The Role of Immune Checkpoints after Cellular Therapy

Abstract
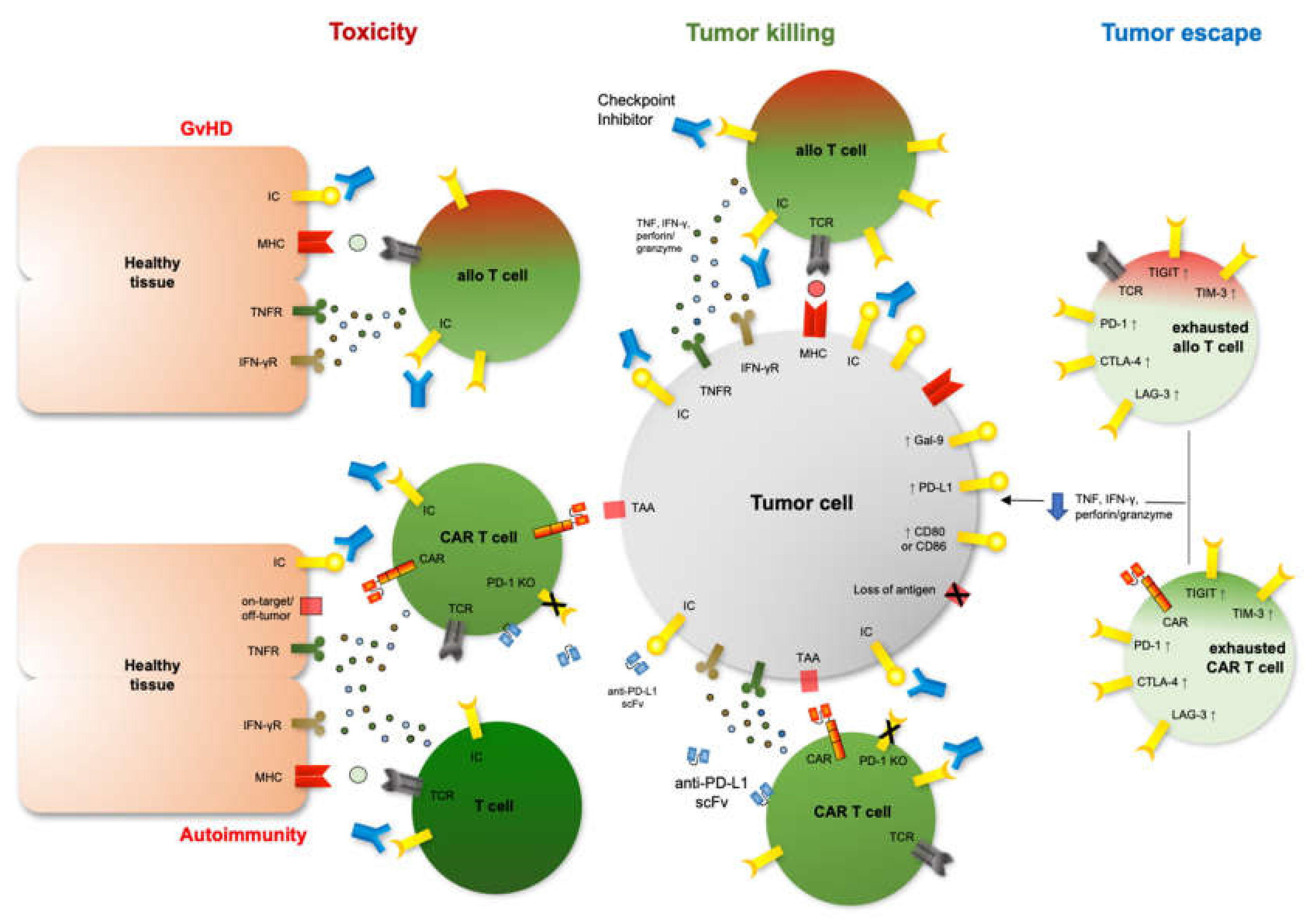
:1. Introduction
1.1. Allogeneic Hematopoietic Stem Cell Transplantation (Allo-HCT)
1.2. Chimeric Antigen Receptor T Cells (Cars)
1.3. Immune Checkpoint Inhibitors (CI)
2. Immune Checkpoints and Checkpoint Inhibition after Allo-HCT
3. Immune Checkpoints and Checkpoint Inhibition after Cars
4. Conclusions
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| aGvHD | acute Graft-versus-Host Disease |
| allo-HCT | allogeneic hematopoietic stem cell transplantation |
| ALL | acute lymphocytic leukemia |
| AML | acute myeloid leukemia |
| CAIX | carbonic anhydrase IX |
| CAR | chimeric antigen receptor |
| CD | cluster of differentiation |
| cGvHD | chronic Graft-versus-Host Disease |
| CI | checkpoint inhibition/inhibitor |
| CR | complete remission |
| CRS | cytokine release syndrome |
| CRISPR/Cas9 | clustered regularly interspaced short palindromic repeats associated with Cas9 endonuclease |
| CTLA-4 | cytotoxic T-lymphocyte-associated protein-4 |
| DLBCL | diffuse large B-cell lymphoma |
| DLI | donor lymphocyte infusion |
| EGFR | epidermal growth factor receptor |
| EMA | European Medicines Agency |
| FDA | Food and Drug Administration |
| FL | follicular lymphoma |
| FOXP3 GD2 | Forkhead-box-protein P3 disialoganglioside |
| GvHD | Graft-versus-Host Disease |
| GvL | Graft-versus-leukemia |
| GvT | Graft-versus-tumor |
| HD | Hodgkin’s disease |
| (c)HL | (classical) Hodgkin lymphoma |
| ICANS | immune cell associated neurotoxicity syndrome |
| ICOS | inducible T cell costimulator |
| irAEs | immune-related adverse events |
| KLRG-1 | killer cell lectin-like receptor subfamily member 1 |
| LAG-3 | lymphocyte-activation gene 3 |
| mAbs | monoclonal antibody |
| MDS | myelodysplastic syndrome |
| MGMT | 0-6-Methylguanine DNA-methyltransferase |
| MiHA | minor histocompatibility antigen |
| MF | myelofibrosis |
| NHL | Non-Hodgkin-Lymphoma |
| ORR | overall response rate |
| OS | overall survival |
| PD-1 | programmed death-1 |
| PD-L1 | programmed death- ligand 1 |
| PFS | progression free survival |
| PMBCL | primary mediastinal B cell lymphoma |
| PR | partial remission |
| RNA | ribonucleotide acid |
| scFv | single chain variable fragment |
| SD | stable disease |
| TIGIT | T cell immunoglobulin and ITIM domains |
| TIM-3 | T-cell immunoglobulin mucin-3 |
References
- Gajewski, T.F.; Schreiber, H.; Fu, Y.-X. Innate and adaptive immune cells in the tumor microenvironment. Nat. Immunol. 2013, 14, 1014–1022. [Google Scholar] [CrossRef] [Green Version]
- Beatty, G.L.; Gladney, W.L. Immune escape mechanisms as a guide for cancer immunotherapy. Clin. Cancer Res. 2015, 21, 687–692. [Google Scholar] [CrossRef] [Green Version]
- Iwai, Y.; Ishida, M.; Tanaka, Y.; Okazaki, T.; Honjo, T.; Minato, N. Involvement of PD-L1 on tumor cells in the escape from host immune system and tumor immunotherapy by PD-L1 blockade. Proc. Natl. Acad. Sci. USA 2002, 99, 12293–12297. [Google Scholar] [CrossRef] [Green Version]
- Zang, X.; Allison, J.P. The B7 Family and Cancer Therapy: Costimulation and Coinhibition. Clin. Cancer Res. 2007, 13, 5271–5279. [Google Scholar] [CrossRef] [Green Version]
- Leach, D.R.; Krummel, M.F.; Allison, J.P. Enhancement of Antitumor Immunity by CTLA-4 Blockade. Science 1996, 271, 1734–1736. [Google Scholar] [CrossRef] [Green Version]
- Kusmartsev, S.; Gabrilovich, D.I. Role Of Immature Myeloid Cells in Mechanisms of Immune Evasion in Cancer. Cancer Immunol. Immunother. 2006, 55, 237–245. [Google Scholar] [CrossRef]
- Thomas, E.D.; Lochte, H.L.; Lu, W.C.; Ferrebee, J.W. Intravenous Infusion of Bone Marrow in Patients Receiving Radiation and Chemotherapy. New Engl. J. Med. 1957, 257, 491–496. [Google Scholar] [CrossRef]
- Weiden, P.L.; Flournoy, N.; Thomas, E.D.; Prentice, R.; Fefer, A.; Buckner, C.D.; Storb, R. Antileukemic Effect of Graft-versus-Host Disease in Human Recipients of Allogeneic-Marrow Grafts. New Engl. J. Med. 1979, 300, 1068–1073. [Google Scholar] [CrossRef]
- Billingham, R.E. The biology of graft-versus-host reactions. Harvey Lect. 1966, 62, 21–78. [Google Scholar]
- Horowitz, M.M.; Gale, R.P.; Sondel, P.M.; Goldman, J.M.; Kersey, J.; Kolb, H.J.; A Rimm, A.; Ringden, O.; Rozman, C.; Speck, B. Graft-versus-leukemia reactions after bone marrow transplantation. Blood 1990, 75, 555–562. [Google Scholar] [CrossRef] [Green Version]
