Unexpected Impact of a Hepatitis C Virus Inhibitor on 17β-Estradiol Signaling in Breast Cancer



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
2.1. Telaprevir Affects Intracellular ERα Levels
2.2. Telaprevir-Dependent Mechanism for the Control of ERα Levels
2.3. Effect of Telaprevir on ERα Transcriptional Activity
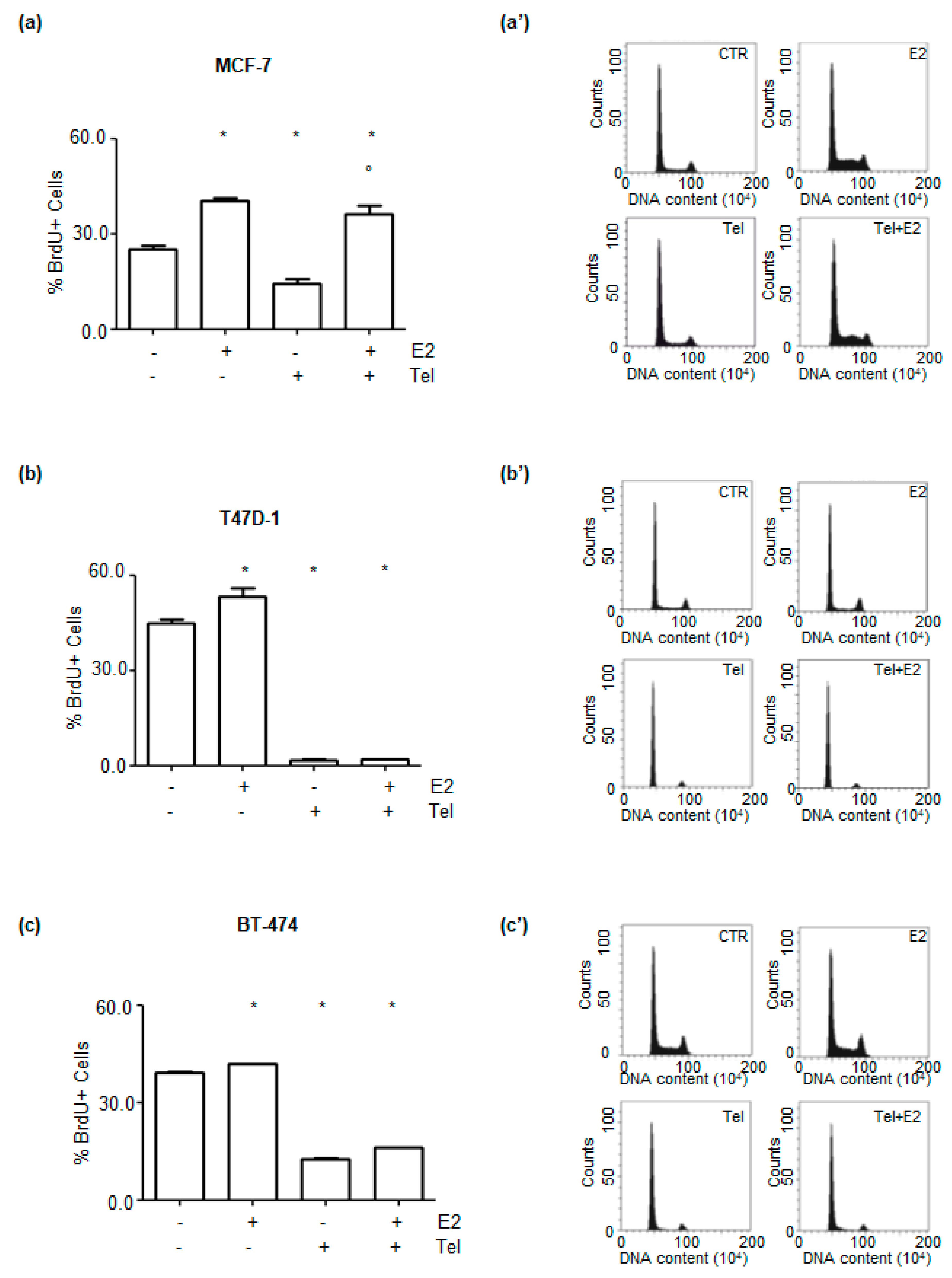
2.4. Effect of Telaprevir on E2-Dependent Breast Cancer Cell Proliferation
3. Discussion
4. Materials and Methods
4.1. Cell Culture and Reagents
4.2. Western Blot Analysis
4.3. Growth Curves
4.4. Real-Time Measurement of ERα Transcriptional Activity
4.5. In Vitro Binding Assay
4.6. RNA Isolation and qPCR Analysis
4.7. Bromodeoxyuridine Incorporation Assay
4.8. Cell Cycle Analysis
4.9. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Ascenzi, P.; Bocedi, A.; Marino, M. Structure-function relationship of estrogen receptor alpha and beta: Impact on human health. Mol. Asp. Med. 2006, 27, 299–402. [Google Scholar] [CrossRef] [PubMed]
- Fuentes, N.; Silveyra, P. Estrogen receptor signaling mechanisms. Adv. Protein Chem. Struct. Biol. 2019, 116, 135–170. [Google Scholar] [CrossRef] [PubMed]
- Busonero, C.; Leone, S.; Bartoloni, S.; Acconcia, F. Strategies to degrade estrogen receptor alpha in primary and ESR1 mutant-expressing metastatic breast cancer. Mol. Cell. Endocrinol. 2019, 480, 107–121. [Google Scholar] [CrossRef] [PubMed]
- Acconcia, F.; Ascenzi, P.; Bocedi, A.; Spisni, E.; Tomasi, V.; Trentalance, A.; Visca, P.; Marino, M. Palmitoylation-dependent estrogen receptor alpha membrane localization: Regulation by 17 beta-estradiol. Mol. Biol. Cell 2005, 16, 231–237. [Google Scholar] [CrossRef] [PubMed]
- La Rosa, P.; Pesiri, V.; Leclercq, G.; Marino, M.; Acconcia, F. Palmitoylation Regulates 17beta-Estradiol-Induced Estrogen Receptor-alpha Degradation and Transcriptional Activity. Mol. Endocrinol. 2012, 26, 762–774. [Google Scholar] [CrossRef] [Green Version]
- Pedram, A.; Razandi, M.; Lewis, M.; Hammes, S.; Levin, E.R. Membrane-localized estrogen receptor alpha is required for normal organ development and function. Dev. Cell 2014. [Google Scholar] [CrossRef] [Green Version]
- Adlanmerini, M.; Solinhac, R.; Abot, A.; Fabre, A.; Raymond-Letron, I.; Guihot, A.L.; Boudou, F.; Sautier, L.; Vessieres, E.; Kim, S.H.; et al. Mutation of the palmitoylation site of estrogen receptor alpha in vivo reveals tissue-specific roles for membrane versus nuclear actions. Proc. Natl. Acad. Sci. USA 2014, 111, E283–E290. [Google Scholar] [CrossRef] [Green Version]
- Acconcia, F.; Marino, M. The Effects of 17beta-estradiol in Cancer are Mediated by Estrogen Receptor Signaling at the Plasma Membrane. Front. Physiol. 2011, 2, 30. [Google Scholar] [CrossRef] [Green Version]
