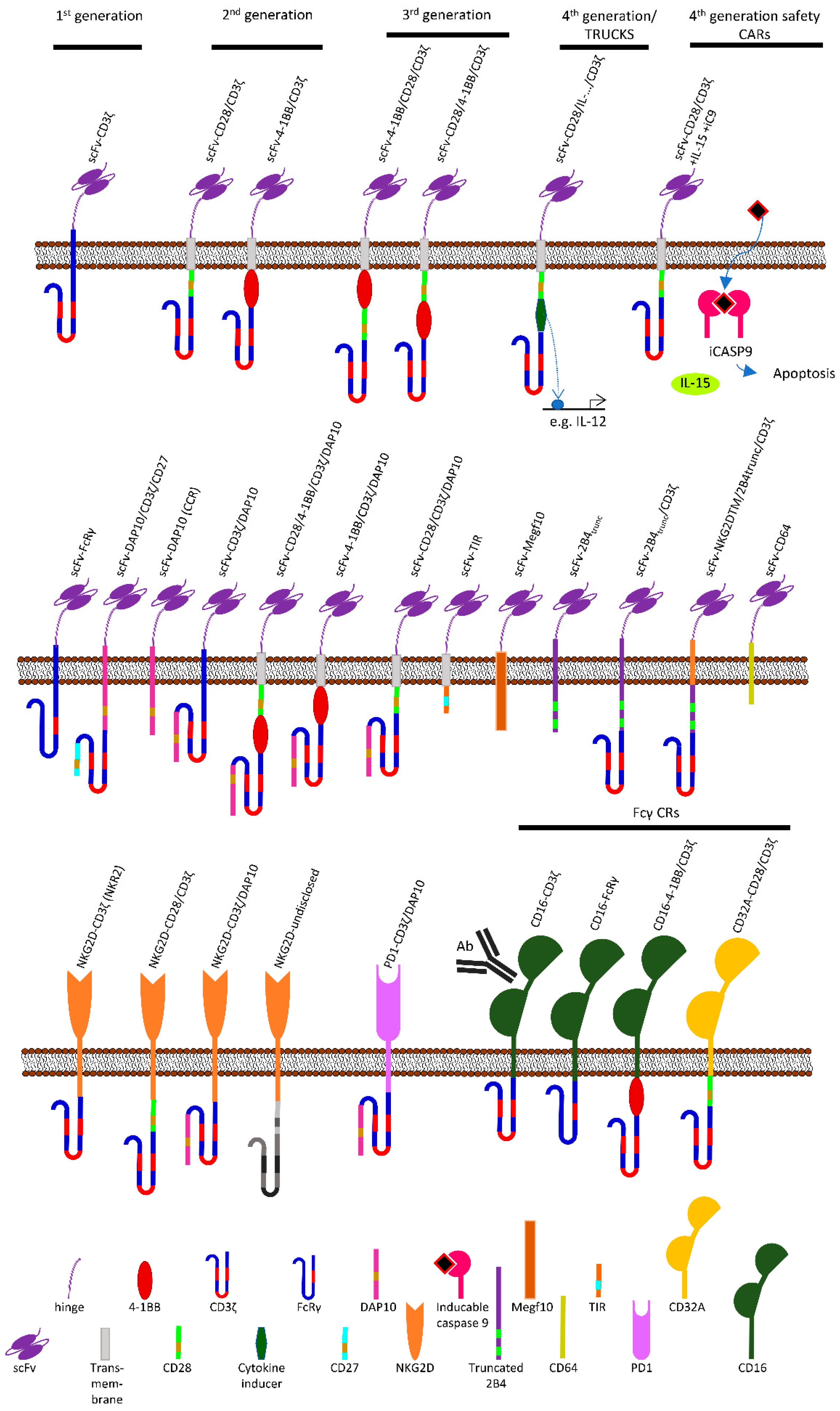
CARs: Beyond T Cells and T Cell-Derived Signaling Domains



Abstract
:1. Conventional T Cells Are the Pioneers of Chimeric Antigen Receptor (CAR) Therapy
2. Alternative Cell Types Suitable for CAR Cell Therapy
2.1. γ/δ T Cells
Clinical Trials Employing CAR-γ/δ T Cells
2.2. NKT Cells
Clinical Trials Employing CAR-NKT Cells
2.3. NK Cells
Clinical Trials Employing CAR-NK Cells
2.4. Myeloid Cells
2.4.1. Neutrophils
2.4.2. Monocytes
2.4.3. Macrophages
2.4.4. Myeloid Dendritic Cells
3. Alternative Extracellular and Intracellular Domains Used in CARs
3.1. Classical Signaling Domains
3.1.1. CD3ζ
3.1.2. CD28
3.1.3. 4-1BB (CD137)
3.2. Non-Classical Extracellular and Signaling Domains
3.2.1. NKG2D
3.2.2. DAP10
3.2.3. Fcγ-receptors
FcγRIII (CD16)
FcγRIIA (CD32A)
3.2.4. 2B4 (CD244)
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| CAR | Chimeric antigen receptor |
| TCR | T cell receptor |
| MHC | Major histocompatibility complex |
| APC | Antigen-presenting cell |
| TE | Effector T cell |
| TM | Memory T cell |
| TCM | Central memory T cell |
| TEM | Effector memory T cell |
| ALL | Acute lymphoblastic leukemia |
| TME | Tumor microenvironment |
| CRS | Cytokine release syndrome |
| GvHD | Graft versus host disease |
| HSP | Heat shock protein |
| PPR | Pattern recognition receptor |
| IEL | Intraepithelial lymphocyte |
| ADCC | Antibody-dependent cellular cytotoxicity |
| NCR | Natural cytotoxicity receptor |
| KIR | Killer cell immunoglobulin-like receptor |
| CLL | Chronic lymphocytic leukemia |
| HSC | Hematopoietic stem cell |
| EGFR | Epidermal growth factor receptor |
| TAM | Tumor-associated macrophage |
| BMDM | Bone marrow-derived macrophage |
| CAR-P | Chimeric antigen receptor for phagocytosis |
| CARMA | CAR macrophage |
| TIR | Toll interleukin receptor |
| DC | Dendritic cell |
| DCvac | DC vaccination |
| Eps8 | EGFR pathway substrate 8 |
| EV | Extracellular vesicle |
| scFv | Single-chain variable fragment |
| TM | Transmembrane |
| ITAM | Immunoreceptor tyrosine-based activation motif |
| AICD | Activation-induced cell death |
| AML | Acute myeloid leukemia |
| MM | Multiple myeloma |
| meso | Mesothelin |
| iPSC | Induced pluripotent stem cell |
| DAP10 | DNAX-activating protein of 10 kDa |
| CCR | Chimeric co-stimulatory receptor |
| uniCAR | Universal CAR |
| Fcγ CR | Fcγ chimeric receptor |
| mAb | Monoclonal antibody |
| EGFR | Epidermal growth factor receptor |
| CRC | Colorectal cancer |
| SLAM | Signaling lymphocyte activation molecule |
| ITSM | Immunoreceptor tyrosine-based switch motif |
| SAP | SLAM-associated protein |
| T-ALL | T cell acute lymphoblastic leukemia |
References
- Kaech, S.M.; Wherry, E.J.; Ahmed, R. Effector and memory T-cell differentiation: Implications for vaccine development. Nat. Rev. Immunol. 2002, 2, 251–262. [Google Scholar] [CrossRef] [PubMed]
- Stemberger, C.; Neuenhahn, M.; Buchholz, V.R.; Busch, D.H. Origin of CD8+ effector and memory T cell subsets. Cell. Mol. Immunol. 2007, 4, 399–405. [Google Scholar] [PubMed]
- Gattinoni, L.; Lugli, E.; Ji, Y.; Pós, Z.; Paulos, C.M.; Quigley, M.F.; De Almeida, J.R.; Gostick, E.; Yu, Z.; Carpenito, C.; et al. A human memory T cell subset with stem cell–like properties. Nat. Med. 2011, 17, 1290–1297. [Google Scholar] [CrossRef]
- Gerlach, C.; Rohr, J.C.; Perie, L.; Van Rooij, N.; Van Heijst, J.W.J.; Velds, A.; Urbanus, J.; Naik, S.H.; Jacobs, H.; Beltman, J.; et al. Heterogeneous Differentiation Patterns of Individual CD8+ T Cells. Science 2013, 340, 635–639. [Google Scholar] [CrossRef] [PubMed]
- Buchholz, V.R.; Flossdorf, M.; Hensel, I.; Kretschmer, L.; Weissbrich, B.; Gräf, P.; Verschoor, A.; Schiemann, M.; Höfer, T.; Busch, D.H. Disparate Individual Fates Compose Robust CD8+ T Cell Immunity. Science 2013, 340, 630–635. [Google Scholar] [CrossRef] [PubMed]
- Graef, P.; Buchholz, V.R.; Stemberger, C.; Flossdorf, M.; Henkel, L.; Schiemann, M.; Drexler, I.; Höfer, T.; Riddell, S.R.; Busch, D.H. Serial Transfer of Single-Cell-Derived Immunocompetence Reveals Stemness of CD8+ Central Memory T Cells. Immunity 2014, 41, 116–126. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sommermeyer, D.; Hudecek, M.; Kosasih, P.L.; Gogishvili, T.; Maloney, D.G.; Turtle, C.J.; Riddell, S.R. Chimeric antigen receptor-modified T cells derived from defined CD8+ and CD4+ subsets confer superior antitumor reactivity in vivo. Leukemia 2015, 30, 492–500. [Google Scholar] [CrossRef] [Green Version]
- Gross, G.; Gorochov, G.; Waks, T.; Eshhar, Z. Generation of effector T cells expressing chimeric T cell receptor with antibody type-specificity. Transplant. Proc. 1989, 21, 127–130. [Google Scholar]
- FDA Approves Second CAR T-cell Therapy. Cancer Discov. 2017, 8, 5–6. [CrossRef] [Green Version]
- Yip, A.; Webster, R.M. The market for chimeric antigen receptor T cell therapies. Nat. Rev. Drug Discov. 2018, 17, 161–162. [Google Scholar] [CrossRef]
- Mackall, C.L.; Fleisher, T.A.; Brown, M.R.; Andrich, M.P.; Chen, C.C.; Feuerstein, I.M.; Horowitz, M.E.; Magrath, I.T.; Shad, A.T.; Steinberg, S.M.; et al. Age, Thymopoiesis, and CD4+ T-Lymphocyte Regeneration after Intensive Chemotherapy. N. Engl. J. Med. 1995, 332, 143–149. [Google Scholar] [CrossRef] [PubMed]
- Hakim, F.T.; Cepeda, R.; Kaimei, S.; Mackall, C.L.; McAtee, N.; Zujewski, J.; Cowan, K.; Gress, R.E. Constraints on CD4 Recovery Postchemotherapy in Adults: Thymic Insufficiency and Apoptotic Decline of Expanded Peripheral CD4 Cells. Blood 1997, 90, 3789–3798. [Google Scholar] [CrossRef] [PubMed]
- Brentjens, R.J.; Rivière, I.; Park, J.H.; Davila, M.L.; Wang, X.; Stefanski, J.; Taylor, C.; Yeh, R.; Bartido, S.; Borquez-Ojeda, O.; et al. Safety and persistence of adoptively transferred autologous CD19-targeted T cells in patients with relapsed or chemotherapy refractory B-cell leukemias. Blood 2011, 118, 4817–4828. [Google Scholar] [CrossRef] [PubMed]
- Kalos, M.; Levine, B.L.; Porter, D.L.; Katz, S.I.; Grupp, S.A.; Bagg, A.; June, C.H. T Cells with Chimeric Antigen Receptors Have Potent Antitumor Effects and Can Establish Memory in Patients with Advanced Leukemia. Sci. Transl. Med. 2011, 3, 95ra73. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kochenderfer, J.N.; Dudley, M.E.; Feldman, S.A.; Wilson, W.H.; Spaner, D.E.; Maric, I.; Stetler-Stevenson, M.; Phan, G.Q.; Hughes, M.S.; Sherry, R.M.; et al. B-cell depletion and remissions of malignancy along with cytokine-associated toxicity in a clinical trial of anti-CD19 chimeric-antigen-receptor–transduced T cells. Blood 2012, 119, 2709–2720. [Google Scholar] [CrossRef]
- Davila, M.L.; Bouhassira, D.C.G.; Park, J.H.; Curran, K.J.; Smith, E.L.; Pegram, H.J.; Brentjens, R.J. Chimeric antigen receptors for the adoptive T cell therapy of hematologic malignancies. Int. J. Hematol. 2013, 99, 361–371. [Google Scholar] [CrossRef]
- E Walker, R.; Bechtel, C.M.; Natarajan, V.; Baseler, M.; Hege, K.M.; A Metcalf, J.; Stevens, R.; Hazen, A.; Blaese, R.M.; Chen, C.C.; et al. Long-term in vivo survival of receptor-modified syngeneic T cells in patients with human immunodeficiency virus infection. Blood 2000, 96, 467–474. [Google Scholar]
- Mitsuyasu, R.T.; A Anton, P.; Deeks, S.G.; Scadden, D.T.; Connick, E.; Downs, M.T.; Bakker, A.; Roberts, M.R.; June, C.H.; Jalali, S.; et al. Prolonged survival and tissue trafficking following adoptive transfer of CD4zeta gene-modified autologous CD4(+) and CD8(+) T cells in human immunodeficiency virus-infected subjects. Blood 2000, 96, 785–793. [Google Scholar] [CrossRef]
- Hudecek, M.; Sommermeyer, D.; Kosasih, P.L.; Silva-Benedict, A.; Liu, L.; Rader, C.; Jensen, M.C.; Riddell, S.R. The Nonsignaling Extracellular Spacer Domain of Chimeric Antigen Receptors Is Decisive for In Vivo Antitumor Activity. Cancer Immunol. Res. 2014, 3, 125–135. [Google Scholar] [CrossRef] [Green Version]
- Berger, C.; Jensen, M.C.; Lansdorp, P.M.; Gough, M.; Elliott, C.; Riddell, S.R. Adoptive transfer of effector CD8+ T cells derived from central memory cells establishes persistent T cell memory in primates. J. Clin. Investig. 2008, 118, 294–305. [Google Scholar] [CrossRef] [Green Version]
- Berger, C.; Berger, M.; Anderson, D.; Riddell, S.R. A non-human primate model for analysis of safety, persistence, and function of adoptively transferred T cells. J. Med Primatol. 2010, 40, 88–103. [Google Scholar] [CrossRef] [PubMed]
