Cardiac Protective Role of Heat Shock Protein 27 in the Stress Induced by Drugs of Abuse

 ,
, {kind=link}
{kind=link}
Abstract
:1. Introduction
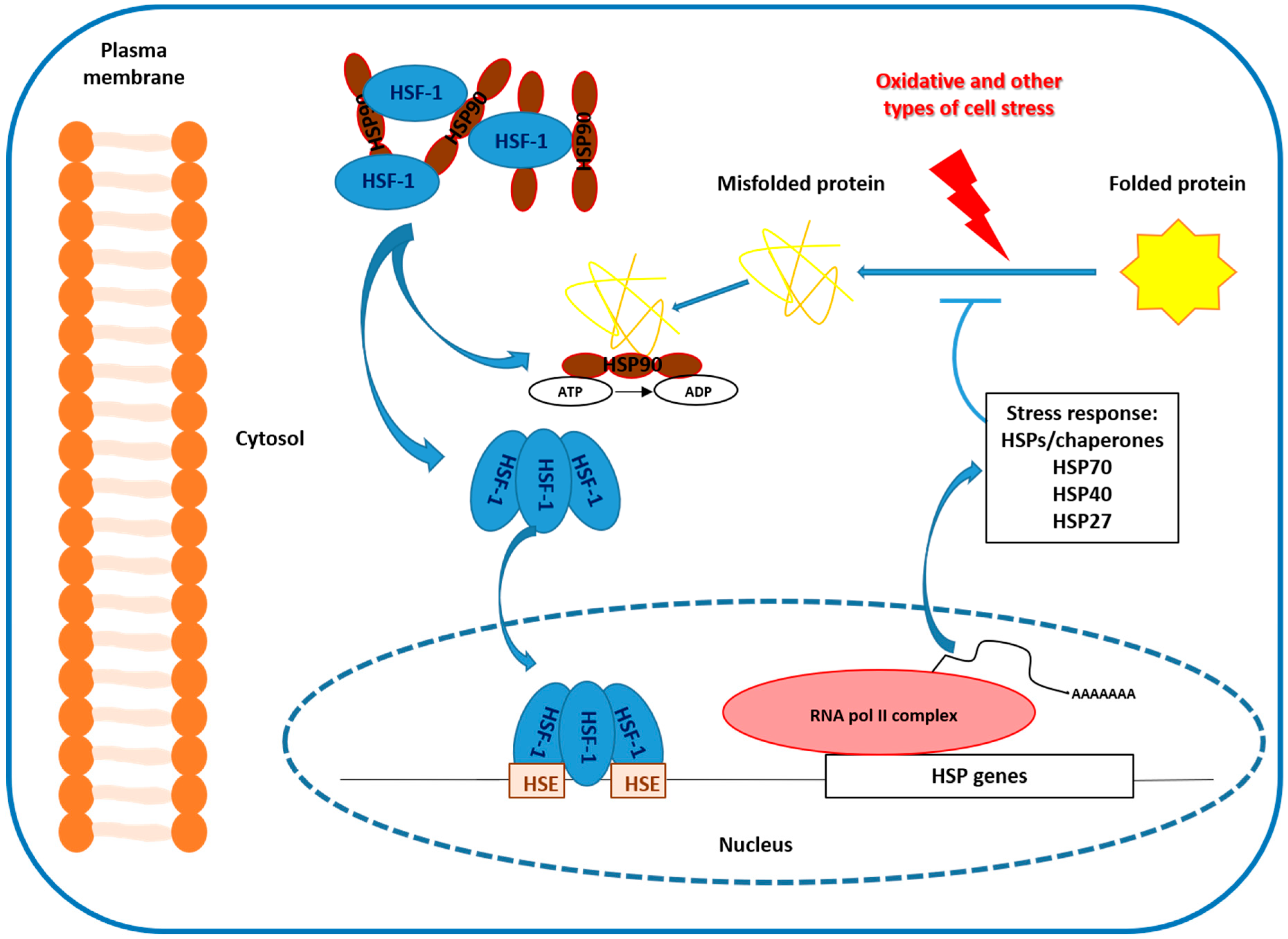
2. Heat Shock Protein 27 (HSP-27) and Stress
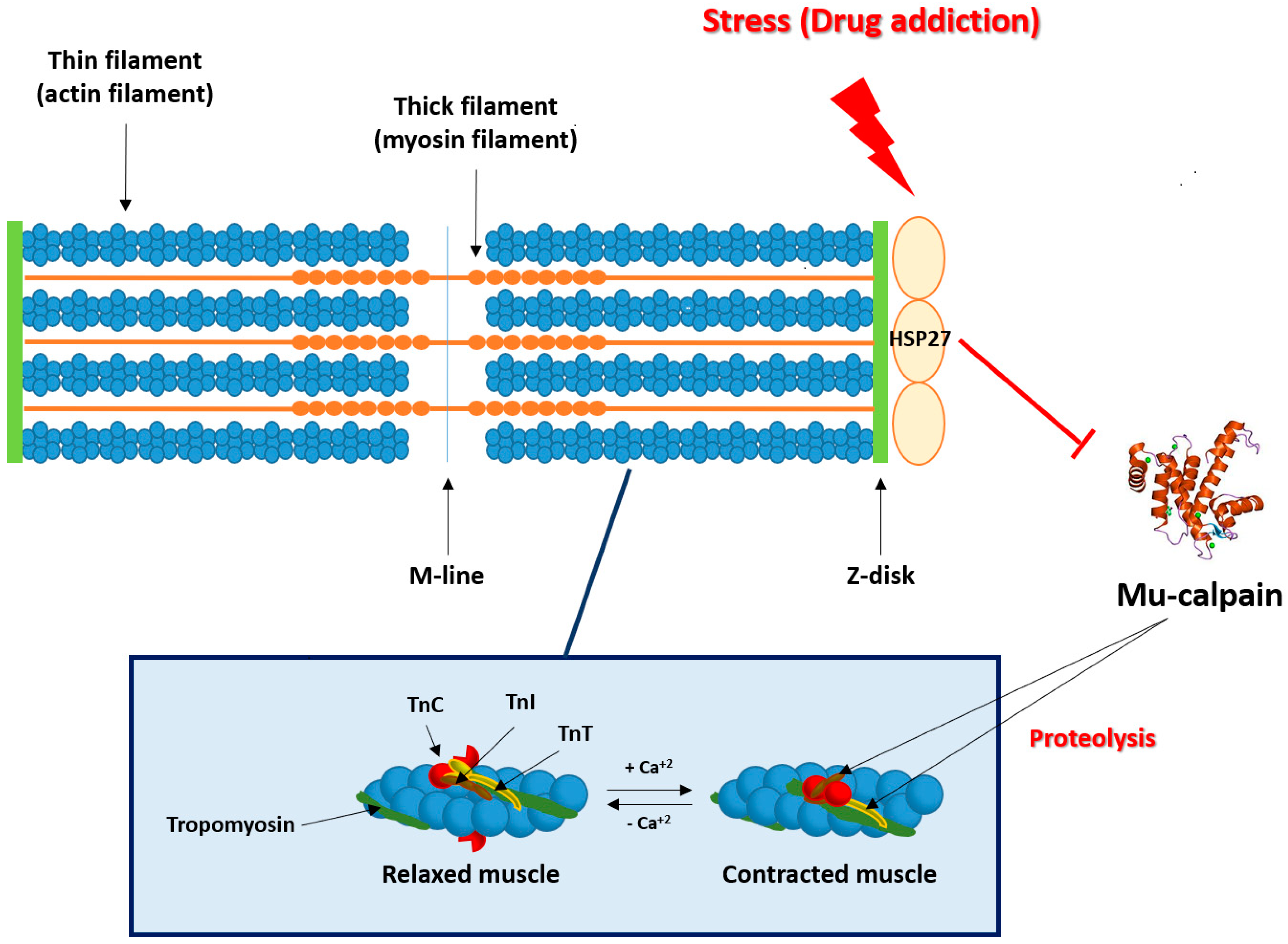
3. Stress/Addiction and Heart
4. Morphine Addiction and HSP-27
5. Conclusions and Future Perspective
Funding
Conflicts of Interest
Abbreviations
| ACTH | adrenocorticotrophin hormone |
| CNS | central nervous system |
| COMT | catechol-O-methyl transferase |
| CRF | corticotrophin releasing factor |
| cTnI | cardiac troponin I |
| cTnT | cardiac troponin T |
| ERK | extracellular signal-regulated kinase |
| HPA | hypothalamic-pituitary adrenal |
| HSP | heat shock proteins |
| PKC | protein kinase C |
| ROS | reactive oxygen species |
References
- Latchman, D.S. Protective effect of heat shock proteins. Cardiovascular 2004, 51, 637–646. [Google Scholar] [CrossRef] [Green Version]
- Rogalla, T.; Ehrnsperger, M.; Preville, X.; Kotlyarov, A.; Lutsch, G.; Ducasse, C.; Paul, C.; Wieske, M.; Arrigo, A.-P.; Buchner, J.; et al. Regulation of HSP-27 oligomerization, chaperone function, and protective activity against oxidative stress/tumor necrosis factor alpha by phosphorylation. J. Biol. Chem. 1999, 274, 18947–18956. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lindquist, S.; Craig, E.A. The heat-shock proteins. Annu. Rev. Genet. 1988, 22, 631–677. [Google Scholar] [CrossRef] [PubMed]
- Ahn, S.G.; Yoon, J.H.; Vacratsis, P. Heat-shock cognate 70 is required for the activation of heat-shock factor 1 in mammalian cells. Biochem. J. 1991, 392, 145–152. [Google Scholar] [CrossRef] [Green Version]
- Lis, J.; Wu, C. Protein traffic on the heat shock promoter: Parking, stalling, and trucking along. Cell 1993, 74, 1–4. [Google Scholar] [CrossRef]
- Guettouche, T.; Boellmann, F.; Lane, W.S.; Voellmy, R. Analysis of phosphorylation of human heat shock factor 1 in cells experiencing a stress. BMC Biochem. 2005, 11, 6–14. [Google Scholar]
- Wegele, H.; Müller, L.; Buchner, J. HSP70 and HSP90—A relay team for protein folding. Rev. Physiol. Biochem. Pharmacol. 2004, 151, 1–44. [Google Scholar]
- Kato, K.; Shinohara, H.; Goto, S.; Inaguma, Y.; Morishita, R.; Asano, T. Copurification of small heat shock protein alpha B crystallin from human skeletal muscle. J. Biol. Chem. 1992, 267, 7718–7725. [Google Scholar]
- Huxley, H.; Hanson, J. Changes in the cross-striations of muscle during contraction and stretch and their structural interpretation. Nature 1954, 173, 973–976. [Google Scholar] [CrossRef]
- Kobayashi, M.; Debold, E.P.; Turner, M.A.; Kobayashi, T. Cardiac muscle activation blunted by a mutation to the regulatory component, troponin T. J. Biol. Chem. 2013, 288, 26335–26349. [Google Scholar] [CrossRef] [Green Version]