- Acconcia, F.; Fiocchetti, M.; Marino, M. Xenoestrogen regulation of ERalpha/ERbeta balance in hormone-associated cancers. Mol. Cell. Endocrinol. 2016. [Google Scholar] [CrossRef]
- Leclercq, G.; Lacroix, M.; Laios, I.; Laurent, G. Estrogen receptor alpha: Impact of ligands on intracellular shuttling and turnover rate in breast cancer cells. Curr. Cancer Drug Targets 2006, 6, 39–64. [Google Scholar] [CrossRef] [Green Version]
- Totta, P.; Busonero, C.; Leone, S.; Marino, M.; Acconcia, F. Dynamin II is required for 17beta-estradiol signaling and autophagy-based ERalpha degradation. Sci. Rep. 2016, 6, 23727. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lumachi, F.; Luisetto, G.; Basso, S.M.; Basso, U.; Brunello, A.; Camozzi, V. Endocrine therapy of breast cancer. Curr. Med. Chem. 2011, 18, 513–522. [Google Scholar] [CrossRef] [PubMed]
- Busonero, C.; Leone, S.; Acconcia, F. Emetine induces estrogen receptor alpha degradation and prevents 17beta-estradiol-induced breast cancer cell proliferation. Cell. Oncol. 2017. [Google Scholar] [CrossRef] [PubMed]
- Busonero, C.; Leone, S.; Klemm, C.; Acconcia, F. A functional drug re-purposing screening identifies carfilzomib as a drug preventing 17beta-estradiol: ERalpha signaling and cell proliferation in breast cancer cells. Mol. Cell. Endocrinol. 2018, 460, 229–237. [Google Scholar] [CrossRef]
- Leone, S.; Busonero, C.; Acconcia, F. A high throughput method to study the physiology of E2:ERalpha signaling in breast cancer cells. J. Cell. Physiol. 2018, 233, 3713–3722. [Google Scholar] [CrossRef]
- Totta, P.; Gionfra, F.; Busonero, C.; Acconcia, F. Modulation of 17beta-Estradiol Signaling on Cellular Proliferation by Caveolin-2. J. Cell. Physiol. 2015. [Google Scholar] [CrossRef]
- Totta, P.; Pesiri, V.; Enari, M.; Marino, M.; Acconcia, F. Clathrin Heavy Chain Interacts With Estrogen Receptor alpha and Modulates 17beta-Estradiol Signaling. Mol. Endocrinol. 2015, 29, 739–755. [Google Scholar] [CrossRef] [Green Version]
- Menendez, J.A.; Lupu, R. Fatty acid synthase regulates estrogen receptor-alpha signaling in breast cancer cells. Oncogenesis 2017, 6, e299. [Google Scholar] [CrossRef]
- Vethakanraj, H.S.; Sesurajan, B.P.; Padmanaban, V.P.; Jayaprakasam, M.; Murali, S.; Sekar, A.K. Anticancer effect of acid ceramidase inhibitor ceranib-2 in human breast cancer cell lines MCF-7, MDA MB-231 by the activation of SAPK/JNK, p38 MAPK apoptotic pathways, inhibition of the Akt pathway, downregulation of ERalpha. Anti-Cancer Drugs 2018, 29, 50–60. [Google Scholar] [CrossRef]
- Busonero, C.; Leone, S.; Bianchi, F.; Acconcia, F. In silico screening for ERα downmodulators identifies thioridazine as an anti-proliferative agent in primary, 4OH-tamoxifen-resistant and Y537S ERα-expressing breast cancer cells. Cell. Oncol. 2018. [Google Scholar] [CrossRef]