- Klebanoff, C.A.; Gattinoni, L.; Torabi-Parizi, P.; Kerstann, K.; Cardones, A.R.; Finkelstein, S.E.; Palmer, D.C.; Antony, P.A.; Hwang, S.T.; Rosenberg, S.A.; et al. Central memory self/tumor-reactive CD8+ T cells confer superior antitumor immunity compared with effector memory T cells. Proc. Natl. Acad. Sci. USA 2005, 102, 9571–9576. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hinrichs, C.S.; Borman, Z.A.; Cassard, L.; Gattinoni, L.; Spolski, R.; Yu, Z.; Sanchez-Perez, L.; Muranski, P.; Kern, S.J.; Logun, C.; et al. Adoptively transferred effector cells derived from naïve rather than central memory CD8+ T cells mediate superior antitumor immunity. Proc. Natl. Acad. Sci. USA 2009, 106, 17469–17474. [Google Scholar] [CrossRef] [Green Version]
- Westwood, J.A.; Smyth, M.J.; Teng, M.W.; Moeller, M.; Trapani, J.A.; Scott, A.M.; Smyth, F.E.; Cartwright, G.A.; Power, B.; Hönemann, D.; et al. Adoptive transfer of T cells modified with a humanized chimeric receptor gene inhibits growth of Lewis-Y-expressing tumors in mice. Proc. Natl. Acad. Sci. USA 2005, 102, 19051–19056. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moeller, M.; Kershaw, M.; Cameron, R.; A Westwood, J.; Trapani, J.A.; Smyth, M.J.; Darcy, P.K. Sustained Antigen-Specific Antitumor Recall Response Mediated by Gene-Modified CD4+ T Helper-1 and CD8+ T Cells. Cancer Res. 2007, 67, 11428–11437. [Google Scholar] [CrossRef] [Green Version]
- Turtle, C.J.; Hanafi, L.-A.; Berger, C.; Gooley, T.A.; Cherian, S.; Hudecek, M.; Sommermeyer, D.; Melville, K.; Pender, B.; Budiarto, T.M.; et al. CD19 CAR-T cells of defined CD4+:CD8+ composition in adult B cell ALL patients. J. Clin. Investig. 2016, 126, 2123–2138. [Google Scholar] [CrossRef] [Green Version]
- Lim, W.A.; June, C.H. The Principles of Engineering Immune Cells to Treat Cancer. Cell 2017, 168, 724–740. [Google Scholar] [CrossRef] [Green Version]
- Danilova, L.V.; Anagnostou, V.; Caushi, J.X.; Sidhom, J.-W.; Guo, H.; Chan, H.Y.; Suri, P.; Tam, A.J.; Zhang, J.; El Asmar, M.; et al. The Mutation-Associated Neoantigen Functional Expansion of Specific T Cells (MANAFEST) Assay: A Sensitive Platform for Monitoring Antitumor Immunity. Cancer Immunol. Res. 2018. [Google Scholar] [CrossRef] [Green Version]
- Brudno, J.N.; Kochenderfer, J.N. Chimeric antigen receptor T-cell therapies for lymphoma. Nat. Rev. Clin. Oncol. 2017, 15, 31–46. [Google Scholar] [CrossRef]
- Smith, M.; Zakrzewski, J.; James, S.; Sadelain, M. Posttransplant chimeric antigen receptor therapy. Blood 2018, 131, 1045–1052. [Google Scholar] [CrossRef] [Green Version]
- Dotti, G.; Gottschalk, S.; Savoldo, B.; Brenner, M.K. Design and development of therapies using chimeric antigen receptor-expressing T cells. Immunol. Rev. 2014, 257, 107–126. [Google Scholar] [CrossRef] [PubMed]
- Watanabe, K.; Kuramitsu, S.; Posey, A.D.; June, C.H. Expanding the Therapeutic Window for CAR T Cell Therapy in Solid Tumors: The Knowns and Unknowns of CAR T Cell Biology. Front. Immunol. 2018, 9, 2486. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- D’Aloia, M.M.; Zizzari, I.G.; Sacchetti, B.; Pierelli, L.; Alimandi, M. CAR-T cells: The long and winding road to solid tumors. Cell Death Dis. 2018, 9, 282. [Google Scholar] [CrossRef]
- Lamers, C.H.J.; Sleijfer, S.; Van Steenbergen, S.; Van Elzakker, P.; Van Krimpen, B.; Groot, C.; Vulto, A.; Bakker, M.D.; Oosterwijk, E.; Debets, R.; et al. Treatment of Metastatic Renal Cell Carcinoma With CAIX CAR-engineered T cells: Clinical Evaluation and Management of On-target Toxicity. Mol. Ther. 2013, 21, 904–912. [Google Scholar] [CrossRef]
- Perales, M.A.; Kebriaei, P.; Kean, L.S.; Sadelain, M. Building a Safer and Faster CAR: Seatbelts, Airbags, and CRISPR. Boil. Blood Marrow Transplant. 2017, 24, 27–31. [Google Scholar] [CrossRef] [Green Version]
- Harrer, D.C.; Dörrie, J.; Schaft, N. Chimeric Antigen Receptors in Different Cell Types: New Vehicles Join the Race. Hum. Gene Ther. 2018, 29, 547–558. [Google Scholar] [CrossRef]
- Vantourout, P.; Hayday, A. Six-of-the-best: Unique contributions of gammadelta T cells to immunology. Nat. Rev. Immunol. 2013, 13, 88–100. [Google Scholar] [CrossRef] [Green Version]
- Morita, C.; Mariuzza, R.A.; Brenner, M.B. Antigen recognition by human γδ T cells: Pattern recognition by the adaptive immune system. Springer Semin. Immunopathol. 2000, 22, 191–217. [Google Scholar] [CrossRef]
- Holtmeier, W.; Kabelitz, D. γδ T Cells Link Innate and Adaptive Immune Responses. Chem. Immunol. Allergy 2005, 86, 151–183. [Google Scholar] [CrossRef]
- Hilden, J.M.; Dinndorf, P.A.; Meerbaum, S.O.; Sather, H.; Villaluna, D.; Heerema, N.A.; McGlennen, R.; Smith, F.O.; Woods, W.G.; Salzer, W.L.; et al. Analysis of prognostic factors of acute lymphoblastic leukemia in infants: Report on CCG 1953 from the Children’s Oncology Group. Blood 2006, 108, 441–451. [Google Scholar] [CrossRef]
- Mirzaei, H.R.; Mirzaei, H.; Lee, S.Y.; Hadjati, J.; Till, B. Prospects for chimeric antigen receptor (CAR) γδ T cells: A potential game changer for adoptive T cell cancer immunotherapy. Cancer Lett. 2016, 380, 413–423. [Google Scholar] [CrossRef] [Green Version]
- Zhao, Y.; Niu, C.; Cui, J. Gamma-delta (γδ) T cells: Friend or foe in cancer development? J. Transl. Med. 2018, 16, 3. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Silva-Santos, B. Promoting angiogenesis within the tumor microenvironment: The secret life of murine lymphoid IL-17-producing γδ T cells. Eur. J. Immunol. 2010, 40, 1873–1876. [Google Scholar] [CrossRef] [PubMed]
- Paul, S.; Lal, G. Regulatory and effector functions of gamma-delta (γδ) T cells and their therapeutic potential in adoptive cellular therapy for cancer. Int. J. Cancer 2016, 139, 976–985. [Google Scholar] [CrossRef] [PubMed]
- Peng, W.; Chen, J.Q.; Liu, C.; Malu, S.; Creasy, C.; Tetzlaff, M.T.; Xu, C.; McKenzie, J.A.; Zhang, C.; Liang, X.; et al. Loss of PTEN Promotes Resistance to T Cell-Mediated Immunotherapy. Cancer Discov. 2015, 6, 202–216. [Google Scholar] [CrossRef] [Green Version]
- Qu, P.; Wang, L.-Z.; Lin, P.C. Expansion and functions of myeloid-derived suppressor cells in the tumor microenvironment. Cancer Lett. 2016, 380, 253–256. [Google Scholar] [CrossRef]
- Oevermann, L.; Lang, P.; Feuchtinger, T.; Schumm, M.; Teltschik, H.-M.; Schlegel, P.; Handgretinger, R. Immune reconstitution and strategies for rebuilding the immune system after haploidentical stem cell transplantation. Ann. New York Acad. Sci. 2012, 1266, 161–170. [Google Scholar] [CrossRef]
- Critchley-Thorne, R.J.; Simons, D.L.; Yan, N.; Miyahira, A.K.; Dirbas, F.M.; Johnson, D.L.; Swetter, S.M.; Carlson, R.W.; Fisher, G.A.; Koong, A.; et al. Impaired interferon signaling is a common immune defect in human cancer. Proc. Natl. Acad. Sci. USA 2009, 106, 9010–9015. [Google Scholar] [CrossRef] [Green Version]
- Brandes, M. Professional Antigen-Presentation Function by Human gd T Cells. Science 2005, 309, 264–268. [Google Scholar] [CrossRef]
- Muto, M.; Baghdadi, M.; Maekawa, R.; Wada, H.; Seino, K.-I. Myeloid molecular characteristics of human γδ T cells support their acquisition of tumor antigen-presenting capacity. Cancer Immunol. Immunother. 2015, 64, 941–949. [Google Scholar] [CrossRef] [Green Version]
- Godfrey, D.; Stankovic, S.; Baxter, A. Raising the NKT cell family. Nat. Immunol. 2010, 11, 197–206. [Google Scholar] [CrossRef]
- Godfrey, D.; Macdonald, H.R.; Kronenberg, M.; Smyth, M.J.; Van Kaer, L. NKT cells: What’s in a name? Nat. Rev. Immunol. 2004, 4, 231–237. [Google Scholar] [CrossRef] [PubMed]
- Gumperz, J.E.; Miyake, S.; Yamamura, T.; Brenner, M.B. Functionally Distinct Subsets of CD1d-restricted Natural Killer T Cells Revealed by CD1d Tetramer Staining. J. Exp. Med. 2002, 195, 625–636. [Google Scholar] [CrossRef]
- Tachibana, T.; Onodera, H.; Tsuruyama, T.; Imamura, M.; Mori, A.; Nagayama, S.; Hiai, H. Increased Intratumor V?24-Positive Natural Killer T Cells: A Prognostic Factor for Primary Colorectal Carcinomas. Clin. Cancer Res. 2005, 11, 7322–7327. [Google Scholar] [CrossRef] [Green Version]
- Terabe, M.; Matsui, S.; Noben-Trauth, N.; Chen, H.; Watson, C.; Donaldson, D.D.; Carbone, D.P.; Paul, W.E.; Berzofsky, J.A. NKT cell–mediated repression of tumor immunosurveillance by IL-13 and the IL-4R–STAT6 pathway. Nat. Immunol. 2000, 1, 515–520. [Google Scholar] [CrossRef]