- Communal, C.; Sumandea, M.; de Tombe, P.; Narula, J.; Solaro, R.J.; Hajjar, R.J. Functional consequences of caspase activation in cardiac myocytes. Proc. Natl. Acad. Sci. USA 2002, 99, 6252–6256. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Z.; Biesiadecki, B.J.; Jin, J. Selective deletion of the NH2-terminal variable region of cardiac troponin T in ischemia reperfusion by myofibril-associated mu-calpain cleavage. Biochemistry 2006, 45, 11681–11694. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McDonough, J.L.; Arrell, D.K.; Van Eyk, J.E. Troponin I degradation and covalent complex formation accompanies myocardial ischemia/reperfusion injury. Cir. Res. 1999, 84, 9–20. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Di Lisa, F.; De Tullio, R.; Salamino, F.; Barbato, R.; Melloni, E.; Siliprandi, N.; Schiaffino, S.; Pontremoli, S. Specific degradation of troponin T and I by mu-calpain and its modulation by substrate phosphorylation. Biochem. J. 1995, 308, 57–61. [Google Scholar] [CrossRef] [PubMed]
- Greyson, C.; Schwartz, G.G.; Lu, L.; Ye, S.; Helmke, S.; Xu, Y.; Ahmad, H. Calpain inhibition attenuates right ventricular contractile dysfunction after acute pressure overload. J. Mol. Cell. Cardiol. 2008, 44, 59–68. [Google Scholar] [CrossRef] [Green Version]
- Mani, S.K.; Shiraishi, H.; Balasubramanian, S.; Yamane, K.; Chellaiah, M.; Cooper, G.; Banik, N.; Zile, M.R.; Kuppuswamy, D. In vivo administration of calpeptin attenuates calpain activation and cardiomyocyte loss in pressure-overloaded feline myocardium. Am. J. Physiol. Heart. Circ. Physiol. 2008, 295, 314–326. [Google Scholar] [CrossRef] [Green Version]
- Yoshida, K.; Maaieh, M.M.; Shipley, J.B.; Doloresco, M.; Bernardo, N.L.; Qian, Y.Z.; Elliott, G.T.; Kukreja, R.C. Monophosphoryl lipid A induces pharmacologic ‘preconditioning’ in rabbit hearts without concomitant expression of 70-kDa heat shock protein. Mol. Cell. Biochem. 1996, 156, 1–8. [Google Scholar] [CrossRef]
- White, M.Y.; Hambly, B.D.; Jeremy, R.W.; Cordwell, S.J. Ischemia-specific phosphorylation and myofilament translocation of heat shock protein 27 precede alpha B-crystallin and occurs independently of reactive oxygen species in rabbit myocardium. J. Mol. Cell. Cardiol. 2006, 40, 761–774. [Google Scholar] [CrossRef]
- Lu, X.Y.; Chen, L.; Cai, X.L.; Yang, H.T. Overexpression of heat shock protein 27 protects against ischaemia/reperfusion-induced cardiac dysfunction via stabilization of troponin I and T. Cardiovasc. Res. 2008, 79, 500–508. [Google Scholar] [CrossRef] [Green Version]
- Valentim, L.M.; Rodnight, R.; Geyer, A.B.; Horn, A.P.; Tavares, A.; Cimarosti, H.; Netto, C.A.; Salbego, C.G. Changes in heat shock protein 27 phosphorylation and immunocontent in response to preconditioning to oxygen and glucose deprivation in organotypic hippocampal cultures. Neuroscience 2003, 118, 379–386. [Google Scholar] [CrossRef]
- Guo, Y.; Ziesch, A.; Hocke, S.; Kampmann, E.; Ochs, S.; De Toni, E.N.; Göke, B.; Gallmeier, E. Overexpression of heat shock protein 27 (HSP-27) increases gemcitabine sensitivity in pancreatic cancer cells through S-phase arrest and apoptosis. J. Cell. Mol. Med. 2015, 1, 340–350. [Google Scholar] [CrossRef] [PubMed]