- Lee, C.J.; Savani, B.N.; Mohty, M.; Gorin, N.C.; Labopin, M.; Ruggeri, A.; Schmid, C.; Baron, F.; Esteve, J.; Giebel, S.; et al. Post-remission strategies for the prevention of relapse following allogeneic hematopoietic cell transplantation for high-risk acute myeloid leukemia: expert review from the Acute Leukemia Working Party of the European Society for Blood and Marrow Transplantation. Bone Marrow Transplant. 2019, 54, 519–530. [Google Scholar] [CrossRef]
- Parmar, S.; Ritchie, D.S. Allogeneic transplantation as anticancer immunotherapy. Curr. Opin. Immunol. 2014, 27, 38–45. [Google Scholar] [CrossRef]
- Kolb, H.J.; Schattenberg, A.V.; Goldman, J.M.; Hertenstein, B.; Jacobsen, N.; Arcese, W.; Ljungman, P.; Ferrant, A.; Verdonck, L.; Niederwieser, D.; et al. Graft-versus-leukemia effect of donor lymphocyte transfusions in marrow grafted patients. Blood 1995, 86, 2041–2050. [Google Scholar] [CrossRef] [Green Version]
- Gross, G.; Waks, T.; Eshhar, Z. Expression of immunoglobulin-T-cell receptor chimeric molecules as functional receptors with antibody-type specificity. Proc. Natl. Acad. Sci. USA 1989, 86, 10024–10028. [Google Scholar] [CrossRef] [Green Version]
- Eshhar, Z.; Waks, T.; Gross, G.; Schindler, D.G. Specific activation and targeting of cytotoxic lymphocytes through chimeric single chains consisting of antibody-binding domains and the gamma or zeta subunits of the immunoglobulin and T-cell receptors. Proc. Natl. Acad. Sci. USA 1993, 90, 720–724. [Google Scholar] [CrossRef] [Green Version]
- Eshhar, Z.; Gross, G.; Waks, T.; Lustgarten, J.; Bach, N.; Ratner, A.; Treisman, J.; Schindler, D.G. T-Bodies: Chimeric T-Cell Receptors with Antibody-Type Specificity. Methods 1995, 8, 133–142. [Google Scholar] [CrossRef]
- Porter, D.L.; Levine, B.L.; Kalos, M.; Bagg, A.; June, C.H. Chimeric antigen receptor-modified T cells in chronic lymphoid leukemia. New Engl. J. Med. 2011, 365, 725–733. [Google Scholar] [CrossRef] [Green Version]
- Maude, S.L.; Frey, N.; Shaw, P.A.; Aplenc, R.; Barrett, D.M.; Bunin, N.J.; Chew, A.; Gonzalez, V.E.; Zheng, Z.; Lacey, S.F.; et al. Chimeric antigen receptor T cells for sustained remissions in leukemia. New Engl. J. Med. 2014, 371, 1507–1517. [Google Scholar] [CrossRef] [Green Version]
- Locke, F.L.; Ghobadi, A.; A Jacobson, C.; Miklos, D.B.; Lekakis, L.J.; O Oluwole, O.; Lin, Y.; Braunschweig, I.; Hill, B.T.; Timmerman, J.M.; et al. Long-term safety and activity of axicabtagene ciloleucel in refractory large B-cell lymphoma (ZUMA-1): A single-arm, multicentre, phase 1–2 trial. Lancet Oncol. 2019, 20, 31–42. [Google Scholar] [CrossRef]
- Grupp, S.A.; Maude, S.L.; Rives, S.; Baruchel, A.; Boyer, M.W.; Bittencourt, H.; Bader, P.; Büchner, J.; Laetsch, T.W.; Stefanski, H.; et al. Updated Analysis of the Efficacy and Safety of Tisagenlecleucel in Pediatric and Young Adult Patients with Relapsed/Refractory (r/r) Acute Lymphoblastic Leukemia. Blood 2018, 132, 895. [Google Scholar] [CrossRef]
- Schuster, S.J.; Bishop, M.R.; Tam, C.; Borchmann, P.; Jaeger, U.; Waller, E.K.; Holte, H.; McGuirk, J.P.; Jaglowski, S.; Tobinai, K.; et al. Sustained Disease Control for Adult Patients with Relapsed or Refractory Diffuse Large B-Cell Lymphoma: An Updated Analysis of Juliet, a Global Pivotal Phase 2 Trial of Tisagenlecleucel. Blood 2018, 132, 1684. [Google Scholar] [CrossRef]
- Maude, S.L.; Laetsch, T.W.; Buechner, J.; Rives, S.; Boyer, M.; Bittencourt, H.; Bader, P.; Verneris, M.R.; Stefanski, H.E.; Myers, G.D.; et al. Tisagenlecleucel in Children and Young Adults with B-Cell Lymphoblastic Leukemia. New Engl. J. Med. 2018, 378, 439–448. [Google Scholar] [CrossRef]
- Fitzgerald, J.C.; Weiss, S.L.; Maude, S.L.; Barrett, D.M.; Lacey, S.F.; Melenhorst, J.J.; Shaw, P.; Berg, R.A.; June, C.H.; Porter, D.L.; et al. Cytokine Release Syndrome After Chimeric Antigen Receptor T Cell Therapy for Acute Lymphoblastic Leukemia. Crit. Care Med. 2017, 45, e124–e131. [Google Scholar] [CrossRef]
- Brudno, J.N.; Kochenderfer, J.N. Recent advances in CAR T-cell toxicity: Mechanisms, manifestations and management. Blood Rev. 2019, 34, 45–55. [Google Scholar] [CrossRef]
- Cheng, J.; Zhao, L.; Zhang, Y.; Qin, Y.; Guan, Y.; Zhang, T.; Liu, C.; Zhou, J. Understanding the Mechanisms of Resistance to CAR T-Cell Therapy in Malignancies. Front. Oncol. 2019, 9, 1237. [Google Scholar] [CrossRef] [Green Version]