- Yu, J. Antitumor Activity of T Cells Generated from Lymph Nodes Draining the SEA-expressing Murine B16 Melanoma and Secondarily Activated with Dendritic Cells. Int. J. Boil. Sci. 2009, 5, 135–146. [Google Scholar] [CrossRef] [PubMed]
- Gottschalk, C.; Mettke, E.; Kurts, C. The Role of Invariant Natural Killer T Cells in Dendritic Cell Licensing, Cross-Priming, and Memory CD8+ T Cell Generation. Front. Immunol. 2015, 6, 379. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morvan, M.G.; Lanier, L.L. NK cells and cancer: You can teach innate cells new tricks. Nat. Rev. Cancer 2015, 16, 7–19. [Google Scholar] [CrossRef]
- Long, G.V.; Stroyakovskiy, D.; Gogas, H.; Levchenko, E.V.; De Braud, F.; Larkin, J.; Garbe, C.; Jouary, T.; Hauschild, A.; Grob, J.-J.; et al. Dabrafenib and trametinib versus dabrafenib and placebo for Val600 BRAF-mutant melanoma: A multicentre, double-blind, phase 3 randomised controlled trial. Lancet 2015, 386, 444–451. [Google Scholar] [CrossRef]
- Bollino, D.; Webb, T.J. Chimeric antigen receptor-engineered natural killer and natural killer T cells for cancer immunotherapy. Transl. Res. 2017, 187, 32–43. [Google Scholar] [CrossRef]
- Ruggeri, L.; Capanni, M.; Urbani, E.; Shlomchik, W.D.; Posati, S.; Rogaia, D.; Martelli, M.F.; Velardi, A.; Perruccio, K.; Tosti, A.; et al. Effectiveness of Donor Natural Killer Cell Alloreactivity in Mismatched Hematopoietic Transplants. Science 2002, 295, 2097–2100. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miller, J.S.; Soignier, Y.; Panoskaltsis-Mortari, A.; McNearney, S.A.; Yun, G.H.; Fautsch, S.K.; McKenna, D.; Le, C.; DeFor, T.; Burns, L.J.; et al. Successful adoptive transfer and in vivo expansion of human haploidentical NK cells in patients with cancer. Blood 2005, 105, 3051–3057. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Klöß, S.; Oberschmidt, O.; A Morgan, M.; Dahlke, J.; Arseniev, L.; Huppert, V.; Granzin, M.; Gardlowski, T.; Matthies, N.; Soltenborn, S.; et al. Optimization of Human NK Cell Manufacturing: Fully Automated Separation, ImprovedEx VivoExpansion Using IL-21 with Autologous Feeder Cells, and Generation of Anti-CD123-CAR-Expressing Effector Cells. Hum. Gene Ther. 2017, 28, 897–913. [Google Scholar] [CrossRef]
- Bosch-Voskens, C.; Watanabe, R.; Rollins, S.; Campana, D.; Hasumi, K.; Mann, D. Ex-vivo expanded human NK cells express activating receptors that mediate cytotoxicity of allogeneic and autologous cancer cell lines by direct recognition and antibody directed cellular cytotoxicity. J. Exp. Clin. Cancer Res. 2010, 29, 134. [Google Scholar] [CrossRef] [Green Version]
- Sutlu, T.; Stellan, B.; Gilljam, M.; Quezada, H.C.; Nahi, H.H.; Gahrton, G.; Alici, E. Clinical-grade, large-scale, feeder-free expansion of highly active human natural killer cells for adoptive immunotherapy using an automated bioreactor. Cytotherapy 2010, 12, 1044–1055. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, E.; Tong, Y.; Dotti, G.; Shaim, H.; Savoldo, B.; Mukherjee, M.; Orange, J.; Wan, X.; Lu, X.; Reynolds, A.; et al. Cord blood NK cells engineered to express IL-15 and a CD19-targeted CAR show long-term persistence and potent antitumor activity. Leukemia 2017, 32, 520–531. [Google Scholar] [CrossRef]
- Lowe, E.; Truscott, L.C.; De Oliveira, S. In Vitro Generation of Human NK Cells Expressing Chimeric Antigen Receptor Through Differentiation of Gene-Modified Hematopoietic Stem Cells. Breast Cancer 2016, 1441, 241–251. [Google Scholar] [CrossRef] [Green Version]
- Li, Y.; Hermanson, D.L.; Moriarity, B.S.; Kaufman, D. Human iPSC-Derived Natural Killer Cells Engineered with Chimeric Antigen Receptors Enhance Anti-Tumor Activity. Cell Stem Cell 2018, 23, 181–192. [Google Scholar] [CrossRef] [Green Version]
- Oelsner, S.; Friede, M.E.; Zhang, C.; Wagner, J.; Badura, S.; Bader, P.; Ullrich, E.; Ottmann, O.; Klingemann, H.; Tonn, T.; et al. Continuously expanding CAR NK-92 cells display selective cytotoxicity against B-cell leukemia and lymphoma. Cytotherapy 2017, 19, 235–249. [Google Scholar] [CrossRef] [Green Version]
- Klingemann, H.; Boissel, L.; Toneguzzo, F. Natural Killer Cells for Immunotherapy—Advantages of the NK-92 Cell Line over Blood NK Cells. Front. Immunol. 2016, 7, 177. [Google Scholar] [CrossRef] [Green Version]
- Yong, C.S.; John, L.B.; Devaud, C.; Prince, M.H.; Johnstone, R.W.; Trapani, J.A.; Darcy, P.K.; Kershaw, M. A role for multiple chimeric antigen receptor-expressing leukocytes in antigen-specific responses to cancer. Oncotarget 2016, 7, 34582–34598. [Google Scholar] [CrossRef] [PubMed]
- Hu, Y.; Tian, Z.-G.; Zhang, C. Chimeric antigen receptor (CAR)-transduced natural killer cells in tumor immunotherapy. Acta Pharmacol. Sin. 2017, 39, 167–176. [Google Scholar] [CrossRef]
- Ramos, C.A.; Rouce, R.; Robertson, C.S.; Reyna, A.; Narala, N.; Vyas, G.; Mehta, B.; Zhang, H.; Dakhova, O.; Carrum, G.; et al. In Vivo Fate and Activity of Second- versus Third-Generation CD19-Specific CAR-T Cells in B Cell Non-Hodgkin’s Lymphomas. Mol. Ther. 2018, 26, 2727–2737. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, E.; Marin, D.; Banerjee, P.; Macapinlac, H.A.; Thompson, P.; Basar, R.; Kerbauy, L.N.; Overman, B.; Thall, P.; Kaplan, M.; et al. Use of CAR-Transduced Natural Killer Cells in CD19-Positive Lymphoid Tumors. N. Engl. J. Med. 2020, 382, 545–553. [Google Scholar] [CrossRef] [PubMed]
- De Oliveira, S.; Ryan, C.; Giannoni, F.; Hardee, C.L.; Tremcinska, I.; Katebian, B.; Wherley, J.; Sahaghian, A.; Tu, A.; Grogan, T.; et al. Modification of Hematopoietic Stem/Progenitor Cells with CD19-Specific Chimeric Antigen Receptors as a Novel Approach for Cancer Immunotherapy. Hum. Gene Ther. 2013, 24, 824–839. [Google Scholar] [CrossRef] [PubMed]
- Larson, S.; De Oliveira, S. Gene-modified hematopoietic stem cells for cancer immunotherapy. Hum. Vaccines Immunother. 2014, 10, 982–985. [Google Scholar] [CrossRef] [Green Version]
- Gschweng, E.; De Oliveira, S.; Kohn, D.B. Hematopoietic stem cells for cancer immunotherapy. Immunol. Rev. 2014, 257, 237–249. [Google Scholar] [CrossRef] [Green Version]
- Larson, S.M.; Truscott, L.C.; Chiou, T.-T.; Patel, A.; Kao, R.; Tu, A.; Tyagi, T.; Lu, X.; Elashoff, D.; De Oliveira, S. Pre-clinical development of gene modification of haematopoietic stem cells with chimeric antigen receptors for cancer immunotherapy. Hum. Vaccines Immunother. 2017, 13, 1094–1104. [Google Scholar] [CrossRef]
- Kao, R.L.; Truscott, L.C.; Chiou, T.-T.; Tsai, W.; Wu, A.M.; De Oliveira, S. A Cetuximab-Mediated Suicide System in Chimeric Antigen Receptor-Modified Hematopoietic Stem Cells for Cancer Therapy. Hum. Gene Ther. 2019, 30, 413–428. [Google Scholar] [CrossRef]
- Hege, K.M.; Cooke, K.S.; Finer, M.H.; Zsebo, K.M.; Roberts, M.R. Systemic T Cell–independent Tumor Immunity after Transplantation of Universal Receptor–modified Bone Marrow into SCID Mice. J. Exp. Med. 1996, 184, 2261–2270. [Google Scholar] [CrossRef] [Green Version]
- Roberts, M.R.; Cooke, K.S.; Tran, A.C.; A Smith, K.; Lin, W.Y.; Wang, M.; Dull, T.J.; Farson, D.; Zsebo, K.M.; Finer, M.H. Antigen-specific cytolysis by neutrophils and NK cells expressing chimeric immune receptors bearing zeta or gamma signaling domains. J. Immunol. 1998, 161, 375–384. [Google Scholar] [PubMed]
- Lin, W.Y.; Roberts, M.R. Developmental dissociation of T cells from B, NK, and myeloid cells revealed by MHC class II–specific chimeric immune receptors bearing TCR-ζ or FcR-γ chain signaling domains. Blood 2002, 100, 3045–3048. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Witko-Sarsat, V.; Rieu, P.; Descamps-Latscha, B.; Lesavre, P.; Halbwachs-Mecarelli, L. Neutrophils: Molecules, functions and pathophysiological aspects. Lab. Investig. 2000, 80, 617–653. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nichols, B.A.; Bainton, D.F.; Farquhar, M.G. Differentiation of Monocytes. J. Cell Boil. 1971, 50, 498–515. [Google Scholar] [CrossRef] [PubMed]
- Biglari, A.; Southgate, T.D.; Fairbairn, L.J.; E Gilham, D. Human monocytes expressing a CEA-specific chimeric CD64 receptor specifically target CEA-expressing tumour cells in vitro and in vivo. Gene Ther. 2006, 13, 602–610. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.-X.; Zhang, S.-X.; Wu, H.-J.; Rong, X.-L.; Guo, J. M2b macrophage polarization and its roles in diseases. J. Leukoc. Boil. 2018, 106, 345–358. [Google Scholar] [CrossRef] [Green Version]