- Cuerrier, C.M.; Chen, Y.-X.; Tremblay, D.; Rayner, K.J.; McNulty, M.; Zhao, X.; Kennedy, C.R.J.; De Belleroche, J.; Pelling, A.E.; O’Brien, E. Chronic over-expression of heat shock protein 27 attenuates atherogenesis and enhances plaque remodeling: A combined histological and mechanical assessment of aortic lesions. PLoS ONE 2013, 8, e55867. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guo, Z.; Cooper, L.F. An N-terminal 33-amino-acid-delection variant of HSP25 retains oligomerization and functional properties. Biochem. Biophys. Res. Commun. 2000, 270, 183–189. [Google Scholar] [CrossRef]
- Parcellier, A.; Schmitt, E.; Brunet, M.; Hammann, A.; Solary, E.; Garrido, C. Small heat shock proteins HSP-27 and alphaB-crystallin: Cytoprotective and oncogenic functions. Antioxid. Redox. Signal. 2005, 7, 404–413. [Google Scholar] [CrossRef] [PubMed]
- Bruey, J.-M.; Paul, C.; Fromentin, A.; Hilpert, S.; Arrigo, A.-P.; Solary, E.; Garrido, C. Differential regulation of HSP-27 oligomerization in tumor cells grown in vitro and in vivo. Oncogene 2000, 19, 4855–4863. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Burban, A.; Sharanek, A.; Guguen-Guillouzo, C.; Guillouzo, A. Endoplasmic reticulum stress precedes oxidative stress in antibiotic-induced cholestasis and cytotoxicity in human hepatocytes. Free Radic. Biol. Med. 2018, 115, 166–178. [Google Scholar] [CrossRef]
- Stetler, R.A.; Gao, Y.; Signore, A.P.; Cao, G.; Chen, J. HSP-27: Mechanisms of cellular protection against neuronal injury. Curr. Mol. Med. 2009, 9, 863–872. [Google Scholar] [CrossRef] [Green Version]
- Efthymiou, C.A.; Mocanu, M.M.; De Belleroche, J.; Wells, D.J.; Latchmann, D.S.; Yellon, D.M. Heat shock protein 27 protects the heart against myocardial infarction. Basic Res. Cardiol. 2004, 99, 392–394. [Google Scholar] [CrossRef]
- Horváth, I.; Multhoff, G.; Sonnleitner, A.; Vígh, L. Membrane associated stress proteins: More than simply chaperones. Biochem. Biophys. Acta 2008, 1778, 1653–1664. [Google Scholar] [CrossRef] [Green Version]
- Bolli, R. Myocardial ‘stunning’ in man. Circulation 1992, 86, 1671–1691. [Google Scholar] [CrossRef] [Green Version]
- Lochner, A.; Marais, E.; Genade, S.; Huisamen, B.; Dutoit, E.F.; Moolman, J.A. Protection of the ischaemic heart: Investigations into the phenomenon of ischaemic preconditioning. Cardiovasc. J. Afr. 2009, 20, 43–51. [Google Scholar] [PubMed]
- Hao, X.; Zhang, S.; Timakov, B.; Zhang, P. The HSP-27 gene is not required for Drosophila development but its activity is associated with starvation resistance. Cell Stress Chaperones 2007, 12, 364–372. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koob, G.F.; Kreek, M.J. Stress, dysregulation of drug reward pathways, and the transition to drug dependence. Am. J. Psychiatry. 2007, 164, 1149–1159. [Google Scholar] [CrossRef] [PubMed]