- Wherry, E.J.; Kurachi, M. Molecular and cellular insights into T cell exhaustion. Nat. Rev. Immunol. 2015, 15, 486–499. [Google Scholar] [CrossRef]
- Keir, M.E.; Butte, M.J.; Freeman, G.J.; Sharpe, A.H. PD-1 and Its Ligands in Tolerance and Immunity. Annu. Rev. Immunol. 2008, 26, 677–704. [Google Scholar] [CrossRef] [Green Version]
- Pardoll, D.M. The blockade of immune checkpoints in cancer immunotherapy. Nat. Rev. Cancer 2012, 12, 252–264. [Google Scholar] [CrossRef] [Green Version]
- Buchbinder, E.I.; Desai, A. CTLA-4 and PD-1 Pathways. Am. J. Clin. Oncol. 2016, 39, 98–106. [Google Scholar] [CrossRef] [Green Version]
- Haanen, J.B. Immune checkpoint inhibitors for the treatment of cancer. Ann. Oncol. 2015, 26, vii7. [Google Scholar] [CrossRef]
- Motzer, R.J.; Escudier, B.; McDermott, D.F.; George, S.; Hammers, H.J.; Srinivas, S.; Tykodi, S.S.; Sosman, J.A.; Procopio, G.; Plimack, E.R.; et al. Nivolumab versus Everolimus in Advanced Renal-Cell Carcinoma. New Engl. J. Med. 2015, 373, 1803–1813. [Google Scholar] [CrossRef] [PubMed]
- Aguilar, E.; Ricciuti, B.; Gainor, J.; Kehl, K.; Kravets, S.; Dahlberg, S.; Nishino, M.; Sholl, L.; Adeni, A.; Subegdjo, S.; et al. Outcomes to first-line pembrolizumab in patients with non-small-cell lung cancer and very high PD-L1 expression. Ann. Oncol. 2019, 30, 1653–1659. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Antonia, S.J.; Borghaei, H.; Ramalingam, S.S.; Horn, L.; Carpeño, J.D.C.; Pluzanski, A.; A Burgio, M.; Garassino, M.; Chow, L.Q.M.; Gettinger, S.; et al. Four-year survival with nivolumab in patients with previously treated advanced non-small-cell lung cancer: a pooled analysis. Lancet Oncol. 2019, 20, 1395–1408. [Google Scholar] [CrossRef]
- Paz-Ares, L.; Luft, A.; Vicente, D.; Tafreshi, A.; Gümüş, M.; Mazieres, J.; Hermes, B.; Şenler, F.Ç.; Csőszi, T.; Fülöp, A.; et al. Pembrolizumab plus Chemotherapy for Squamous Non–Small-Cell Lung Cancer. New Engl. J. Med. 2018, 379, 2040–2051. [Google Scholar] [CrossRef] [PubMed]
- Sharpe, A.H.; Pauken, K.E. The diverse functions of the PD1 inhibitory pathway. Nat. Rev. Immunol. 2018, 18, 153–167. [Google Scholar] [CrossRef] [PubMed]
- Seidel, J.; Otsuka, A.; Kabashima, K. Anti-PD-1 and Anti-CTLA-4 Therapies in Cancer: Mechanisms of Action, Efficacy, and Limitations. Front. Oncol. 2018, 8. [Google Scholar] [CrossRef]
- Hanahan, D.; A Weinberg, R. Hallmarks of Cancer: The Next Generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [Green Version]
- Haanen, J.B.A.G.; Carbonnel, F.; Robert, C.; Kerr, K.M.; Peters, S.; Larkin, J.; Jordan, K. on behalf of the ESMO Guidelines Committee Management of toxicities from immunotherapy: ESMO Clinical Practice Guidelines for diagnosis, treatment and follow-up. Ann. Oncol. 2017, 28, iv119–iv142. [Google Scholar] [CrossRef]
- Zeiser, R.; Vago, L. Mechanisms of immune escape after allogeneic hematopoietic cell transplantation. Blood 2019, 133, 1290–1297. [Google Scholar] [CrossRef]
- Blazar, B.R.; Hill, G.R.; Murphy, W.J. Dissecting the biology of allogeneic HSCT to enhance the GvT effect whilst minimizing GvHD. Nat. Rev. Clin. Oncol. 2020, 2020, 1–18. [Google Scholar] [CrossRef]
- Falkenburg, J.F.; Jedema, I. Graft versus tumor effects and why people relapse. Hematology 2017, 2017, 693–698. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sakuishi, K.; Apetoh, L.; Sullivan, J.M.; Blazar, B.R.; Kuchroo, V.K.; Anderson, A.C. Targeting Tim-3 and PD-1 pathways to reverse T cell exhaustion and restore anti-tumor immunity. J. Exp. Med. 2010, 207, 2187–2194. [Google Scholar] [CrossRef] [PubMed]
- Jain, P.; Tian, X.; Cordes, S.; Chen, J.; Cantilena, C.R.; Bradley, C.; Panjwani, R.; Chinian, F.; Keyvanfar, K.; Battiwalla, M.; et al. Over-expression of PD-1 Does Not Predict Leukemic Relapse after Allogeneic Stem Cell Transplantation. Boil. Blood Marrow Transplant. 2019, 25, 216–222. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hutten, T.; Norde, W.J.; Woestenenk, R.; Wang, R.C.; Maas, F.; Kester, M.; Falkenburg, J.F.; Berglund, S.; Luznik, L.; Jansen, J.H.; et al. Increased Coexpression of PD-1, TIGIT, and KLRG-1 on Tumor-Reactive CD8+ T Cells During Relapse after Allogeneic Stem Cell Transplantation. Biol. Blood Marrow Transplant. 2018, 24, 666–677. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Noviello, M.; Manfredi, F.; Ruggiero, E.; Perini, T.; Oliveira, G.; Cortesi, F.; De Simone, P.; Toffalori, C.; Gambacorta, V.; Greco, R.; et al. Bone marrow central memory and memory stem T-cell exhaustion in AML patients relapsing after HSCT. Nat. Commun. 2019, 10, 1065. [Google Scholar] [CrossRef]