- Mosser, D.; Edwards, J. Exploring the full spectrum of macrophage activation. Nat. Rev. Immunol. 2008, 8, 958–969. [Google Scholar] [CrossRef]
- Velmurugan, R.; Challa, D.K.; Ram, S.; Ober, R.; Ward, E.S. Macrophage-Mediated Trogocytosis Leads to Death of Antibody-Opsonized Tumor Cells. Mol. Cancer Ther. 2016, 15, 1879–1889. [Google Scholar] [CrossRef] [Green Version]
- Klichinsky, M.; Ruella, M.; Shestova, O.; Kenderian, S.S.; Kim, M.Y.; O’Connor, R.; Scholler, J.; June, C.; Gill, S. Abstract 4575: Chimeric antigen receptor macrophages (CARMA) for adoptive cellular immunotherapy of solid tumors. Immunology 2017, 77, 4575. [Google Scholar] [CrossRef]
- Morrissey, M.A.; Williamson, A.P.; Steinbach, A.; Roberts, E.; Kern, N.; Headley, M.B.; Vale, R.D. Chimeric antigen receptors that trigger phagocytosis. eLife 2018, 7, e36688. [Google Scholar] [CrossRef]
- Chanmee, T.; Ontong, P.; Konno, K.; Itano, N. Tumor-Associated Macrophages as Major Players in the Tumor Microenvironment. Cancers 2014, 6, 1670–1690. [Google Scholar] [CrossRef] [Green Version]
- Smith, E.W.C.; Weagel, E.; Smith, C.; Liu, P.G.; Robison, R.; O’Neill, K. Macrophage Polarization and Its Role in Cancer. J. Clin. Cell. Immunol. 2015, 6, 1–8. [Google Scholar] [CrossRef]
- Bingle, L.; Brown, N.J.; Lewis, C. The role of tumour-associated macrophages in tumour progression: Implications for new anticancer therapies. J. Pathol. 2002, 196, 254–265. [Google Scholar] [CrossRef]
- Murdoch, C.; Giannoudis, A.; Lewis, C.E. Mechanisms regulating the recruitment of macrophages into hypoxic areas of tumors and other ischemic tissues. Blood 2004, 104, 2224–2234. [Google Scholar] [CrossRef]
- Valipour, B.; Velaei, K.; Abedelahi, A.; Karimipour, M.; Darabi, M.; Charoudeh, H.N. NK cells: An attractive candidate for cancer therapy. J. Cell. Physiol. 2019, 234, 19352–19365. [Google Scholar] [CrossRef] [PubMed]
- Tseng, D.; Volkmer, J.-P.; Willingham, S.B.; Contreras-Trujillo, H.; Fathman, J.W.; Fernhoff, N.B.; Seita, J.; Inlay, M.A.; Weiskopf, K.; Miyanishi, M.; et al. Anti-CD47 antibody-mediated phagocytosis of cancer by macrophages primes an effective antitumor T-cell response. Proc. Natl. Acad. Sci. USA 2013, 110, 11103–11108. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Asano, K.; Nabeyama, A.; Miyake, Y.; Qiu, C.-H.; Kurita, A.; Tomura, M.; Kanagawa, O.; Fujii, S.-I.; Tanaka, M. CD169-Positive Macrophages Dominate Antitumor Immunity by Crosspresenting Dead Cell-Associated Antigens. Immunity1 2011, 34, 85–95. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Velazquez, E.J.; Lattin, J.E.; Brindley, T.D.; Reinstein, Z.Z.; Chu, R.; Liu, L.; Weagel, E.G.; Townsend, M.H.; Whitley, K.V.; Lawrence, E.L.; et al. Abstract 2563: Macrophage Toll-like receptor-chimeric antigen receptors (MOTO-CARs) as a novel adoptive cell therapy for the treatment of solid malignancies. Immunology 2018, 78, 2563. [Google Scholar] [CrossRef]
- Najafi, M.; Goradel, N.H.; Farhood, B.; Salehi, E.; Nashtaei, M.S.; Khanlarkhani, N.; Khezri, Z.; Majidpoor, J.; Mortezaee, K.; Habibi, M.; et al. Macrophage polarity in cancer: A review. J. Cell. Biochem. 2018, 120, 2756–2765. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Edwards, J.; Mosser, D.M. The Expression of Exogenous Genes in Macrophages: Obstacles and Opportunities. Breast Cancer 2009, 531, 123–143. [Google Scholar] [CrossRef] [Green Version]
- Schuler, G.; Schuler-Thurner, B.; Steinman, R.M. The use of dendritic cells in cancer immunotherapy. Curr. Opin. Immunol. 2003, 15, 138–147. [Google Scholar] [CrossRef]
- Wu, M.; Zhang, L.; Zhang, H.; Ning, J.; Tu, S.; He, Y.; Li, Y. CD19 chimeric antigen receptor–redirected T cells combined with epidermal growth factor receptor pathway substrate 8 peptide–derived dendritic cell vaccine in leukemia. Cytotherapy 2019, 21, 659–670. [Google Scholar] [CrossRef] [PubMed]
- Squadrito, M.L.; Cianciaruso, C.; Hansen, S.K.; De Palma, M. EVIR: Chimeric receptors that enhance dendritic cell cross-dressing with tumor antigens. Nat. Methods 2018, 15, 183–186. [Google Scholar] [CrossRef] [PubMed]
- Eshhar, Z.; Waks, T.; Gross, G.; Schindler, D.G. Specific activation and targeting of cytotoxic lymphocytes through chimeric single chains consisting of antibody-binding domains and the gamma or zeta subunits of the immunoglobulin and T-cell receptors. Proc. Natl. Acad. Sci. USA 1993, 90, 720–724. [Google Scholar] [CrossRef] [Green Version]
- Savoldo, B.; Ramos, C.A.; Liu, E.; Mims, M.P.; Keating, M.J.; Carrum, G.; Kamble, R.T.; Bollard, C.M.; Gee, A.P.; Mei, Z.; et al. CD28 costimulation improves expansion and persistence of chimeric antigen receptor-modified T cells in lymphoma patients. J. Clin. Investig. 2011, 121, 1822–1826. [Google Scholar] [CrossRef] [Green Version]
- Long, A.H.; Haso, W.M.; Shern, J.F.; Wanhainen, K.; Murgai, M.; Ingaramo, M.; Smith, J.P.; Walker, A.J.; Kohler, M.E.; Venkateshwara, V.R.; et al. 4-1BB costimulation ameliorates T cell exhaustion induced by tonic signaling of chimeric antigen receptors. Nat. Med. 2015, 21, 581–590. [Google Scholar] [CrossRef] [Green Version]
- Zhao, Z.; Condomines, M.; Van Der Stegen, S.J.; Perna, F.; Kloss, C.C.; Gunset, G.; Plotkin, J.; Sadelain, M. Structural Design of Engineered Costimulation Determines Tumor Rejection Kinetics and Persistence of CAR T Cells. Cancer Cell 2015, 28, 415–428. [Google Scholar] [CrossRef] [Green Version]
- Zabel, M.; Tauber, P.A.; Pickl, W.F. The making and function of CAR cells. Immunol. Lett. 2019, 212, 53–69. [Google Scholar] [CrossRef]
- Loskog, A.; Giandomenico, V.; Rossig, C.; Pule, M.; Dotti, G.; Brenner, M.K. Addition of the CD28 signaling domain to chimeric T-cell receptors enhances chimeric T-cell resistance to T regulatory cells. Leukemia 2006, 20, 1819–1828. [Google Scholar] [CrossRef]
- Song, D.-G.; Ye, Q.; Poussin, M.; Harms, G.M.; Figini, M.; Powell, D.J. CD27 costimulation augments the survival and antitumor activity of redirected human T cells in vivo. Blood 2012, 119, 696–706. [Google Scholar] [CrossRef]
- Cheadle, E.J.; Sheard, V.; Hombach, A.A.; Chmielewski, M.; Riet, T.; Berrevoets, C.; Schooten, E.; Lamers, C.; Abken, H.; Debets, R.; et al. Chimeric Antigen Receptors for T-Cell Based Therapy. Adv. Struct. Saf. Stud. 2012, 907, 645–666. [Google Scholar] [CrossRef]
- Guedan, S.; Chen, X.; Madar, A.; Carpenito, C.; Mcgettigan, S.E.; Frigault, M.J.; Lee, J.; Posey, A.D.; Scholler, J.; Scholler, N.; et al. ICOS-based chimeric antigen receptors program bipolar TH17/TH1 cells. Blood 2014, 124, 1070–1080. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kawalekar, O.U.; O’Connor, R.S.; Fraietta, J.A.; Guo, L.; Mcgettigan, S.E.; Posey, A.D.; Patel, P.R.; Guedan, S.; Scholler, J.; Keith, B.; et al. Distinct Signaling of Coreceptors Regulates Specific Metabolism Pathways and Impacts Memory Development in CAR T Cells. Immunity 2016, 44, 380–390. [Google Scholar] [CrossRef] [PubMed]
- Gomes-Silva, D.; Mukherjee, M.; Srinivasan, M.; Krenciute, G.; Dakhova, O.; Zheng, Y.; Cabral, J.M.S.; Rooney, C.M.; Orange, J.S.; Brenner, M.K.; et al. Tonic 4-1BB Costimulation in Chimeric Antigen Receptors Impedes T Cell Survival and Is Vector-Dependent. Cell Rep. 2017, 21, 17–26. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, G.; Boucher, J.C.; Kotani, H.; Park, K.; Zhang, Y.; Shrestha, B.; Wang, X.; Guan, L.; Beatty, N.; Abate-Daga, D.; et al. 4-1BB enhancement of CAR T function requires NF-κB and TRAFs. JCI Insight 2018, 3. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mamonkin, M.; Mukherjee, M.; Srinivasan, M.; Sharma, S.; Gomes-Silva, D.; Mo, F.; Krenciute, G.; Orange, J.S.; Brenner, M.K. Reversible Transgene Expression Reduces Fratricide and Permits 4-1BB Costimulation of CAR T Cells Directed to T-cell Malignancies. Cancer Immunol. Res. 2017, 6, 47–58. [Google Scholar] [CrossRef] [Green Version]
- Salter, A.I.; Ivey, R.G.; Kennedy, J.J.; Voillet, V.; Rajan, A.; Alderman, E.J.; Voytovich, U.J.; Lin, C.; Sommermeyer, D.; Liu, L.; et al. Phosphoproteomic analysis of chimeric antigen receptor signaling reveals kinetic and quantitative differences that affect cell function. Sci. Signal. 2018, 11, eaat6753. [Google Scholar] [CrossRef] [Green Version]
- Golumba-Nagy, V.; Kuehle, J.; Hombach, A.A.; Abken, H. CD28-ζ CAR T Cells Resist TGF-β Repression through IL-2 Signaling, Which Can Be Mimicked by an Engineered IL-7 Autocrine Loop. Mol. Ther. 2018, 26, 2218–2230. [Google Scholar] [CrossRef] [Green Version]