- Gentile, I.; Viola, C.; Borgia, F.; Castaldo, G.; Borgia, G. Telaprevir: A promising protease inhibitor for the treatment of hepatitis C virus infection. Curr. Med. Chem. 2009, 16, 1115–1121. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bertino, G.; Ardiri, A.; Proiti, M.; Rigano, G.; Frazzetto, E.; Demma, S.; Ruggeri, M.I.; Scuderi, L.; Malaguarnera, G.; Bertino, N.; et al. Chronic hepatitis C: This and the new era of treatment. World J. Hepatol. 2016, 8, 92–106. [Google Scholar] [CrossRef]
- Weir, H.M.; Bradbury, R.H.; Lawson, M.; Rabow, A.A.; Buttar, D.; Callis, R.J.; Curwen, J.O.; de Almeida, C.; Ballard, P.; Hulse, M.; et al. AZD9496: An Oral Estrogen Receptor Inhibitor That Blocks the Growth of ER-Positive and ESR1-Mutant Breast Tumors in Preclinical Models. Cancer Res. 2016, 76, 3307–3318. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Joseph, J.D.; Darimont, B.; Zhou, W.; Arrazate, A.; Young, A.; Ingalla, E.; Walter, K.; Blake, R.A.; Nonomiya, J.; Guan, Z.; et al. The selective estrogen receptor downregulator GDC-0810 is efficacious in diverse models of ER+ breast cancer. eLife 2016, 5. [Google Scholar] [CrossRef]
- Laios, I.; Journe, F.; Nonclercq, D.; Vidal, D.S.; Toillon, R.A.; Laurent, G.; Leclercq, G. Role of the proteasome in the regulation of estrogen receptor alpha turnover and function in MCF-7 breast carcinoma cells. J. Steroid Biochem. Mol. Biol. 2005, 94, 347–359. [Google Scholar] [CrossRef] [PubMed]
- Pesiri, V.; La Rosa, P.; Stano, P.; Acconcia, F. Identification of an estrogen receptor alpha non-covalent ubiquitin binding surface: Role in 17beta-estradiol-induced transcriptional activity. J. Cell Sci. 2013, 126, 2577–2582. [Google Scholar] [CrossRef] [Green Version]
- Metivier, R.; Penot, G.; Hubner, M.R.; Reid, G.; Brand, H.; Kos, M.; Gannon, F. Estrogen receptor-alpha directs ordered, cyclical, and combinatorial recruitment of cofactors on a natural target promoter. Cell 2003, 115, 751–763. [Google Scholar] [CrossRef] [Green Version]
- Reid, G.; Hubner, M.R.; Metivier, R.; Brand, H.; Denger, S.; Manu, D.; Beaudouin, J.; Ellenberg, J.; Gannon, F. Cyclic, proteasome-mediated turnover of unliganded and liganded ERalpha on responsive promoters is an integral feature of estrogen signaling. Mol. Cell. 2003, 11, 695–707. [Google Scholar] [CrossRef]
- Harrod, A.; Fulton, J.; Nguyen, V.T.M.; Periyasamy, M.; Ramos-Garcia, L.; Lai, C.F.; Metodieva, G.; de Giorgio, A.; Williams, R.L.; Santos, D.B.; et al. Genomic modelling of the ESR1 Y537S mutation for evaluating function and new therapeutic approaches for metastatic breast cancer. Oncogene 2017, 36, 2286–2296. [Google Scholar] [CrossRef] [Green Version]
- Cipolletti, M.; Leone, S.; Bartoloni, S.; Busonero, C.; Acconcia, F. Real-time measurement of E2: ERalpha transcriptional activity in living cells. J. Cell. Physiol. 2020. [Google Scholar] [CrossRef]