- Toffalori, C.; Zito, L.; Gambacorta, V.; Riba, M.; Oliveira, G.; Bucci, G.; Barcella, M.; Spinelli, O.; Greco, R.; Crucitti, L.; et al. Immune signature drives leukemia escape and relapse after hematopoietic cell transplantation. Nat. Med. 2019, 25, 603–611. [Google Scholar] [CrossRef]
- Norde, W.J.; Maas, F.; Hobo, W.; Korman, A.; Quigley, M.; Kester, M.G.; Hebeda, K.; Falkenburg, J.H.; Schaap, N.; de Witte, T.M.; et al. PD-1/PD-L1 interactions contribute to functional T-cell impairment in patients who relapse with cancer after allogeneic stem cell transplantation. Cancer Res. 2011, 71, 5111–5122. [Google Scholar] [CrossRef] [Green Version]
- Kong, Y.; Zhang, J.; Claxton, D.F.; Ehmann, W.C.; Rybka, W.B.; Zhu, L.; Zeng, H.; Schell, T.; Zheng, H. PD-1(hi)TIM-3(+) T cells associate with and predict leukemia relapse in AML patients post allogeneic stem cell transplantation. Blood Cancer J. 2015, 5, e330. [Google Scholar] [CrossRef] [Green Version]
- Schade, H.; Sen, S.; Neff, C.P.; Freed, B.M.; Gao, D.; Gutman, J.A.; Palmer, B.E. Programmed Death 1 Expression on CD4 + T Cells Predicts Mortality after Allogeneic Stem Cell Transplantation. Biol. Blood Marrow Transplant. 2016, 22, 2172–2179. [Google Scholar] [CrossRef]
- Hattori, N.; Nakamaki, T. Natural Killer Immunotherapy for Minimal Residual Disease Eradication Following Allogeneic Hematopoietic Stem Cell Transplantation in Acute Myeloid Leukemia. Int. J. Mol. Sci. 2019, 20, 2057. [Google Scholar] [CrossRef] [Green Version]
- Hattori, N.; Kawaguchi, Y.; Sasaki, Y.; Shimada, S.; Murai, S.; Abe, M.; Baba, Y.; Watanuki, M.; Fujiwara, S.; Arai, N.; et al. Monitoring TIGIT/DNAM-1 and PVR/PVRL2 Immune Checkpoint Expression Levels in Allogeneic Stem Cell Transplantation for Acute Myeloid Leukemia. Biol. Blood Marrow Transplant. 2019, 25, 861–867. [Google Scholar] [CrossRef]
- Michonneau, D.; Sagoo, P.; Breart, B.; Garcia, Z.; Celli, S.; Bousso, P. The PD-1 Axis Enforces an Anatomical Segregation of CTL Activity that Creates Tumor Niches after Allogeneic Hematopoietic Stem Cell Transplantation. Immunity 2016, 44, 143–154. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koestner, W.; Hapke, M.; Herbst, J.; Klein, C.; Welte, K.; Fruehauf, J.; Flatley, A.; Vignali, D.A.; Hardtke-Wolenski, M.; Jaeckel, E.; et al. PD-L1 blockade effectively restores strong graft-versus-leukemia effects without graft-versus-host disease after delayed adoptive transfer of T-cell receptor gene-engineered allogeneic CD8+ T cells. Blood 2011, 117, 1030–1041. [Google Scholar] [CrossRef] [PubMed]
- Asakura, S.; Hashimoto, D.; Takashima, S.; Sugiyama, H.; Maeda, Y.; Akashi, K.; Tanimoto, M.; Teshima, T. Alloantigen expression on non-hematopoietic cells reduces graft-versus-leukemia effects in mice. J. Clin. Investig. 2010, 120, 2370–2378. [Google Scholar] [CrossRef] [PubMed]
- Angenendt, L.; Schliemann, C.; Lutz, M.; Rebber, E.; Schulze, A.B.; Weckesser, M.; Stegger, L.; Schäfers, M.; Groth, C.; Kessler, T.; et al. Nivolumab in a patient with refractory Hodgkin’s lymphoma after allogeneic stem cell transplantation. Bone Marrow Transplant. 2016, 51, 443–445. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- A Yared, J.; Hardy, N.; Singh, Z.; Hajj, S.; Badros, A.Z.; Kocoglu, M.; Yanovich, S.; A Sausville, E.; Ujjani, C.; Ruehle, K.; et al. Major clinical response to nivolumab in relapsed/refractory Hodgkin lymphoma after allogeneic stem cell transplantation. Bone Marrow Transplant. 2016, 51, 850–852. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Villasboas, J.C.; Ansell, S.M.; Witzig, T.E. Targeting the PD-1 pathway in patients with relapsed classic Hodgkin lymphoma following allogeneic stem cell transplant is safe and effective. Oncotarget 2016, 7, 13260–13264. [Google Scholar] [CrossRef] [Green Version]
- Shad, A.; Huo, J.; Darcy, C.; Abu-Ghosh, A.; Esposito, G.; Holuba, M.-J.; Robey, N.; Cooke, K.R.; Symons, H.J.; Chen, A.R.; et al. Tolerance and effectiveness of nivolumab after pediatric T-cell replete, haploidentical, bone marrow transplantation: A case report. Pediatr. Blood Cancer 2017, 64, e26257. [Google Scholar] [CrossRef]