- Priceman, S.J.; Gerdts, E.A.; Tilakawardane, D.; Kennewick, K.T.; Murad, J.; Park, A.K.; Jeang, B.; Yamaguchi, Y.; Yang, X.; Urak, R.; et al. Co-stimulatory signaling determines tumor antigen sensitivity and persistence of CAR T cells targeting PSCA+ metastatic prostate cancer. OncoImmunology 2017, 7, e1380764. [Google Scholar] [CrossRef]
- Ramello, M.C.; Benzaïd, I.; Kuenzi, B.M.; Lienlaf-Moreno, M.; Kandell, W.M.; Santiago, D.N.; Pabón-Saldaña, M.; Darville, L.; Fang, B.; Rix, U.; et al. An immunoproteomic approach to characterize the CAR interactome and signalosome. Sci. Signal. 2019, 12, eaap9777. [Google Scholar] [CrossRef]
- Roselli, E.; Frieling, J.S.; Thorner, K.; Ramello, M.C.; Lynch, C.C.; Abate-Daga, D. CAR-T Engineering: Optimizing Signal Transduction and Effector Mechanisms. BioDrugs 2019, 33, 647–659. [Google Scholar] [CrossRef] [PubMed]
- Guedan, S.; Posey, A.D.; Shaw, C.; Wing, A.; Da, T.; Patel, P.R.; Mcgettigan, S.E.; Casado-Medrano, V.; Kawalekar, O.U.; Uribe-Herranz, M.; et al. Enhancing CAR T cell persistence through ICOS and 4-1BB costimulation. JCI Insight 2018, 3. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Beard, R.E.; Zheng, Z.; Lagisetty, K.H.; Burns, W.R.; Tran, E.; Hewitt, S.M.; Abate-Daga, D.; Rosati, S.F.; Fine, H.A.; Ferrone, S.; et al. Multiple chimeric antigen receptors successfully target chondroitin sulfate proteoglycan 4 in several different cancer histologies and cancer stem cells. J. Immunother. Cancer 2014, 2, 25. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhong, X.-S.; Matsushita, M.; Plotkin, J.; Rivière, I.; Sadelain, M. Chimeric Antigen Receptors Combining 4-1BB and CD28 Signaling Domains Augment PI3kinase/AKT/Bcl-XL Activation and CD8+ T Cell–mediated Tumor Eradication. Mol. Ther. 2009, 18, 413–420. [Google Scholar] [CrossRef]
- Till, B.G.; Jensen, M.C.; Wang, J.; Qian, X.; Gopal, A.K.; Maloney, D.G.; Lindgren, C.G.; Lin, Y.; Pagel, J.M.; Budde, L.E.; et al. CD20-specific adoptive immunotherapy for lymphoma using a chimeric antigen receptor with both CD28 and 4-1BB domains: Pilot clinical trial results. Blood 2012, 119, 3940–3950. [Google Scholar] [CrossRef] [Green Version]
- Lesch, S.; Benmebarek, M.-R.; Cadilha, B.L.; Stoiber, S.; Subklewe, M.; Endres, S.; Kobold, S. Determinants of response and resistance to CAR T cell therapy. Semin. Cancer Boil. 2019. [Google Scholar] [CrossRef]
- Guest, R.D.; E Hawkins, R.; Kirillova, N.; Cheadle, E.J.; Arnold, J.; O??neill, A.; Irlam, J.; A Chester, K.; Kemshead, J.T.; Shaw, D.M.; et al. The Role of Extracellular Spacer Regions in the Optimal Design of Chimeric Immune Receptors. J. Immunother. 2005, 28, 203–211. [Google Scholar] [CrossRef]
- Watanabe, N.; Bajgain, P.; Sukumaran, S.; Ansari, S.; Heslop, H.E.; Rooney, C.M.; Brenner, M.K.; Leen, A.M.; Vera, J.F. Fine-tuning the CAR spacer improves T-cell potency. OncoImmunology 2016, 5, e1253656. [Google Scholar] [CrossRef] [Green Version]
- Alabanza, L.; Pegues, M.; Geldres, C.; Shi, V.; Wiltzius, J.J.; Sievers, S.A.; Yang, S.; Kochenderfer, J.N. Function of Novel Anti-CD19 Chimeric Antigen Receptors with Human Variable Regions Is Affected by Hinge and Transmembrane Domains. Mol. Ther. 2017, 25, 2452–2465. [Google Scholar] [CrossRef] [Green Version]
- Feucht, J.; Sun, J.; Eyquem, J.; Ho, Y.-J.; Zhao, Z.; Leibold, J.; Dobrin, A.; Cabriolu, A.; Hamieh, M.; Sadelain, M. Calibration of CAR activation potential directs alternative T cell fates and therapeutic potency. Nat. Med. 2018, 25, 82–88. [Google Scholar] [CrossRef]
- Call, M.E.; Pyrdol, J.; Wiedmann, M.; Wucherpfennig, K.W. The Organizing Principle in the Formation of the T Cell Receptor-CD3 Complex. Cell 2002, 111, 967–979. [Google Scholar] [CrossRef] [Green Version]
- Dong, D.; Zheng, L.; Lin, J.; Zhang, B.; Zhu, Y.; Li, N.; Xie, S.; Wang, Y.; Gao, N.; Huang, Z. Structural basis of assembly of the human T cell receptor–CD3 complex. Nature 2019, 573, 546–552. [Google Scholar] [CrossRef] [PubMed]
- Dushek, O.; Goyette, J.; Van Der Merwe, P.A. Non-catalytic tyrosine-phosphorylated receptors. Immunol. Rev. 2012, 250, 258–276. [Google Scholar] [CrossRef] [PubMed]
- Abram, C.L.; Lowell, C.A. The Expanding Role for ITAM-Based Signaling Pathways in Immune Cells. Sci. STKE 2007, 2007, re2. [Google Scholar] [CrossRef] [PubMed]
- Reddy, M.; Eirikis, E.; Davis, C.; Davis, H.M.; Prabhakar, U. Comparative analysis of lymphocyte activation marker expression and cytokine secretion profile in stimulated human peripheral blood mononuclear cell cultures: An in vitro model to monitor cellular immune function. J. Immunol. Methods 2004, 293, 127–142. [Google Scholar] [CrossRef] [PubMed]
- Bridgeman, J.S.; Hawkins, R.E.; Bagley, S.; Blaylock, M.; Holland, M.; Gilham, D.E. The Optimal Antigen Response of Chimeric Antigen Receptors Harboring the CD3ζ Transmembrane Domain Is Dependent upon Incorporation of the Receptor into the Endogenous TCR/CD3 Complex. J. Immunol. 2010, 184, 6938–6949. [Google Scholar] [CrossRef] [Green Version]
- Stoiber, S.; Cadilha, B.L.; Benmebarek, M.-R.; Lesch, S.; Endres, S.; Kobold, S. Limitations in the Design of Chimeric Antigen Receptors for Cancer Therapy. Cells 2019, 8, 472. [Google Scholar] [CrossRef] [Green Version]
- Woerly, G.; Lacy, P.; Ben Younes, A.; Roger, N.; Loiseau, S.; Moqbel, R.; Capron, M. Human eosinophils express and release IL-13 following CD28-dependent activation. J. Leukoc. Boil. 2002, 72, 769–779. [Google Scholar]
- Venuprasad, K.; Parab, P.; Prasad, D.V.R.; Sharma, S.; Banerjee, P.P.; Deshpande, M.; Mitra, D.K.; Pal, S.; Bhadra, R.; Mitra, D.; et al. Immunobiology of CD28 expression on human neutrophils. I. CD28 regulates neutrophil migration by modulating CXCR-1 expression. Eur. J. Immunol. 2001, 31, 1536–1543. [Google Scholar] [CrossRef]
- Williams, J.A.; Lumsden, J.M.; Yu, X.; Feigenbaum, L.; Zhang, J.; Steinberg, S.M.; Hodes, R.J. Regulation of thymic NKT cell development by the B7-CD28 costimulatory pathway. J. Immunol. 2008, 181, 907–917. [Google Scholar] [CrossRef] [Green Version]
- Michel, F.M.; Attal-Bonnefoy, G.; Mangino, G.; Mise-Omata, S.; Acuto, O. CD28 as a molecular amplifier extending TCR ligation and signaling capabilities. Immunity 2001, 15, 935–945. [Google Scholar] [CrossRef] [Green Version]
- Acuto, O.; Cantrell, D. T Cell Activation and the Cytoskeleton. Annu. Rev. Immunol. 2000, 18, 165–184. [Google Scholar] [CrossRef] [PubMed]
- Porciello, N.; Tuosto, L. CD28 costimulatory signals in T lymphocyte activation: Emerging functions beyond a qualitative and quantitative support to TCR signalling. Cytokine Growth Factor Rev. 2016, 28, 11–19. [Google Scholar] [CrossRef] [PubMed]
- Boomer, J.S.; Green, J.M. An Enigmatic Tail of CD28 Signaling. Cold Spring Harb. Perspect. Boil. 2010, 2, a002436. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Milone, M.C.; Fish, J.D.; Carpenito, C.; Carroll, R.G.; Binder, G.K.; Teachey, D.; Samanta, M.; Lakhal, M.; Gloss, B.; Danet-Desnoyers, G.; et al. Chimeric Receptors Containing CD137 Signal Transduction Domains Mediate Enhanced Survival of T Cells and Increased Antileukemic Efficacy In Vivo. Mol. Ther. 2009, 17, 1453–1464. [Google Scholar] [CrossRef]
- Lawson, D.H.; Lee, S.; Zhao, F.; Tarhini, A.A.; Margolin, K.A.; Ernstoff, M.S.; Atkins, M.B.; Cohen, G.I.; Whiteside, T.L.; Butterfield, L.H.; et al. Randomized, Placebo-Controlled, Phase III Trial of Yeast-Derived Granulocyte-Macrophage Colony-Stimulating Factor (GM-CSF) Versus Peptide Vaccination Versus GM-CSF Plus Peptide Vaccination Versus Placebo in Patients With No Evidence of Disease After Complete Surgical Resection of Locally Advanced and/or Stage IV Melanoma: A Trial of the Eastern Cooperative Oncology Group–American College of Radiology Imaging Network Cancer Research Group (E4697). J. Clin. Oncol. 2015, 33, 4066–4076. [Google Scholar] [CrossRef]
- Hombach, A.; Holzinger, A.; Abken, H. The Weal and Woe of Costimulation in the Adoptive Therapy of Cancer with Chimeric Antigen Receptor (CAR)-Redirected T Cells. Curr. Mol. Med. 2013, 13, 1079–1088. [Google Scholar] [CrossRef]
- Sadelain, M.; Brentjens, R.; Rivière, I. The basic principles of chimeric antigen receptor design. Cancer Discov. 2013, 3, 388–398. [Google Scholar] [CrossRef] [Green Version]