- Kasch, S. Serum catecholamines in cocaine intoxicated patients with cardiac symptoms. Ann. Emerg. Med. 1987, 16, 481–486. [Google Scholar]
- Dunn, A.J.; Swiergiel, A.H. Effects of acute and chronic stressors and CRF in rat and mouse tests for depression. Ann. N. Y. Acad. Sci. 2008, 1148, 118–126. [Google Scholar] [CrossRef]
- Holsboer, F.; Ising, M. Central CRH system in depression and anxiety—evidencefrom clinical studies with CRH1 receptor antagonists. Eur. J. Pharmacol. 2008, 583, 350–357. [Google Scholar] [CrossRef]
- Wood, S.K.; Woods, J.H. Corticotropin-releasing factor receptor-1: A therapeutic target for cardiac autonomic disturbances. Expert Opin. Ther. Targets 2007, 11, 1401–1413. [Google Scholar] [CrossRef]
- Koob, G.F. Corticotropin-releasing factor, norepinephrine, and stress. Biol. Psychiatry 1999, 46, 1167–1180. [Google Scholar] [CrossRef]
- Stinus, M.; Cador, M.; Zorrilla, E.P.; Koob, G.F. Buprenorphine and CRF1R antagonists block the acquisition of opiate withdrawal-induced conditioned place aversion in rats. Neuropsychopharmacology 2005, 30, 90–98. [Google Scholar] [CrossRef] [Green Version]
- Rosen, S.D. From heart to brain: The genesis and processing of cardiac pain. Can. J. Cardiol. 2012, 28, S7–S19. [Google Scholar] [CrossRef]
- González-Cuello, A.; Milanés, M.V.; Castells, M.T.; Laorden, M.L. Morphine withdrawal-induced c-fos expression in the heart: A peripheral mechanism. Eur. J. Pharmacol. 2004, 487, 117–124. [Google Scholar] [CrossRef] [PubMed]
- Milanés, M.V.; Martínez, M.D.; González-Cuello, A.; Laorden, M.L. Evidence for a peripheral mechanism in cardiac opioid withdrawal. Naunyn-Schmiedeberg’s Arch. Pharmacol. 2001, 364, 193–198. [Google Scholar]
- Navarro-Zaragoza, J.; Martínez-Laorden, E.; Mora, L.; Hidalgo, J.; Milanés, M.V.; Laorden, M.L. Cardiac adverse effects of naloxone-precipitated morphine withdrawal on right ventricle: Role of corticotropin-releasing factor (CRF1) receptor. Toxicol. Appl. Pharmacol. 2014, 275, 28–35. [Google Scholar] [CrossRef] [PubMed]
- Frishman, W.H.; Del Vecchio, A.; Sanal, S.; Ismail, A. Cardiovascular manifestations of substance abuse: Part 2: Alcohol, amphetamines, heroin, cannabis, and caffeine. Heart Dis. 2003, 5, 253–271. [Google Scholar] [CrossRef]
- Nerantzis, C.E.; Koulouris, S.N.; Marianou, S.K.; Pastromas, S.C.; Koutsaftis, P.N.; Agapitos, E.B. Histologic findings of the sinus node and the perinodal area in street heroin addicts, victims of sudden unexpected death. J. Forensic Sci. 2011, 56, 645–648. [Google Scholar] [CrossRef]
- Navarro-Zaragoza, J.; Ros-Simó, C.; Milanés, M.V.; Valverde, O.; Laorden, M.L. Binge Ethanol and MDMA combination exacerbates toxic cardiac effects by inducing cellular stress. PLoS ONE 2015, 10, e0141502. [Google Scholar]
- Navarro-Zaragoza, J.; Ros-Simó, C.; Milanés, M.V.; Valverde, O.; Laorden, M.L. Binge ethanol and MDMA combination exacerbates HSP-27 and Trx-1 (biomarkers of toxic cardiac effects) expression in right ventricle. Life Sci. 2019, 220, 50–57. [Google Scholar] [CrossRef]