- Onizuka, M.; Kojima, M.; Matsui, K.; Machida, S.; Toyosaki, M.; Aoyama, Y.; Kawai, H.; Amaki, J.; Hara, R.; Ichiki, A.; et al. Successful treatment with low-dose nivolumab in refractory Hodgkin lymphoma after allogeneic stem cell transplantation. Int. J. Hematol. 2017, 106, 141–145. [Google Scholar] [CrossRef]
- Covut, F.; Pinto, R.; Cooper, B.W.; Tomlinson, B.; Metheny, L.; Malek, E.; Lazarus, H.M.; De Lima, M.; Caimi, P.F. Nivolumab before and after allogeneic hematopoietic cell transplantation. Bone Marrow Transplant. 2017, 52, 1054–1056. [Google Scholar] [CrossRef] [Green Version]
- El Cheikh, J.; Massoud, R.; Abudalle, I.; Haffar, B.; Mahfouz, R.; A Kharfan-Dabaja, M.; Jisr, T.; Mougharbel, A.; Ibrahim, A.; Bazarbachi, A. Nivolumab salvage therapy before or after allogeneic stem cell transplantation in Hodgkin lymphoma. Bone Marrow Transplant. 2017, 52, 1074–1077. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Singh, A.K.; Porrata, L.F.; Aljitawi, O.; Lin, T.; Shune, L.; Ganguly, S.; McGuirk, J.P.; Abhyankar, S. Fatal GvHD induced by PD-1 inhibitor pembrolizumab in a patient with Hodgkin’s lymphoma. Bone Marrow Transplant. 2016, 51, 1268–1270. [Google Scholar] [CrossRef] [PubMed]
- Haverkos, B.M.; Abbott, D.; Hamadani, M.; Armand, P.; Flowers, M.E.; Merryman, R.; Kamdar, M.; Kanate, A.S.; Saad, A.; Mehta, A.; et al. PD-1 blockade for relapsed lymphoma post–allogeneic hematopoietic cell transplant: high response rate but frequent GVHD. Blood 2017, 130, 221–228. [Google Scholar] [CrossRef] [PubMed]
- Herbaux, C.; Gauthier, J.; Brice, P.; Drumez, E.; Ysebaert, L.; Doyen, H.; Fornecker, L.; Bouabdallah, K.; Manson, G.; Ghesquières, H.; et al. Efficacy and tolerability of nivolumab after allogeneic transplantation for relapsed Hodgkin lymphoma. Blood 2017, 129, 2471–2478. [Google Scholar] [CrossRef] [Green Version]
- Chan, T.S.Y.; Khong, P.-L.; Kwong, Y. Pembrolizumab for relapsed anaplastic large cell lymphoma after allogeneic haematopoietic stem cell transplantation: efficacy and safety. Ann. Hematol. 2016, 95, 1913–1915. [Google Scholar] [CrossRef]
- Albring, J.C.; Inselmann, S.; Sauer, T.; Schliemann, C.; Altvater, B.; Kailayangiri, S.; Rössig, C.; Hartmann, W.; Knorrenschild, J.R.; Sohlbach, K.; et al. PD-1 checkpoint blockade in patients with relapsed AML after allogeneic stem cell transplantation. Bone Marrow Transplant. 2017, 52, 317–320. [Google Scholar] [CrossRef]
- Holderried, T.A.W.; Fraccaroli, A.; Schumacher, M.; Heine, A.; Brossart, P.; Stelljes, M.; Klobuch, S.; Kröger, N.; Apostolova, P.; Finke, J.; et al. The role of checkpoint blockade after allogeneic stem cell transplantation in diseases other than Hodgkin’s Lymphoma. Bone Marrow Transplant. 2019, 54, 1662–1667. [Google Scholar] [CrossRef]
- Davids, M.S.; Kim, H.T.; Costello, C.L.; Herrera, A.F.; Locke, F.L.; Maegawa, R.O.; Savell, A.; Mazzeo, M.; Avigan, D.E.; Chen, Y.-B.; et al. A Phase I/Ib Study of Nivolumab for Relapsed Hematologic Malignancies after Allogeneic Hematopoietic Cell Transplantation (alloHCT). Blood 2018, 132, 705. [Google Scholar] [CrossRef]
- Kline, J.; Liu, H.; Michael, T.; Artz, A.S.; Godfrey, J.; Curran, E.K.; Stock, W.; Smith, S.M.; Bishop, M.R. Pembrolizumab for the Treatment of Disease Relapse Following Allogeneic Hematopoietic Cell Transplantation. Blood 2018, 132, 3415. [Google Scholar] [CrossRef]
- Bashey, A.; Medina, B.; Corringham, S.; Pasek, M.; Carrier, E.; Vrooman, L.; Lowy, I.; Solomon, S.R.; Morris, L.E.; Holland, H.K.; et al. CTLA4 blockade with ipilimumab to treat relapse of malignancy after allogeneic hematopoietic cell transplantation. Blood 2009, 113, 1581–1588. [Google Scholar] [CrossRef]
- Davids, M.S.; Kim, H.T.; Bachireddy, P.; Costello, C.; Liguori, R.; Savell, A.; Lukez, A.P.; Avigan, D.; Chen, Y.-B.; McSweeney, P.; et al. Ipilimumab for Patients with Relapse after Allogeneic Transplantation. New Engl. J. Med. 2016, 375, 143–153. [Google Scholar] [CrossRef] [PubMed]
- Khouri, I.F.; Scutti, J.A.B.; Milton, D.R.; Nicholas, C.; Pineda, M.; Jabbour, E.J.; Bassett, R.L.; Yadav, S.S.; Vence, L.M.; Allison, J.P.; et al. Durable Responses with Ipilimumab Plus Lenalidomide after Allogeneic and Autologous Stem Cell Transplantation for Patients with Lymphoid Malignancies. Blood 2018, 132, 4585. [Google Scholar] [CrossRef]