- Redeker, A.; Arens, R. Improving Adoptive T Cell Therapy: The Particular Role of T Cell Costimulation, Cytokines, and Post-Transfer Vaccination. Front. Immunol. 2016, 7, 189. [Google Scholar] [CrossRef] [Green Version]
- Faitschuk, E.; Hombach, A.; Frenzel, L.P.; Wendtner, C.-M.; Abken, H. Chimeric antigen receptor T cells targeting Fc μ receptor selectively eliminate CLL cells while sparing healthy B cells. Blood 2016, 128, 1711–1722. [Google Scholar] [CrossRef] [Green Version]
- Weinkove, R.; George, P.; Dasyam, N.; McLellan, A.D. Selecting costimulatory domains for chimeric antigen receptors: Functional and clinical considerations. Clin. Transl. Immunol. 2019, 8, e1049. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kintz, H.; Nylen, E.; Barber, A. Inclusion of Dap10 or 4-1BB costimulation domains in the chPD1 receptor enhances anti-tumor efficacy of T cells in murine models of lymphoma and melanoma. Cell. Immunol. 2020, 351, 104069. [Google Scholar] [CrossRef] [PubMed]
- Zhang, T.; Wu, M.-R.; Sentman, C.L. An NKp30-based chimeric antigen receptor promotes T cell effector functions and antitumor efficacy in vivo. J. Immunol. 2012, 189, 2290–2299. [Google Scholar] [CrossRef] [PubMed]
- Krug, C.; Birkholz, K.; Paulus, A.; Schwenkert, M.; Schmidt, P.; Hoffmann, N.; Hombach, A.; Fey, G.; Abken, H.; Schuler, G.; et al. Stability and activity of MCSP-specific chimeric antigen receptors (CARs) depend on the scFv antigen-binding domain and the protein backbone. Cancer Immunol. Immunother. 2015, 64, 1623–1635. [Google Scholar] [CrossRef] [PubMed]
- Frigault, M.J.; Lee, J.; Basil, M.C.; Carpenito, C.; Motohashi, S.; Scholler, J.; Kawalekar, O.U.; Guedan, S.; Mcgettigan, S.E.; Posey, A.D.; et al. Identification of chimeric antigen receptors that mediate constitutive or inducible proliferation of T cells. Cancer Immunol. Res. 2015, 3, 356–367. [Google Scholar] [CrossRef] [Green Version]
- Lee, D.W.; Kochenderfer, J.N.; Stetler-Stevenson, M.; Cui, Y.K.; Delbrook, C.; A Feldman, S.; Fry, T.J.; Orentas, R.; Sabatino, M.; Shah, N.N.; et al. T cells expressing CD19 chimeric antigen receptors for acute lymphoblastic leukaemia in children and young adults: A phase 1 dose-escalation trial. Lancet 2015, 385, 517–528. [Google Scholar] [CrossRef]
- Park, J.H.; Riviere, I.; Gonen, M.; Wang, X.; Sénéchal, B.; Curran, K.J.; Sauter, C.; Wang, Y.; Santomasso, B.; Mead, E.; et al. Long-Term Follow-up of CD19 CAR Therapy in Acute Lymphoblastic Leukemia. N. Engl. J. Med. 2018, 378, 449–459. [Google Scholar] [CrossRef]
- Lang, S.; Vujanovic, N.L.; Wollenberg, B.; Whiteside, T.L. Absence of B7.1-CD28/CTLA-4-mediated co-stimulation in human NK cells. Eur. J. Immunol. 1998, 28, 780–786. [Google Scholar] [CrossRef]
- Goodier, M.R.; Londei, M. CD28 is not directly involved in the response of human CD3-CD56+ natural killer cells to lipopolysaccharide: A role for T cells. Immunology 2004, 111, 384–390. [Google Scholar] [CrossRef]
- Verma, I.M.; Van Antwerp, D.; Stevenson, J.K.; Schwarz, E.M.; Miyamoto, S. Rel/NF-kappa B/I kappa B family: Intimate tales of association and dissociation. Genes Dev. 1995, 9, 2723–2735. [Google Scholar] [CrossRef] [Green Version]
- Melero, I.; Johnston, J.V.; Shufford, W.W.; Mittler, R.S.; Chen, L. NK1.1 Cells Express 4-1BB (CDw137) Costimulatory Molecule and Are Required for Tumor Immunity Elicited by Anti-4-1BB Monoclonal Antibodies. Cell. Immunol. 1998, 190, 167–172. [Google Scholar] [CrossRef] [PubMed]
- Drenkard, D.; Becke, F.M.; Langstein, J.; Spruss, T.; Kunz-Schughart, L.A.; Tan, T.E.; Lim, Y.C.; Schwarz, H. CD137 is expressed on blood vessel walls at sites of inflammation and enhances monocyte migratory activity. FASEB J. 2006, 21, 456–463. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Von Kempis, J.; Schwarz, H.; Lotz, M. Differentiation-dependent and stimulus-specific expression of ILA, the human 4-1BB-homologue, in cells of mesenchymal origin. Osteoarthr. Cartil. 1997, 5, 394–406. [Google Scholar] [CrossRef] [Green Version]
- Cannons, J.L.; Choi, Y.; Watts, T.H. Role of TNF receptor-associated factor 2 and p38 mitogen-activated protein kinase activation during 4-1BB-dependent immune response. J. Immunol. 2000, 165, 6193–6204. [Google Scholar] [CrossRef] [Green Version]
- Lee, H.-W.; Nam, K.-O.; Park, S.-J.; Kwon, B.S. 4-1BB enhances CD8+ T cell expansion by regulating cell cycle progression through changes in expression of cyclins D and E and cyclin-dependent kinase inhibitor p27kip1. Eur. J. Immunol. 2003, 33, 2133–2141. [Google Scholar] [CrossRef]
- Imai, C.; Iwamoto, S.; Campana, D. Genetic modification of primary natural killer cells overcomes inhibitory signals and induces specific killing of leukemic cells. Blood 2005, 106, 376–383. [Google Scholar] [CrossRef] [Green Version]
- Fraietta, J.A.; Lacey, S.F.; Orlando, E.J.; Pruteanu-Malinici, I.; Gohil, M.; Lundh, S.; Boesteanu, A.C.; Wang, Y.; O’Connor, R.S.; Hwang, W.-T.; et al. Determinants of response and resistance to CD19 chimeric antigen receptor (CAR) T cell therapy of chronic lymphocytic leukemia. Nat. Med. 2018, 24, 563–571. [Google Scholar] [CrossRef]
- Maude, S.L.; Laetsch, T.W.; Buechner, J.; Rives, S.; Boyer, M.; Bittencourt, H.; Bader, P.; Verneris, M.R.; Stefanski, H.E.; Myers, G.D.; et al. Tisagenlecleucel in Children and Young Adults with B-Cell Lymphoblastic Leukemia. N. Engl. J. Med. 2018, 378, 439–448. [Google Scholar] [CrossRef]
- Houchins, J.P.; Yabe, T.; McSherry, C.; Bach, F.H. DNA sequence analysis of NKG2, a family of related cDNA clones encoding type II integral membrane proteins on human natural killer cells. J. Exp. Med. 1991, 173, 1017–1020. [Google Scholar] [CrossRef] [Green Version]
- Bauer, S. Activation of NK Cells and T Cells by NKG2D, a Receptor for Stress-Inducible MICA. Science 1999, 285, 727–729. [Google Scholar] [CrossRef]
- Raulet, D.H. Roles of the NKG2D immunoreceptor and its ligands. Nat. Rev. Immunol. 2003, 3, 781–790. [Google Scholar] [CrossRef] [PubMed]
- González, S.; López-Soto, A.; Suárez-Álvarez, B.; López-Vázquez, A.; López-Soto, A.; Gonzalez, S. NKG2D ligands: Key targets of the immune response. Trends Immunol. 2008, 29, 397–403. [Google Scholar] [CrossRef] [PubMed]
- Zafirova, B.; Wensveen, F.M.; Gulin, M.; Polić, B. Regulation of immune cell function and differentiation by the NKG2D receptor. Cell. Mol. Life Sci. 2011, 68, 3519–3529. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Serrano, A.E.; Menares-Castillo, E.; Garrido-Tapia, M.; Ribeiro, C.H.; Hernández, C.J.; Mendoza-Naranjo, A.; Gatica-Andrades, M.; Valenzuela-Diaz, R.; Zuñiga, R.; López, M.N.; et al. Interleukin 10 decreases MICA expression on melanoma cell surface. Immunol. Cell Boil. 2010, 89, 447–457. [Google Scholar] [CrossRef] [PubMed]
- Jamieson, A.M.; Diefenbach, A.; McMahon, C.W.; Xiong, N.; Carlyle, J.R.; Raulet, D.H. The Role of the NKG2D Immunoreceptor in Immune Cell Activation and Natural Killing. Immunity 2002, 17, 19–29. [Google Scholar] [CrossRef] [Green Version]
- Diefenbach, A.; Tomasello, E.; Lucas, M.; Jamieson, A.M.; Hsia, J.K.; Vivier, E.; Raulet, D.H. Selective associations with signaling proteins determine stimulatory versus costimulatory activity of NKG2D. Nat. Immunol. 2002, 3, 1142–1149. [Google Scholar] [CrossRef]
- Zhang, T.; Lemoi, B.A.; Sentman, C.L. Chimeric NK-receptor–bearing T cells mediate antitumor immunotherapy. Blood 2005, 106, 1544–1551. [Google Scholar] [CrossRef]
- Demoulin, B.; Cook, W.J.; Murad, J.; Graber, D.J.; Sentman, M.-L.; Lonez, C.; E Gilham, D.; Sentman, C.L.; Agaugue, S. Exploiting natural killer group 2D receptors for CAR T-cell therapy. Futur. Oncol. 2017, 13, 1593–1605. [Google Scholar] [CrossRef] [Green Version]
- Zhang, T.; Barber, A.; Sentman, C.L. Generation of Antitumor Responses by Genetic Modification of Primary Human T Cells with a Chimeric NKG2D Receptor. Cancer Res. 2006, 66, 5927–5933. [Google Scholar] [CrossRef] [Green Version]
- Chang, Y.-H.; Connolly, J.; Shimasaki, N.; Mimura, K.; Kono, K.; Campana, D. A Chimeric Receptor with NKG2D Specificity Enhances Natural Killer Cell Activation and Killing of Tumor Cells. Cancer Res. 2013, 73, 1777–1786. [Google Scholar] [CrossRef] [Green Version]
- Wood, L.D.; Parsons, D.W.; Jones, S.; Lin, J.; Sjöblom, T.; Leary, R.J.; Shen, N.; Boca, S.M.; Barber, T.; Ptak, J.; et al. The Genomic Landscapes of Human Breast and Colorectal Cancers. Science 2007, 318, 1108–1113. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barber, A.; Sentman, C.L. NKG2D receptor regulates human effector T-cell cytokine production. Blood 2011, 117, 6571–6581. [Google Scholar] [CrossRef] [Green Version]