- García-Carmona, J.A.; Georgiou, P.; Zanos, P.; Bailey, A.; Laorden, M.L. Methamphetamine withdrawal induces activation of CRF neurons in the brain stress system in parallel with an increased activity of cardiac sympathetic pathways. Naunyn-Schmiedeberg’s Arch. Pharmacol. 2018, 391, 423–434. [Google Scholar] [CrossRef]
- Alarcón, S.; Hernández, J.; Laorden, M.L. Electrophysiological effects of opioid receptor selective agonists on guinea-pig papillary muscle. Reg. Pep. 1995, 5, 149–154. [Google Scholar] [CrossRef]
- Llobel, F.; Laorden, M.L. Effects of morphine on atrial preparations obtained from nonfailing and failing human hearts. Br. J. Anaesth. 1996, 76, 106–110. [Google Scholar] [CrossRef]
- Gross, E.R.; Hsu, A.K.; Gross, G.J. GSβ inhibition and KATP channel opening mediate acute opioid-induced cardioprotection at reperfusion. Basic Res. Cardiol. 2007, 102, 341–349. [Google Scholar] [CrossRef]
- Peart, J.N.; Gross, E.R.; Reichelt, M.E.; Hsu, A.; Headrick, J.P.; Gross, G.J. Activation of kappaopioid receptors at reperfusion affords cardioprotection in both rat and mouse hearts. Basic Res. Cardiol. 2008, 103, 454–463. [Google Scholar] [CrossRef] [PubMed]
- Peart, J.N.; Gross, G.J. Cardioprotective effects of acute and chronic opioid treatment is mediated via different signalling pathways. Am. J. Physiol. Heart Circ. Physiol. 2006, 291, H1746–H1753. [Google Scholar] [CrossRef] [PubMed]
- Kienbaum, P.; Heuter, T.; Michel, M.C.; Scherbaum, N.; Gastpar, M.; Peters, J. Chronic μ-opioid receptor stimulation in humans decreases muscle sympathetic nerve activity. Circulation 2001, 103, 850–855. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kienbaum, P.; Heuter, T.; Scherbaum, N.; Gastpar, M.; Peters, J. Chronic μ-opioid receptor stimulation alters cardiovascular regulation in humans: Differential effects on muscle sympathetic and heart rate responses to arterial hypotension. J. Cardiovasc. Pharmacol. 2002, 40, 363–369. [Google Scholar] [CrossRef]
- Rabadan, J.V.; Milanés, M.V.; Laorden, M.L. Effects of acute administration of morphine on right atrial catecholamine content and heart rate in chronically morphine-treated rats. Br. J. Anaesth. 1997, 78, 439–441. [Google Scholar] [CrossRef]
- Almela, P.; Milanés, M.V.; Laorden, M.L. The PKs PKA and ERK 1/2 are involved in phosphorylation of TH at serine 40 and 31 during morphine withdrawal in rat hearts. Br. J. Pharmacol. 2008, 155, 73–83. [Google Scholar] [CrossRef] [Green Version]
- Almela, P.; Milanés, M.V.; Laorden, M.L. Tyrosine hydroxylase phsophorylation after naloxone-induced morphine withdrawal in the left ventricle. Basic Res. Cardiol. 2009, 104, 366–376. [Google Scholar] [CrossRef]
- Martínez-Laorden, E.; Hurle, M.A.; Milanés, M.V.; Laorden, M.L.; Almela, P. Morphine withdrawal activates hypothalamic-pituitary-adrenal axis and heat shock protein 27 in the left ventricle: The role of extracellular signal-regulated kinase. J. Pharmacol. Exp. Ther. 2012, 342, 665–674. [Google Scholar] [CrossRef] [Green Version]
