 35, 2019–2030. [Google Scholar] [CrossRef] [PubMed]
- Alberti-Servera, L.; Von Muenchow, L.; Tsapogas, P.; Capoferri, G.; Eschbach, K.; Beisel, C.; Ceredig, R.; Ivanek, R.; Rolink, A. Single-cell RNA sequencing reveals developmental heterogeneity among early lymphoid progenitors. EMBO J. 2017, 36, 3619–3633. [Google Scholar] [CrossRef] [PubMed]
- Ceredig, R.; Rolink, A.G.; Brown, G. Models of hematopoiesis: Seeing the wood for the trees. Nat. Rev. Immunol. 2009, 9, 93–300. [Google Scholar] [CrossRef] [PubMed]
- Brown, G.; Tsapogas, P.; Ceredig, R. The changing face of hematopoiesis: A spectrum of options is available to stem cells. Immunol. Cell Biol. 2018, 96, 898–911. [Google Scholar] [CrossRef]
- Brown, G.; Ceredig, R. Modelling the hematopoietic landscape. Front. Cell Dev. Biol. 2019, 7, 104. [Google Scholar] [CrossRef]
- Psaila, B.; Mead, A.J. Single-cell approaches reveal novel cellular pathways for megakaryocyte and erythroid differentiation. Blood 2019, 133, 1427–1435. [Google Scholar] [CrossRef]
- Nestorowa, S.; Hamey, F.K.; Sala, B.P.; Diamanti, E.; Shepherd, M.; Laurenti, E.; Wilson, N.K.; Kent, D.G.; Gottens, B. A single-cell resolution map of mouse hematopoietic stem and progenitor cell differentiation. Blood 2016, 128, e20–e31. [Google Scholar] [CrossRef] [Green Version]
- Balciunaite, G.; Ceredig, R.; Rolink, A.G. The earliest subpopulation of mouse thymocytes contains potent T, significant macrophage, and natural killer but no B-lymphocyte potential. Blood 2005, 105, 1930–1936. [Google Scholar] [CrossRef] [Green Version]
- Olsson, A.; Venkatasubramanian, M.; Chaudhri, V.K.; Aronow, B.J.; Salomonis, N.; Singh, H.; Grimes, H.L. Single-cell analysis of mixed-lineage states leading to a binary cell fate choice. Nature 2016, 537, 698–702. [Google Scholar] [CrossRef] [PubMed]
- Grover, A.; Mancini, E.; Moore, S.; Mead, A.J.; Atkinson, D.; Rasmussen, K.D.; O’Carrol, D.; Jacobsen, S.E.; Nerlov, C. Erythropoietin guides multipotent hematopoietic progenitor cells toward an erythroid fate. J. Exp. Med. 2014, 211, 181–188. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rieger, M.A.; Hoppe, P.S.; Smejkal, B.M.; Eitelhuber, A.C.; Schroeder, T. Hematopoietic cytokines can instruct lineage choice. Science 2009, 325, 217–218. [Google Scholar] [CrossRef] [PubMed]
- Metcalf, D.; Burgess, A.W. Clonal analysis of progenitor cell commitment of granulocyte or macrophage production. J. Cell Physiol. 1982, 111, 275–283. [Google Scholar] [CrossRef]
- Tsapogas, P.; Swee, L.K.; Nusser, A.; Nuber, N.; Kreuzaler, M.; Capoferri, G.; Rolink, H.; Ceredig, R.; Rolink, A. In vivo evidence for an instructive role of fms-like tyrosine kinase-3 (flt3) ligand in hematopoietic development. Haematologica 2014, 99, 638–646. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Griffin, J.D.; Lowenberg, B. Clonogenic cells in acute myeloblastic leukaemia. Blood 1986, 68, 1185–1195. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Singh, R.P.; Grinenko, T.; Ramasz, B.; Franke, K.; Lesche, M.; Dahl, A.; Gassmann, M.; Chavakis, T.; Henry, I.; Wielockx, B. Hematopoietic stem cells but not multipotent progenitors drive erythropoiesis during chromic erythroid stress in epo transgenic mice. Stem Cell Rep. 2018, 10, 1908–1919. [Google Scholar] [CrossRef]
- Brown, G.; Sanchez-Garcia, I. Is lineage decision-making restricted during tumoral reprograming of haematopoietic stem cells? Oncotarget 2015, 6, 43326–43341. [Google Scholar] [CrossRef] [Green Version]


© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Brown, G.; Sánchez, L.; Sánchez-García, I. Are Leukaemic Stem Cells Restricted to a Single Cell Lineage? Int. J. Mol. Sci. 2020, 21, 45. https://doi.org/10.3390/ijms21010045
Brown G, Sánchez L, Sánchez-García I. Are Leukaemic Stem Cells Restricted to a Single Cell Lineage? International Journal of Molecular Sciences. 2020; 21(1):45. https://doi.org/10.3390/ijms21010045
Chicago/Turabian StyleBrown, Geoffrey, Lucía Sánchez, and Isidro Sánchez-García. 2020. "Are Leukaemic Stem Cells Restricted to a Single Cell Lineage?" International Journal of Molecular Sciences 21, no. 1: 45. https://doi.org/10.3390/ijms21010045
APA StyleBrown, G., Sánchez, L., & Sánchez-García, I. (2020). Are Leukaemic Stem Cells Restricted to a Single Cell Lineage? International Journal of Molecular Sciences, 21(1), 45. https://doi.org/10.3390/ijms21010045