- Zhang, T.; Sentman, C.L. Mouse tumor vasculature expresses NKG2D ligands and can be targeted by chimeric NKG2D-modified T cells. J. Immunol. 2013, 190, 2455–2463. [Google Scholar] [CrossRef] [Green Version]
- Lehner, M.; Gotz, G.; Proff, J.; Schaft, N.; Dorrie, J.; Full, F.; Ensser, A.; Muller, Y.A.; Cerwenka, A.; Abken, H.; et al. Redirecting T cells to Ewing’s sarcoma family of tumors by a chimeric NKG2D receptor expressed by lentiviral transduction or mRNA transfection. PLoS ONE 2012, 7, e31210. [Google Scholar] [CrossRef] [PubMed]
- Maasho, K.; Opoku-Anane, J.; Marusina, A.I.; Coligan, J.E.; Borrego, F. Cutting Edge: NKG2D Is a Costimulatory Receptor for Human Naive CD8+ T Cells. J. Immunol. 2005, 174, 4480–4484. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Markiewicz, M.A.; Carayannopoulos, L.N.; Naidenko, O.V.; Matsui, K.; Burack, W.R.; Wise, E.L.; Fremont, D.H.; Allen, P.M.; Yokoyama, W.; Colonna, M.; et al. Costimulation through NKG2D enhances murine CD8+ CTL function: Similarities and differences between NKG2D and CD28 costimulation. J. Immunol. 2005, 175, 2825–2833. [Google Scholar] [CrossRef] [Green Version]
- Upshaw, J.L.; Leibson, P.J. NKG2D-mediated activation of cytotoxic lymphocytes: Unique signaling pathways and distinct functional outcomes. Semin. Immunol. 2006, 18, 167–175. [Google Scholar] [CrossRef]
- Lanier, L.L. DAP10- and DAP12-associated receptors in innate immunity. Immunol. Rev. 2009, 227, 150–160. [Google Scholar] [CrossRef] [Green Version]
- Rajasekaran, K.; Xiong, V.; Fong, L.; Gorski, J.; Malarkannan, S. Functional Dichotomy between NKG2D and CD28-Mediated Co-Stimulation in Human CD8+ T Cells. PLoS ONE 2010, 5, e12635. [Google Scholar] [CrossRef] [Green Version]
- Whitman, E.; Barber, A. NKG2D receptor activation of NF-κB enhances inflammatory cytokine production in murine effector CD8+ T cells. Mol. Immunol. 2015, 63, 268–278. [Google Scholar] [CrossRef]
- McQueen, B.; Trace, K.; Whitman, E.; Bedsworth, T.; Barber, A. Natural killer group 2D and CD28 receptors differentially activate mammalian/mechanistic target of rapamycin to alter murine effector CD8+T-cell differentiation. Immunology 2016, 147, 305–320. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lynch, A.; Hawk, W.; Nylen, E.; Ober, S.; Autin, P.; Barber, A. Adoptive transfer of murine T cells expressing a chimeric-PD1-Dap10 receptor as an immunotherapy for lymphoma. Immunology 2017, 152, 472–483. [Google Scholar] [CrossRef] [PubMed]
- Parriott, G.; Deal, K.; Crean, S.; Richardson, E.; Nylen, E.; Barber, A. T cells expressing a chimeric-PD1-Dap10-CD3zeta receptor reduce tumor burden in multiple murine syngeneic models of solid cancer. Immunology 2020. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Qiao, G.; Wang, X.; Song, Y.; Zhou, X.; Jiang, N.; Zhou, L.; Huang, H.; Zhao, J.; Morse, M.A.; et al. Combination of DC/CIK adoptive T cell immunotherapy with chemotherapy in advanced non-small-cell lung cancer (NSCLC) patients: A prospective patients’ preference-based study (PPPS). Clin. Transl. Oncol. 2018, 21, 721–728. [Google Scholar] [CrossRef] [PubMed]
- Lv, J.; Zhao, R.; Wu, D.; Zheng, D.; Wu, Z.; Shi, J.; Wei, X.; Wu, Q.; Long, Y.; Lin, S.; et al. Mesothelin is a target of chimeric antigen receptor T cells for treating gastric cancer. J. Hematol. Oncol. 2019, 12, 18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Duong, C.P.; Westwood, J.A.; Yong, C.S.M.; Murphy, A.; Devaud, C.; John, L.B.; Darcy, P.K.; Kershaw, M. Engineering T Cell Function Using Chimeric Antigen Receptors Identified Using a DNA Library Approach. PLoS ONE 2013, 8, e63037. [Google Scholar] [CrossRef] [Green Version]
- Fisher, J.; Abramowski, P.; Don, N.D.W.; Flutter, B.; Capsomidis, A.; Cheung, G.W.-K.; Gustafsson, K.; Anderson, J. Avoidance of On-Target Off-Tumor Activation Using a Co-stimulation-Only Chimeric Antigen Receptor. Mol. Ther. 2017, 25, 1234–1247. [Google Scholar] [CrossRef] [Green Version]
- Ribas, A.; Lawrence, D.; Atkinson, V.; Agarwal, S.; Miller, W.H.; Carlino, M.S.; Fisher, R.; Long, G.V.; Hodi, F.S.; Tsoi, J.; et al. Combined BRAF and MEK inhibition with PD-1 blockade immunotherapy in BRAF-mutant melanoma. Nat. Med. 2019, 25, 936–940. [Google Scholar] [CrossRef]
- Marin, V.; Kakuda, H.; Dander, E.; Imai, C.; Campana, D.; Biondi, A.; D’Amico, G. Enhancement of the anti-leukemic activity of cytokine induced killer cells with an anti-CD19 chimeric receptor delivering a 4-1BB-ζ activating signal. Exp. Hematol. 2007, 35, 1388–1397. [Google Scholar] [CrossRef]
- Wu, J.; Song, Y.; Bakker, A.B.H.; Bauer, S.; Spies, T.; Lanier, L.L.; Phillips, J.H. An Activating Immunoreceptor Complex Formed by NKG2D and DAP10. Science 1999, 285, 730–732. [Google Scholar] [CrossRef]
- Billadeau, D.D.; Upshaw, J.L.; A Schoon, R.; Dick, C.J.; Leibson, P.J. NKG2D-DAP10 triggers human NK cell–mediated killing via a Syk-independent regulatory pathway. Nat. Immunol. 2003, 4, 557–564. [Google Scholar] [CrossRef]
- Biagi, E.; Marin, V.; Attianese, G.M.P.G.; Dander, E.; D’Amico, G.; Biondi, A. Chimeric T-cell receptors: New challenges for targeted immunotherapy in hematologic malignancies. Haematology 2007, 92, 381–388. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fridman, W.H. Fc receptors and immunoglobulin binding factors 1. FASEB J. 1991, 5, 2684–2690. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Raghavan, M.; Bjorkman, P.J. Fc Receptors and Their Interactions with Immunoglobulins. Annu. Rev. Cell Dev. Boil. 1996, 12, 181–220. [Google Scholar] [CrossRef] [Green Version]
- Nimmerjahn, F.; Ravetch, J.V. Fcγ receptors as regulators of immune responses. Nat. Rev. Immunol. 2008, 8, 34–47. [Google Scholar] [CrossRef] [PubMed]
- Takai, T.; Ono, M.; Hikida, M.; Ohmori, H.; Ravetch, J.V. Augmented humoral and anaphylactic responses in FcγRII-deficient mice. Nature 1996, 379, 346–349. [Google Scholar] [CrossRef] [PubMed]
- Vivier, E.; Rochet, N.; Ackerly, M.; Petrini, J.; Levine, H.; Daley, J.; Anderson, P. Signaling function of reconstituted CD16: ζ: γ receptor complex isoforms. Int. Immunol. 1992, 4, 1313–1323. [Google Scholar] [CrossRef]
- Schumann, G.; Dasgupta, J.D. Specificity of signal transduction through CD16, TCR-CD3 and BCR receptor chains containing the tyrosine-associated activation motif. Int. Immunol. 1994, 6, 1383–1392. [Google Scholar] [CrossRef]
- Indik, Z.; Park, J.; Hunter, S.; Schreiber, A. The molecular dissection of Fc gamma receptor mediated phagocytosis. Blood 1995, 86, 4389–4399. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.; Boesen, C.C.; Radaev, S.; Brooks, A.G.; Fridman, W.H.; Sautès-Fridman, C.; Sun, P.D. Crystal Structure of the Extracellular Domain of a Human FcγRIII. Immunity 2000, 13, 387–395. [Google Scholar] [CrossRef] [Green Version]
- Caratelli, S.; Sconocchia, G.; Arriga, R.; Coppola, A.; Lanzilli, G.; Lauro, D.; Venditti, A.; Del Principe, M.I.; Lococo, F.; Maurillo, L.; et al. FCγ Chimeric Receptor-Engineered T Cells: Methodology, Advantages, Limitations, and Clinical Relevance. Front. Immunol. 2017, 8, 457. [Google Scholar] [CrossRef] [PubMed]
- Sacchetti, B.; Botticelli, A.; Pierelli, L.; Nuti, M.; Alimandi, M. CAR-T with License to Kill Solid Tumors in Search of a Winning Strategy. Int. J. Mol. Sci. 2019, 20, 1903. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ochi, F.; Fujiwara, H.; Tanimoto, K.; Asai, H.; Miyazaki, Y.; Okamoto, S.; Mineno, J.; Kuzushima, K.; Shiku, H.; Barrett, J.; et al. Gene-Modified Human / -T Cells Expressing a Chimeric CD16-CD3 Receptor as Adoptively Transferable Effector Cells for Anticancer Monoclonal Antibody Therapy. Cancer Immunol. Res. 2014, 2, 249–262. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kudo, K.; Imai, C.; Lorenzini, P.; Kamiya, T.; Kono, K.; Davidoff, A.M.; Chng, W.J.; Campana, D. T Lymphocytes Expressing a CD16 Signaling Receptor Exert Antibody-Dependent Cancer Cell Killing. Cancer Res. 2013, 74, 93–103. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Clémenceau, B.; Valsesia-Wittmann, S.; Jallas, A.-C.; Vivien, R.; Rousseau, R.; Marabelle, A.; Caux, C.; Vié, H. In Vitro and In Vivo Comparison of Lymphocytes Transduced with a Human CD16 or with a Chimeric Antigen Receptor Reveals Potential Off-Target Interactions due to the IgG2 CH2-CH3 CAR-Spacer. J. Immunol. Res. 2015, 2015, 1–13. [Google Scholar] [CrossRef] [Green Version]