- Tunbridge, E.M.; Huber, A.; Farrell, S.M.; Stumpenhorst, K.; Harrison, P.J.; Walton, M.E. The role of cateho-o-methyltransferase in reward processing and addiction. CNS Neurol. Disord. Drug Targets 2013, 11, 306–323. [Google Scholar] [CrossRef]
- Li, T.; Chen, C.K.; Hu, X.; Ball, D.; Lin, S.K.; Chen, W. Association analysis of the DRD4 and COMT genes in methamphetamine abuse. Am. J. Med. Genet. B Neuropsychiatr. Genet. 2004, 129, 120–124. [Google Scholar] [CrossRef] [PubMed]
- Beuten, J.; Payne, T.J.; Ma, J.Z.; Li, M.D. Significant association of catechol-O-methyltransferase (COMT) haplotypes with nicotine dependence in male and female smokers of two ethnic populations. Neuropsychopharmacology 2005, 31, 675–684. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oliviera, M.T.; Rego, A.C.; Morgadinho, M.T.; Macedo, T.R.; Oliveira, C.R. Toxic effects of opioid and stimulant drugs on undifferentiated PC12 cells. Ann. N. Y. Acad. Sci. 2002, 965, 487–496. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Udelsman, R.; Holbrook, N.J. Endocrine and molecular responses to surgical stress. Curr. Probl. Surg. 1994, 31, 653–720. [Google Scholar] [CrossRef]
- Almela, P.; Martínez-Laorden, E.; Atucha, N.M.; Milanés, M.V.; Laorden, M.L. Naloxone-precipitated morphine withdrawal evoques phosphorylation of heat shock protein 27 in rat heart through extracellular signal-regulated kinase. J. Mol. Cell. Cardiol. 2011, 51, 129–139. [Google Scholar] [CrossRef]
- Robinson, A.A.; Dunn, M.J.; McCormack, A.; dos Remedios, C.; Rose, M.L. Protective effect of phosphorylated HSP-27 in coronary arteries through actin stabilization. J. Mol. Cell. Cardiol. 2010, 49, 370–379. [Google Scholar] [CrossRef]
- Mehelen, P.; Hickey, E.; Weber, I.A.; Arrigo, A.P. Large unphosphorylated aggregates as the active form of HSP-27 which controls intracellular reactive oxygen species and glutathione levels and generates a protection against TNFalpha in NIH-3T3-ras cells. Biochem. Biophys. Res. Commun. 1997, 241, 187–192. [Google Scholar] [CrossRef]
- Duverger, O.; Paslaru, L.; Morange, M. HSP25 is involved in two steps of the differentiation of PAM212 keratinocytes. J. Biol. Chem. 2004, 279, 10252–10260. [Google Scholar] [CrossRef] [Green Version]
- O’Shaughnessy, R.F.; Welti, J.; Cooke, J.C.; Avilion, A.A.; Monks, B.; Birnbaum, M.; Byrne, C.R. AKT-dependent HSPB1 (HSP-27) activity in epidermal differentiation. J. Biol. Chem. 2007, 282, 17297–17305. [Google Scholar] [CrossRef] [Green Version]
- Peart, J.N.; Gross, E.R.; Headrick, J.P.; Gross, G.J. Impaired p38 MAPK/HSP-27 signaling underlies aging-related failure in opioid-mediated cardioprotection. J. Mol. Cell. Cardiol. 2007, 42, 972–980. [Google Scholar] [CrossRef] [Green Version]
- Blake, M.J.; Buckley, D.J.; Buckley, A.R. Dopaminergic regulation of heat shock protein-70 expression in adrenal gland and aorta. Endocrinology 1993, 132, 1063–1070. [Google Scholar] [CrossRef] [PubMed]