- Davila, M.L.; Riviere, I.; Wang, X.; Bartido, S.; Park, J.; Curran, K.; Chung, S.S.; Stefanski, J.; Borquez-Ojeda, O.; Olszewska, M.; et al. Efficacy and Toxicity Management of 19-28z CAR T Cell Therapy in B Cell Acute Lymphoblastic Leukemia. Sci. Transl. Med. 2014, 6, 224ra25. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, D.W.; Kochenderfer, J.N.; Stetler-Stevenson, M.; Cui, Y.K.; Delbrook, C.; A Feldman, S.; Fry, T.J.; Orentas, R.; Sabatino, M.; Shah, N.N.; et al. T cells expressing CD19 chimeric antigen receptors for acute lymphoblastic leukaemia in children and young adults: a phase 1 dose-escalation trial. Lancet 2015, 385, 517–528. [Google Scholar] [CrossRef]
- Gardner, R.; Wu, D.; Cherian, S.; Fang, M.; Hanafi, L.-A.; Finney, O.; Smithers, H.; Jensen, M.C.; Riddell, S.R.; Maloney, D.G.; et al. Acquisition of a CD19-negative myeloid phenotype allows immune escape of MLL-rearranged B-ALL from CD19 CAR-T-cell therapy. Blood 2016, 127, 2406–2410. [Google Scholar] [CrossRef] [Green Version]
- Park, J.H.; Geyer, M.B.; Brentjens, R.J. CD19-targeted CAR T-cell therapeutics for hematologic malignancies: interpreting clinical outcomes to date. Blood 2016, 127, 3312–3320. [Google Scholar] [CrossRef]
- Turtle, C.J.; Hanafi, L.-A.; Berger, C.; Gooley, T.A.; Cherian, S.; Hudecek, M.; Sommermeyer, D.; Melville, K.; Pender, B.; Budiarto, T.M.; et al. CD19 CAR-T cells of defined CD4+:CD8+ composition in adult B cell ALL patients. J. Clin. Investig. 2016, 126, 2123–2138. [Google Scholar] [CrossRef] [Green Version]
- Fraietta, J.A.; Lacey, S.F.; Orlando, E.J.; Pruteanu-Malinici, I.; Gohil, M.; Lundh, S.; Boesteanu, A.C.; Wang, Y.; O’Connor, R.S.; Hwang, W.-T.; et al. Determinants of response and resistance to CD19 chimeric antigen receptor (CAR) T cell therapy of chronic lymphocytic leukemia. Nat. Med. 2018, 24, 563–571. [Google Scholar] [CrossRef]
- Shah, N.N.; Fry, T.J. Mechanisms of resistance to CAR T cell therapy. Nat. Rev. Clin. Oncol. 2019, 16, 372–385. [Google Scholar] [CrossRef]
- Hamieh, M.; Dobrin, A.; Cabriolu, A.; Van Der Stegen, S.J.C.; Giavridis, T.; Mansilla-Soto, J.; Eyquem, J.; Zhao, Z.; Whitlock, B.M.; Miele, M.M.; et al. CAR T cell trogocytosis and cooperative killing regulate tumour antigen escape. Nature 2019, 568, 112–116. [Google Scholar] [CrossRef]
- Joyce, J.A.; Fearon, D.T. T cell exclusion, immune privilege, and the tumor microenvironment. Science 2015, 348, 74–80. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kochenderfer, J.N.; Dudley, M.E.; Carpenter, R.O.; Kassim, S.H.; Rose, J.J.; Telford, W.G.; Hakim, F.T.; Halverson, D.C.; Fowler, D.H.; Hardy, N.M.; et al. Donor-derived CD19-targeted T cells cause regression of malignancy persisting after allogeneic hematopoietic stem cell transplantation. Blood 2013, 122, 4129–4139. [Google Scholar] [CrossRef] [PubMed]
- Kochenderfer, J.N.; Dudley, M.E.; Kassim, S.H.; Somerville, R.P.; Carpenter, R.O.; Stetler-Stevenson, M.; Yang, J.C.; Phan, G.Q.; Hughes, M.S.; Sherry, R.M.; et al. Chemotherapy-Refractory Diffuse Large B-Cell Lymphoma and Indolent B-Cell Malignancies Can Be Effectively Treated With Autologous T Cells Expressing an Anti-CD19 Chimeric Antigen Receptor. J. Clin. Oncol. 2015, 33, 540–549. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brudno, J.N.; Somerville, R.P.; Shi, V.; Rose, J.J.; Halverson, D.C.; Fowler, D.H.; Gea-Banacloche, J.C.; Pavletic, S.Z.; Hickstein, D.D.; Lu, T.L.; et al. Allogeneic T Cells That Express an Anti-CD19 Chimeric Antigen Receptor Induce Remissions of B-Cell Malignancies That Progress After Allogeneic Hematopoietic Stem-Cell Transplantation Without Causing Graft-Versus-Host Disease. J. Clin. Oncol. 2016, 34, 1112–1121. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Galon, J.; Rossi, J.; Turcan, S.; Danan, C.; Locke, F.L.; Neelapu, S.S.; Miklos, D.B.; Bartlett, N.L.; Jacobson, C.A.; Braunschweig, I.; et al. Characterization of anti-CD19 chimeric antigen receptor (CAR) T cell-mediated tumor microenvironment immune gene profile in a multicenter trial (ZUMA-1) with axicabtagene ciloleucel (axi-cel, KTE-C19). J. Clin. Oncol. 2017, 35, 3025. [Google Scholar] [CrossRef]
- Gargett, T.; Yu, W.; Dotti, G.; Yvon, E.S.; Christo, S.N.; Hayball, J.D.; Lewis, I.D.; Brenner, M.K.; Brown, M. GD2-specific CAR T Cells Undergo Potent Activation and Deletion Following Antigen Encounter but can be Protected From Activation-induced Cell Death by PD-1 Blockade. Mol. Ther. 2016, 24, 1135–1149. [Google Scholar] [CrossRef] [Green Version]