- Scott, S.C.; Lee, S.S.; Abraham, J. Mechanisms of therapeutic CDK4/6 inhibition in breast cancer. Semin. Oncol. 2017, 44, 385–394. [Google Scholar] [CrossRef] [PubMed]
- La Rosa, P.; Acconcia, F. Signaling functions of ubiquitin in the 17beta-estradiol (E2): Estrogen receptor (ER) alpha network. J. Steroid Biochem. Mol. Biol. 2011, 127, 223–230. [Google Scholar] [CrossRef] [PubMed]
- Totta, P.; Pesiri, V.; Marino, M.; Acconcia, F. Lysosomal Function Is Involved in 17beta-Estradiol-Induced Estrogen Receptor alpha Degradation and Cell Proliferation. PLoS ONE 2014, 9, e94880. [Google Scholar] [CrossRef] [PubMed]
- Kiang, T.K.; Wilby, K.J.; Ensom, M.H. Telaprevir: Clinical pharmacokinetics, pharmacodynamics, and drug-drug interactions. Clin. Pharm. 2013, 52, 487–510. [Google Scholar] [CrossRef]
- Hurtado, A.; Holmes, K.A.; Ross-Innes, C.S.; Schmidt, D.; Carroll, J.S. FOXA1 is a key determinant of estrogen receptor function and endocrine response. Nat. Genet. 2011, 43, 27–33. [Google Scholar] [CrossRef] [Green Version]
- Eeckhoute, J.; Keeton, E.K.; Lupien, M.; Krum, S.A.; Carroll, J.S.; Brown, M. Positive cross-regulatory loop ties GATA-3 to estrogen receptor alpha expression in breast cancer. Cancer Res. 2007, 67, 6477–6483. [Google Scholar] [CrossRef] [Green Version]
- Nassa, G.; Salvati, A.; Tarallo, R.; Gigantino, V.; Alexandrova, E.; Memoli, D.; Sellitto, A.; Rizzo, F.; Malanga, D.; Mirante, T.; et al. Inhibition of histone methyltransferase DOT1L silences ERalpha gene and blocks proliferation of antiestrogen-resistant breast cancer cells. Sci. Adv. 2019, 5, eaav5590. [Google Scholar] [CrossRef] [Green Version]
- Feng, Q.; Zhang, Z.; Shea, M.J.; Creighton, C.J.; Coarfa, C.; Hilsenbeck, S.G.; Lanz, R.; He, B.; Wang, L.; Fu, X.; et al. An epigenomic approach to therapy for tamoxifen-resistant breast cancer. Cell Res. 2014, 24, 809–819. [Google Scholar] [CrossRef]
- Martin, L.A.; Ribas, R.; Simigdala, N.; Schuster, E.; Pancholi, S.; Tenev, T.; Gellert, P.; Buluwela, L.; Harrod, A.; Thornhill, A.; et al. Discovery of naturally occurring ESR1 mutations in breast cancer cell lines modelling endocrine resistance. Nat. Commun. 2017, 8, 1865. [Google Scholar] [CrossRef]
- Fanning, S.W.; Mayne, C.G.; Dharmarajan, V.; Carlson, K.E.; Martin, T.A.; Novick, S.J.; Toy, W.; Green, B.; Panchamukhi, S.; Katzenellenbogen, B.S.; et al. Estrogen receptor alpha somatic mutations Y537S and D538G confer breast cancer endocrine resistance by stabilizing the activating function-2 binding conformation. eLife 2016, 5. [Google Scholar] [CrossRef]
- Bahreini, A.; Li, Z.; Wang, P.; Levine, K.M.; Tasdemir, N.; Cao, L.; Weir, H.M.; Puhalla, S.L.; Davidson, N.E.; Stern, A.M.; et al. Mutation site and context dependent effects of ESR1 mutation in genome-edited breast cancer cell models. Breast Cancer Res. BCR 2017, 19, 60. [Google Scholar] [CrossRef] [Green Version]