- D’Aloia, M.M.; Caratelli, S.; Palumbo, C.; Battella, S.; Arriga, R.; Lauro, D.; Palmieri, G.; Sconocchia, G.; Alimandi, M. T lymphocytes engineered to express a CD16-chimeric antigen receptor redirect T-cell immune responses against immunoglobulin G–opsonized target cells. Cytotherapy 2016, 18, 278–290. [Google Scholar] [CrossRef]
- Tanaka, H.; Fujiwara, H.; Ochi, F.; Tanimoto, K.; Casey, N.; Okamoto, S.; Mineno, J.; Kuzushima, K.; Shiku, H.; Sugiyama, T.; et al. Development of Engineered T Cells Expressing a Chimeric CD16-CD3 Receptor to Improve the Clinical Efficacy of Mogamulizumab Therapy Against Adult T-Cell Leukemia. Clin. Cancer Res. 2016, 22, 4405–4416. [Google Scholar] [CrossRef] [Green Version]
- Rataj, F.; Jacobi, S.J.; Stoiber, S.; Asang, F.; Ogonek, J.; Tokarew, N.; Cadilha, B.L.; Van Puijenbroek, E.; Heise, C.; Duewell, P.; et al. High-affinity CD16-polymorphism and Fc-engineered antibodies enable activity of CD16-chimeric antigen receptor-modified T cells for cancer therapy. Br. J. Cancer 2018, 120, 79–87. [Google Scholar] [CrossRef] [Green Version]
- Arriga, R.; Caratelli, S.; Lanzilli, G.; Ottaviani, A.; Cenciarelli, C.; Sconocchia, T.; Spagnoli, G.C.; Iezzi, G.; Roselli, M.; Lauro, D.; et al. CD16-158-valine chimeric receptor T cells overcome the resistance of KRAS-mutated colorectal carcinoma cells to cetuximab. Int. J. Cancer 2019, 146, 2531–2538. [Google Scholar] [CrossRef]
- Caratelli, S.; Arriga, R.; Sconocchia, T.; Ottaviani, A.; Lanzilli, G.; Pastore, D.; Cenciarelli, C.; Venditti, A.; Del Principe, M.I.; Lauro, D.; et al. In vitro elimination of epidermal growth factor receptor-overexpressing cancer cells by CD32A-chimeric receptor T cells in combination with cetuximab or panitumumab. Int. J. Cancer 2019, 146, 236–247. [Google Scholar] [CrossRef]
- Lee, J.-Y.; Forman, J.P.; McNerney, M.E.; Stepp, S.; Kuppireddi, S.; Guzior, D.; Latchman, Y.E.; Sayegh, M.H.; Yagita, H.; Park, C.-K.; et al. Requirement of homotypic NK-cell interactions through 2B4(CD244)/CD48 in the generation of NK effector functions. Blood 2005, 107, 3181–3188. [Google Scholar] [CrossRef] [Green Version]
- Agresta, L.; Hoebe, K.H.N.; Janssen, E.M. The Emerging Role of CD244 Signaling in Immune Cells of the Tumor Microenvironment. Front. Immunol. 2018, 9, 2809. [Google Scholar] [CrossRef] [PubMed]
- Valiante, N.M.; Trinchieri, G. Identification of a novel signal transduction surface molecule on human cytotoxic lymphocytes. J. Exp. Med. 1993, 178, 1397–1406. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- A Garni-Wagner, B.; Purohit, A.; A Mathew, P.; Bennett, M.; Kumar, V. A novel function-associated molecule related to non-MHC-restricted cytotoxicity mediated by activated natural killer cells and T cells. J. Immunol. 1993, 151, 60–70. [Google Scholar] [PubMed]
- Altvater, B.; Landmeier, S.; Pscherer, S.; Temme, J.; Schweer, K.; Kailayangiri, S.; Campana, D.; Juergens, H.; Pule, M.; Rossig, C. 2B4 (CD244) signaling by recombinant antigen-specific chimeric receptors costimulates natural killer cell activation to leukemia and neuroblastoma cells. Clin. Cancer Res. 2009, 15, 4857–4866. [Google Scholar] [CrossRef] [Green Version]
- Altvater, B.; Landmeier, S.; Pscherer, S.; Temme, J.; Juergens, H.; Pule, M.; Rossig, C. 2B4 (CD244) signaling via chimeric receptors costimulates tumor-antigen specific proliferation and in vitro expansion of human T cells. Cancer Immunol. Immunother. 2009, 58, 1991–2001. [Google Scholar] [CrossRef]
- Chen, L.; Liu, Y.; Zheng, Y.; Xu, J.; Zhang, Y.; Liu, W.; Li, Z.; Huang, G.; Li, W. Furanodienone overcomes temozolomide resistance in glioblastoma through the downregulation of CSPG4-Akt-ERK signalling by inhibiting EGR1-dependent transcription. Phytotherapy Res. 2019, 33, 1736–1747. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
| Cell Type | Notes | Treatment | Cancer Types | Locations | NCT Number 1 |
|---|---|---|---|---|---|
| γ/δ T cells | Not yet recruiting | anti-CD19-CAR γ/δ T cells | Different B-cell lymphomas | Beijing, China | NCT02656147 |
| Recruiting; observational only | determining number and viability of patients’ γ/δ T cells in preparation for a CAR trial | Acute myeloid leukemia. | Sutton, United Kingdom | NCT03885076 | |
| Not yet recruiting | NKG2D-CAR γ/δ T cells | Different solid tumors | Johor Bahru, Malaysia | NCT04107142 | |
| NKT cells | Withdrawn, replaced by NCT03294954 | iC9-anti-GD2-CD28/OX40/CD3ζ CAR NKT (GINAKIT) cells | Neuroblastoma | Houston, Texas, USA | NCT02439788 |
| Recruiting; new CAR construct | anti-GD2-CD28/CD3ζ-IL15 CAR NKT (GINAKIT2) cells | Neuroblastoma | Houston, Texas, USA | NCT03294954 | |
| Not yet recruiting | anti-CD19-CD28/CD3ζ-IL15 CAR NKT (ANCHOR) cells | Different B-cell lymphomas | Houston, Texas, USA | NCT03774654 | |
| Combining CAR-T cells and DC vaccination | Recruiting | CAR-T cells, engineered immune effector (EIE) CTLs, DCvac(no details available) | Neurofibromatosis | Shenzhen, Guangdong, China | NCT04085159 |
| Recruiting | Different CAR-T cells, Eps8 or WT1 DCvac | Relapsed/refractory Leukemia, myelodysplastic syndrome | Guangzhou, Guangdong, China | NCT03291444 |
| Notes | Treatment | Cancer Types | Locations | NCT Number 1 |
|---|---|---|---|---|
| Completed | anti-CD19-4-1BB/CD3ζ CAR-NK cells | Acute lymphoblastic leukemia | Memphis, Tennessee, USA | NCT00995137 |
| Suspended | anti-CD19-CD3ζ/4-1BB CAR-NK cells | B-cell acute lymphoblastic leukemia | Singapore | NCT01974479 |
| Unknown status | anti-CD7-CD3ζ/CD28/4-1BB CAR NK-92 cells | Acute myeloid leukemia and 8 more hematologic cancers | Suzhou, Jiangsu, China | NCT02742727 |
| Unknown status | anti-MUC1 CAR-NK cells | Hepatocellular carcinoma and 6 more solid cancers | Suzhou, Jiangsu, China | NCT02839954 |
| Unknown status | anti-CD19-TCRζ/CD28/4-1BB CAR-NK cells | Acute lymphocytic leukemia and 5 more hematologic cancers | Suzhou, Jiangsu, China | NCT02892695 |
| Unknown status | anti-CD33-CD3ζ/CD28/4-1BB CAR-NK cells | Acute myelogenous leukemia and 4 more hematologic cancers | Suzhou, Jiangsu, China | NCT02944162 |
| Recruiting; results published | anti-CD19-CD28/CD3ζ/iCasp9/ IL-15 CAR-NK cells | Different B-cell lymphomas | Houston, Texas, USA | NCT03056339 |
| Recruiting | NKG2D-CAR-NK cells | Different solid tumors | Guangzhou, Guangdong, China | NCT03415100 |
| Not yet recruiting | anti-PSMA CAR-NK cells | Castration-resistant prostate cancer | Beijing, China | NCT03692663 |
| Not yet recruiting | anti-CD19/CD22 CAR-NK cells | Refractory B-cell lymphoma | Beijing, China | NCT03824964 |
| Not yet recruiting | anti-CD22 CAR-NK Cells | Refractory B-cell lymphoma | Beijing, China | NCT03692767 |
| Not yet recruiting | anti-meso CAR-NK Cells | Epithelial ovarian cancer | Beijing, China | NCT03692637 |
| Not yet recruiting | anti-CD19 CAR-NK Cells | Refractory B-cell lymphoma | Beijing, China | NCT03690310 |
| Recruiting | anti-BCMA CAR-NK-92 cells | Multiple myeloma | Wuxi, Jiangsu, China | NCT03940833 |
| Recruiting | anti-ROBO1 CAR-NK cells | Pancreatic cancer | Shanghai, China | NCT03941457 |
| Recruiting | anti-ROBO1 CAR-NK cells | Different solid tumors | Suzhou, Jiangsu, China | NCT03940820 |
| Withdrawn; lack of funding | anti-CD19-CD28/CD3ζ/iCasp9/IL-15 CAR-NK cells | Different B-cell lymphomas | Houston, Texas, USA | NCT03579927 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sievers, N.M.; Dörrie, J.; Schaft, N. CARs: Beyond T Cells and T Cell-Derived Signaling Domains. Int. J. Mol. Sci. 2020, 21, 3525. https://doi.org/10.3390/ijms21103525
Sievers NM, Dörrie J, Schaft N. CARs: Beyond T Cells and T Cell-Derived Signaling Domains. International Journal of Molecular Sciences. 2020; 21(10):3525. https://doi.org/10.3390/ijms21103525
Chicago/Turabian StyleSievers, Nico M., Jan Dörrie, and Niels Schaft. 2020. "CARs: Beyond T Cells and T Cell-Derived Signaling Domains" International Journal of Molecular Sciences 21, no. 10: 3525. https://doi.org/10.3390/ijms21103525
APA StyleSievers, N. M., Dörrie, J., & Schaft, N. (2020). CARs: Beyond T Cells and T Cell-Derived Signaling Domains. International Journal of Molecular Sciences, 21(10), 3525. https://doi.org/10.3390/ijms21103525