- Blake, M.J.; Udelsman, R.; Feulner, G.J.; Norton, D.D.; Holbrook, N.J. Stress-induced heat shock protein 70 expression in adrenal cortex: An adrenocorticotrophic hormone-sensitive, age-dependent response. Proc. Natl. Acad. Sci. USA 1991, 88, 9873–9877. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pignatelli, D.; Ferreira, J.; Soares, P.; Costa, M.J.; Magalhaes, M.C. Immunohistochemical study of heat shock protein 27, 60 and 70 in the normal human adrenal and in adrenal tumors with suppressed ACTH production. Microsc. Res. Tech. 2003, 61, 315–323. [Google Scholar] [CrossRef] [PubMed]
- Yoshida-Hiroi, M.; Bradbury, M.J.; Eisenhofer, G.; Hiroi, N.; Vale, W.W.; Novotny, G.E.; Hartwig, H.G.; Scherbaum, W.A.; Bornstein, S.R. Chromaffin cell function and structure is impaired in corticotropin-releasing hormone receptor type 1-null mice. Mol. Psychiatry 2002, 7, 967–974. [Google Scholar] [CrossRef]
- Martinez-Laorden, E.; Almela, P.; Milanés, M.V.; Laorden, M.L. Expression of heat shock protein 27 and troponin T and troponin I after naloxone-precipitated morphine withdrawal. Eur. J. Pharmacol. 2015, 766, 142–150. [Google Scholar] [CrossRef]
- Taneike, M.; Mizote, I.; Morita, T.; Watanabe, T.; Hikoso, S.; Yamaguchi, O.; Takeda, T.; Oka, T.; Tamai, T.; Oyabu, J.; et al. Calpain protects the heart from hemodynamic stress. J. Biol. Chem. 2011, 286, 32170–32177. [Google Scholar] [CrossRef] [Green Version]
- Thompson, J.; Hu, Y.; Lesnefsky, E.J.; Chen, K. Activation of mitochondrial calpain and increased cardiac injury beyond AIF release. Am J Physiol Heart Circ Physiol. 2016, 310, H376–H384. [Google Scholar] [CrossRef] [Green Version]
- Chen, Q.; Thompson, J.; Hu, Y.; Dean, J.; Lesnefsky, E.J. Inhibition of the ubiquitous calpains protects complex I activity and enables improved mitophagy in the heart following ischemia-reperfusion. Am J Physiol. Cell Physiol. 2019, 317, C910–C921. [Google Scholar] [CrossRef]
- Hu, X.; Li, J.; van Marion, D.M.S.; Zhang, D.; Brundel, B.J.J.M. Heat shock protein inducer GGA*-59 reverses contractile and structural remodeling via restoration of the microtubule network in experimental Atrial Fibrillation. J. Mol. Cell. Cardiol. 2019, 134, 86–97. [Google Scholar] [CrossRef] [Green Version]
- Zorrilla, E.P.; Koob, G.F. The therapeutic potential of CRF1 antagonists for anxiety. Expert. Opin. Investig. Drugs 2004, 13, 799–828. [Google Scholar] [CrossRef]
- Gallagher, J.P.; Orozco-Cabal, L.F.; Liu, J.; Shinnick-Gallagher, P. Synaptic physiology of central CRH system. Eur. J. Pharmacol. 2008, 583, 215–225. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kimura, Y.; Takahashi, K.; Totsune, K.; Muramatsu, Y.; Kaneko, C.; Darnel, A.D.; Suzuki, T.; Ebina, M.; Nukiwa, T.; Sasano, H. Expression of urocortin and corticotropin-releasing factor receptor subtypes in the human heart. J. Clin. Endocrinol. Metab. 2002, 87, 340–346. [Google Scholar] [CrossRef] [PubMed]