- Williams, M.M.; Cook, R.S. Bcl-2 family proteins in breast development and cancer: Could Mcl-1 targeting overcome therapeutic resistance? Oncotarget 2015, 6, 3519–3530. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Prossnitz, E.R.; Maggiolini, M. Mechanisms of estrogen signaling and gene expression via GPR30. Mol. Cell. Endocrinol. 2009, 308, 32–38. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Molina, L.; Figueroa, C.D.; Bhoola, K.D.; Ehrenfeld, P. GPER-1/GPR30 a novel estrogen receptor sited in the cell membrane: Therapeutic coupling to breast cancer. Expert Opin. Ther. Targets 2017, 21, 755–766. [Google Scholar] [CrossRef] [PubMed]
- Fiocchetti, M.; Cipolletti, M.; Ascenzi, P.; Marino, M. Dissecting the 17β-estradiol pathways necessary for neuroglobin anti-apoptotic activity in breast cancer. J. Cell. Physiol. 2018, 233, 5087–5103. [Google Scholar] [CrossRef]
- Caldon, C.E. Estrogen signaling and the DNA damage response in hormone dependent breast cancers. Front. Oncol. 2014, 4, 106. [Google Scholar] [CrossRef] [Green Version]
- Williamson, L.M.; Lees-Miller, S.P. Estrogen receptor alpha-mediated transcription induces cell cycle-dependent DNA double-strand breaks. Carcinogenesis 2011, 32, 279–285. [Google Scholar] [CrossRef]
- Bardia, A.; Hurvitz, S. Targeted Therapy for Premenopausal Women with HR(+), HER2(-) Advanced Breast Cancer: Focus on Special Considerations and Latest Advances. Clin. Cancer Res. 2018, 24, 5206–5218. [Google Scholar] [CrossRef] [Green Version]
- Meylan, E.; Curran, J.; Hofmann, K.; Moradpour, D.; Binder, M.; Bartenschlager, R.; Tschopp, J. Cardif is an adaptor protein in the RIG-I antiviral pathway and is targeted by hepatitis C virus. Nature 2005, 437, 1167–1172. [Google Scholar] [CrossRef]
- Li, K.; Foy, E.; Ferreon, J.C.; Nakamura, M.; Ferreon, A.C.; Ikeda, M.; Ray, S.C.; Gale, M., Jr.; Lemon, S.M. Immune evasion by hepatitis C virus NS3/4A protease-mediated cleavage of the Toll-like receptor 3 adaptor protein TRIF. Proc. Natl. Acad. Sci. USA 2005, 102, 2992–2997. [Google Scholar] [CrossRef] [Green Version]
- Li, X.D.; Sun, L.; Seth, R.B.; Pineda, G.; Chen, Z.J. Hepatitis C virus protease NS3/4A cleaves mitochondrial antiviral signaling protein off the mitochondria to evade innate immunity. Proc. Natl. Acad. Sci. USA 2005, 102, 17717–17722. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- de Wispelaere, M.; Du, G.; Donovan, K.A.; Zhang, T.; Eleuteri, N.A.; Yuan, J.C.; Kalabathula, J.; Nowak, R.P.; Fischer, E.S.; Gray, N.S.; et al. Small molecule degraders of the hepatitis C virus protease reduce susceptibility to resistance mutations. Nat. Commun. 2019, 10, 3468. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lan, Q.; Peyvandi, S.; Duffey, N.; Huang, Y.T.; Barras, D.; Held, W.; Richard, F.; Delorenzi, M.; Sotiriou, C.; Desmedt, C.; et al. Type I interferon/IRF7 axis instigates chemotherapy-induced immunological dormancy in breast cancer. Oncogene 2019, 38, 2814–2829. [Google Scholar] [CrossRef] [PubMed]