- O’Rourke, D.M.; Nasrallah, M.P.; Desai, A.; Melenhorst, J.J.; Mansfield, K.; Morrissette, J.J.D.; Martinez-Lage, M.; Brem, S.; Maloney, E.; Shen, A.; et al. A single dose of peripherally infused EGFRvIII-directed CAR T cells mediates antigen loss and induces adaptive resistance in patients with recurrent glioblastoma. Sci. Transl. Med. 2017, 9, eaaa0984. [Google Scholar] [CrossRef] [Green Version]
- Moon, E.K.; Ranganathan, R.; Eruslanov, E.; Kim, S.; Newick, K.; O’Brien, S.; Lo, A.; Liu, X.; Zhao, Y.; Albelda, S.M. Blockade of Programmed Death 1 Augments the Ability of Human T Cells Engineered to Target NY-ESO-1 to Control Tumor Growth after Adoptive Transfer. Clin. Cancer Res. 2016, 22, 436–447. [Google Scholar] [CrossRef] [Green Version]
- Prosser, M.E.; E Brown, C.; Shami, A.F.; Forman, S.J.; Jensen, M.C. Tumor PD-L1 co-stimulates primary human CD8+ cytotoxic T cells modified to express a PD1:CD28 chimeric receptor. Mol. Immunol. 2012, 51, 263–272. [Google Scholar] [CrossRef]
- John, L.B.; Devaud, C.; Duong, C.P.; Yong, C.S.; Beavis, P.A.; Haynes, N.M.; Chow, M.T.; Smyth, M.J.; Kershaw, M.; Darcy, P.K. Anti-PD-1 Antibody Therapy Potently Enhances the Eradication of Established Tumors By Gene-Modified T Cells. Clin. Cancer Res. 2013, 19, 5636–5646. [Google Scholar] [CrossRef] [Green Version]
- Zou, F.; Lu, L.; Liu, J.; Xia, B.; Zhang, W.; Hu, Q.; Liu, W.; Zhang, Y.; Lin, Y.; Jing, S.; et al. Engineered triple inhibitory receptor resistance improves anti-tumor CAR-T cell performance via CD56. Nat. Commun. 2019, 10, 4109–4114. [Google Scholar] [CrossRef]
- Su, S.; Hu, B.; Shao, J.; Shen, B.; Du, J.; Du, Y.; Zhou, J.; Yu, L.; Zhang, L.; Chen, F.; et al. CRISPR-Cas9 mediated efficient PD-1 disruption on human primary T cells from cancer patients. Sci. Rep. 2016, 6, 20070. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rupp, L.J.; Schumann, K.; Roybal, K.T.; Gate, R.E.; Ye, C.J.; Lim, W.A.; Marson, A. CRISPR/Cas9-mediated PD-1 disruption enhances anti-tumor efficacy of human chimeric antigen receptor T cells. Sci. Rep. 2017, 7, 737. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Zhang, Y.; Cheng, C.; Cheng, A.; Zhang, X.; Li, N.; Xia, C.; Wei, X.; Liu, X.; Wang, H. CRISPR-Cas9-mediated multiplex gene editing in CAR-T cells. Cell Res. 2017, 27, 154–157. [Google Scholar] [CrossRef] [PubMed]
- Ren, J.; Liu, X.; Fang, C.; Jiang, S.; June, C.H.; Zhao, Y. Multiplex Genome Editing to Generate Universal CAR T Cells Resistant to PD1 Inhibition. Clin. Cancer Res. 2017, 23, 2255–2266. [Google Scholar] [CrossRef] [Green Version]
- Suarez, E.; Chang, D.-K.; Sun, J.; Sui, J.; Freeman, G.J.; Signoretti, S.; Zhu, Q.; Marasco, W.A. Chimeric antigen receptor T cells secreting anti-PD-L1 antibodies more effectively regress renal cell carcinoma in a humanized mouse model. Oncotarget 2016, 7, 34341–34355. [Google Scholar] [CrossRef] [Green Version]
- Rafiq, S.; Yeku, O.O.; Jackson, H.J.; Purdon, T.J.; Van Leeuwen, D.G.; Drakes, D.J.; Song, M.; Miele, M.M.; Li, Z.; Wang, P.; et al. Targeted delivery of a PD-1-blocking scFv by CAR-T cells enhances anti-tumor efficacy in vivo. Nat. Biotechnol. 2018, 36, 847–856. [Google Scholar] [CrossRef]
- Nakajima, M.; Sakoda, Y.; Adachi, K.; Nagano, H.; Tamada, K. Improved survival of chimeric antigen receptor-engineered T (CAR-T) and tumor-specific T cells caused by anti-programmed cell death protein 1 single-chain variable fragment-producing CAR-T cells. Cancer Sci. 2019, 110, 3079–3088. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kenderian, S.S.; Ruella, M.; Shestova, O.; Klichinsky, M.; Kim, M.; Porter, D.L.; June, C.H.; Gill, S. Identification of PD1 and TIM3 As Checkpoints That Limit Chimeric Antigen Receptor T Cell Efficacy in Leukemia. Boil. Blood Marrow Transplant. 2016, 22, S19–S21. [Google Scholar] [CrossRef]
- Li, A.M.; E Hucks, G.; Dinofia, A.M.; Seif, A.E.; Teachey, D.T.; Baniewicz, D.; Callahan, C.; Fasano, C.; McBride, B.; Gonzalez, V.; et al. Checkpoint Inhibitors Augment CD19-Directed Chimeric Antigen Receptor (CAR) T Cell Therapy in Relapsed B-Cell Acute Lymphoblastic Leukemia. Blood 2018, 132, 556. [Google Scholar] [CrossRef]
- Chong, E.A.; Melenhorst, J.J.; Lacey, S.F.; Ambrose, D.E.; Gonzalez, V.; Levine, B.L.; June, C.H.; Schuster, S.J. PD-1 blockade modulates chimeric antigen receptor (CAR)–modified T cells: refueling the CAR. Blood 2017, 129, 1039–1041. [Google Scholar] [CrossRef] [Green Version]