- Kishimoto, T.; Pearse, R.V.; Lin, C.R.; Rosenfeld, M.G. A sauvagine/corticotropin-releasing factor receptor expressed in heart and skeletal muscle. Proc. Natl. Acad. Sci. USA 1995, 92, 1108–1112. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Perrin, M.; Donaldson, C.; Chen, R.; Blount, A.; Berggren, T.; Bilezikjian, L.; Sawchenko, P.; Vale, W. Identification of a second corticotropin-releasing factor receptor gene and characterization of a cDNA expressed in heart. Proc. Natl. Acad. Sci. USA 1195, 92, 2969–2973. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yuan, Z.; McCauley, R.; Chen-Scarabelli, C.; Abounit, K.; Stephanou, A.; Barry, S.P.; Knight, R.; Saravolatz, S.F.; Saravolatz, L.D.; Ulgen, B.O.; et al. Activation of Src proteintyrosine kinase plays an essential role in urocortin-mediated cardioprotection. Mol. Cell. Endocrinol. 2010, 325, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Yang, L.Z.; Tovote, P.; Rayner, M.; Kockskämper, J.; Pieske, B.; Spiess, J. Corticotropin-releasing factor receptors and urocortins, links between the brain and the heart. Eur. J. Pharmacol. 2010, 632, 1–6. [Google Scholar] [CrossRef]
- Kang, Y.M.; Zhang, A.Q.; Zhao, X.F.; Cardinale, J.P.; Elks, C.; Cao, X.M.; Zhang, Z.W.; Francis, J. Paraventricular nucleus corticotrophin releasing hormone contributes to sympathoexcitation via interaction with neurotransmitters in heart failure. Basic. Res. Cardiol. 2011, 106, 473–483. [Google Scholar] [CrossRef] [Green Version]
- Martínez-Laorden, E.; García-Carmona, J.A.; Baroja-Mazo, A.; Romecín, P.; Atucha, N.M.; Milanés, M.V.; Laorden, M.L. Corticotropin-releasing factor (CRF) receptor-1 is involved in cardiac noradrenergic activity observed during naloxone-precipitated morphine withdrawal. Br. J. Pharmacol. 2014, 71, 688–700. [Google Scholar] [CrossRef] [Green Version]


© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Martínez-Laorden, E.; Navarro-Zaragoza, J.; Milanés, M.V.; Laorden, M.L.; Almela, P. Cardiac Protective Role of Heat Shock Protein 27 in the Stress Induced by Drugs of Abuse. Int. J. Mol. Sci. 2020, 21, 3623. https://doi.org/10.3390/ijms21103623
Martínez-Laorden E, Navarro-Zaragoza J, Milanés MV, Laorden ML, Almela P. Cardiac Protective Role of Heat Shock Protein 27 in the Stress Induced by Drugs of Abuse. International Journal of Molecular Sciences. 2020; 21(10):3623. https://doi.org/10.3390/ijms21103623
Chicago/Turabian StyleMartínez-Laorden, Elena, Javier Navarro-Zaragoza, María Victoria Milanés, María Luisa Laorden, and Pilar Almela. 2020. "Cardiac Protective Role of Heat Shock Protein 27 in the Stress Induced by Drugs of Abuse" International Journal of Molecular Sciences 21, no. 10: 3623. https://doi.org/10.3390/ijms21103623
APA StyleMartínez-Laorden, E., Navarro-Zaragoza, J., Milanés, M. V., Laorden, M. L., & Almela, P. (2020). Cardiac Protective Role of Heat Shock Protein 27 in the Stress Induced by Drugs of Abuse. International Journal of Molecular Sciences, 21(10), 3623. https://doi.org/10.3390/ijms21103623