- Bidwell, B.N.; Slaney, C.Y.; Withana, N.P.; Forster, S.; Cao, Y.; Loi, S.; Andrews, D.; Mikeska, T.; Mangan, N.E.; Samarajiwa, S.A.; et al. Silencing of Irf7 pathways in breast cancer cells promotes bone metastasis through immune escape. Nat. Med. 2012, 18, 1224–1231. [Google Scholar] [CrossRef]
- Budhwani, M.; Mazzieri, R.; Dolcetti, R. Plasticity of Type I Interferon-Mediated Responses in Cancer Therapy: From Anti-tumor Immunity to Resistance. Front. Oncol. 2018, 8, 322. [Google Scholar] [CrossRef] [Green Version]
- Weiss, J.; Becker, J.P.; Haefeli, W.E. Telaprevir is a substrate and moderate inhibitor of P-glycoprotein, a strong inductor of ABCG2, but not an activator of PXR in vitro. Int. J. Antimicrob. Agents 2014, 43, 184–188. [Google Scholar] [CrossRef]
- Fujita, Y.; Noguchi, K.; Suzuki, T.; Katayama, K.; Sugimoto, Y. Biochemical interaction of anti-HCV telaprevir with the ABC transporters P-glycoprotein and breast cancer resistance protein. BMC Res. Notes 2013, 6, 445. [Google Scholar] [CrossRef] [Green Version]
- Ambudkar, S.V.; Kim, I.W.; Sauna, Z.E. The power of the pump: Mechanisms of action of P-glycoprotein (ABCB1). Eur. J. Pharm. Sci. Off. J. Eur. Fed. Pharm. Sci. 2006, 27, 392–400. [Google Scholar] [CrossRef]
- Rotroff, D.M.; Dix, D.J.; Houck, K.A.; Kavlock, R.J.; Knudsen, T.B.; Martin, M.T.; Reif, D.M.; Richard, A.M.; Sipes, N.S.; Abassi, Y.A.; et al. Real-time growth kinetics measuring hormone mimicry for ToxCast chemicals in T-47D human ductal carcinoma cells. Chem. Res. Toxicol. 2013, 26, 1097–1107. [Google Scholar] [CrossRef]





© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bartoloni, S.; Leone, S.; Acconcia, F. Unexpected Impact of a Hepatitis C Virus Inhibitor on 17β-Estradiol Signaling in Breast Cancer. Int. J. Mol. Sci. 2020, 21, 3418. https://doi.org/10.3390/ijms21103418
Bartoloni S, Leone S, Acconcia F. Unexpected Impact of a Hepatitis C Virus Inhibitor on 17β-Estradiol Signaling in Breast Cancer. International Journal of Molecular Sciences. 2020; 21(10):3418. https://doi.org/10.3390/ijms21103418
Chicago/Turabian StyleBartoloni, Stefania, Stefano Leone, and Filippo Acconcia. 2020. "Unexpected Impact of a Hepatitis C Virus Inhibitor on 17β-Estradiol Signaling in Breast Cancer" International Journal of Molecular Sciences 21, no. 10: 3418. https://doi.org/10.3390/ijms21103418
APA StyleBartoloni, S., Leone, S., & Acconcia, F. (2020). Unexpected Impact of a Hepatitis C Virus Inhibitor on 17β-Estradiol Signaling in Breast Cancer. International Journal of Molecular Sciences, 21(10), 3418. https://doi.org/10.3390/ijms21103418