- Hill, B.T.; Roberts, Z.J.; Xue, A.; Rossi, J.M.; Smith, M.R. Rapid tumor regression from PD-1 inhibition after anti-CD19 chimeric antigen receptor T-cell therapy in refractory diffuse large B-cell lymphoma. Bone Marrow Transplant. 2019, 2019, 1–4. [Google Scholar] [CrossRef] [Green Version]
- Cao, Y.; Lu, W.; Sun, R.; Jin, X.; Cheng, L.; He, X.; Wang, L.; Yuan, T.; Lyu, C.; Zhao, M. Anti-CD19 Chimeric Antigen Receptor T Cells in Combination With Nivolumab Are Safe and Effective Against Relapsed/Refractory B-Cell Non-hodgkin Lymphoma. Front. Oncol. 2019, 9, 767. [Google Scholar] [CrossRef] [Green Version]
- Jaeger, U.; Worel, N.; McGuirk, J.P.; Riedell, P.A.; Fleury, I.; Borchmann, P.; Chu, J.; Abdelhady, A.M.; Forcina, A.; Pacaud, L.B.; et al. Portia: A Phase 1b Study Evaluating Safety and Efficacy of Tisagenlecleucel and Pembrolizumab in Patients with Relapsed/Refractory Diffuse Large B-Cell Lymphoma. Blood 2019, 134, 5325–5353. [Google Scholar] [CrossRef]
- Jacobson, C.A.; Locke, F.L.; Miklos, D.B.; Herrera, A.F.; Westin, J.R.; Lee, J.; Rossi, J.M.; Sun, J.; Zheng, L.; Avanzi, M.P.; et al. End of Phase 1 Results from Zuma-6: Axicabtagene Ciloleucel (Axi-Cel) in Combination with Atezolizumab for the Treatment of Patients with Refractory Diffuse Large B Cell Lymphoma. Biol. Blood Marrow Transplant. 2019, 25, S173. [Google Scholar] [CrossRef]
- Ardeshna, K.M.; Marzolini, M.A.V.; Norman, J.; Al-Hajj, M.; Thomas, S.; Faulkner, J.; Kotsopoulou, E.; Pule, M.; Peddareddigari, V.G.R.; Khokhar, N.Z.; et al. Phase 1/2 Study of AUTO3 the First Bicistronic Chimeric Antigen Receptor (CAR) Targeting CD19 and CD22 Followed By an Anti-PD1 in Patients with Relapsed/Refractory (r/r) Diffuse Large B Cell Lymphoma (DLBCL): Results of Cohort 1 and 2 of the Alexander Study. Blood 2019, 134, 246. [Google Scholar]
- Stadtmauer, E.A.; Fraietta, J.A.; Davis, M.M.; Cohen, A.D.; Weber, K.L.; Lancaster, E.; Mangan, P.A.; Kulikovskaya, I.; Gupta, M.; Chen, F.; et al. CRISPR-engineered T cells in patients with refractory cancer. Science 2020, 367, eaba7365. [Google Scholar] [CrossRef] [PubMed]
- Zhu, H.; You, Y.; Shen, Z.; Shi, L. EGFRvIII-CAR-T Cells with PD-1 Knockout Have Improved Anti-Glioma Activity. Pathol. Oncol. Res. 2020, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Kambhampati, S.; Gray, L.; Fakhri, B.; Lo, M.; Vu, K.; Arora, S.; Kaplan, L.; Ai, W.Z.; Andreadis, C. Immune-related Adverse Events Associated With Checkpoint Inhibition in the Setting of CAR T Cell Therapy: A Case Series. Clin. Lymphoma Myeloma Leuk. 2020, 20, e118–e123. [Google Scholar] [CrossRef] [Green Version]


{kind=link}
| n | Disease | Characteristics | Intervention | Response | irAEs (Grade) | GvHD (Grade) | Ref. |
|---|---|---|---|---|---|---|---|
31 | r/r HL/FL | retrospective multi-center study HL, n = 29; transformed FL, n = 1; FL + HL, n = 1 85% received ≥ 1 salvage therapy post allo-HCT before anti-PD-1 mAb prior GvHD NOS (19/31) | nivolumab (q2w, 3 mg/kg) n = 28 pembrolizumab (q3w, 200 mg) n = 3 first application 26 mo. after allo-HCT (median) | ORR/CR/PR/SD/PD: 77/50/27/10/13% OS: N/A PFS: 591 days | N/A | aGvHD (N/A, n = 6), overlap (N/A n = 4), cGvHD (N/A, n = 7) In 16/17 GvHD onset after 1–2 doses of CI 8 GvHD related deaths | [63] |
| 20 | r/r HL | retrospective multi-center study HL, n = 20 65% received ≥ 1 salvage therapy post allo-HCT prior aGvHD (10/20), cGvHD (3/20) | nivolumab (q2w, 3 mg/kg) first application 23 mo. after allo-HCT (median) | ORR/CR/PR/PD: 95/42/52/5% OS/PFS: 79/58% | cerebellar ataxia (II, n = 1) hepatitis (II, n = 7) | aGVHD (I, n = 1; III, n = 3; IV, n = 2) In 6/20 GvHD onset after 1 dose of CI 2 GvHD related deaths | [64] |
| 21 | r/r HM | retrospective multi-center study AML/MDS, n = 12; ALL, n = 2; NHL, n = 5; MF, n = 2 relapse after 1st allo-HCT, n = 15; 2nd, n = 5; 3rd, n = 1 prior aGvHD (13/21), cGvHD (7/21) | nivolumab (0.5 mg/kg, 3 mg/kg, 40 mg or 200 mg absolute), n = 5 nivolumab + DLI (0.5 mg/kg, 1 mg/kg, or 40 mg absolute), n = 5 nivo + ipilimumab (both 3 mg/kg), n = 1 ipilimumab (3 mg/kg, or 10 mg/kg), n = 10 first application 4.5 mo. after allo-HCT (median) | ORR/CR/PR/SD/PD: 43/14/29/4/48% ORR (nivolumab): 40% ORR (nivo + DLI): 80% ORR (ipilimumuab): 20% OS: 79 days PFS: N/A | N/A | 10/21 (any grade) 6/21 aGvHD III/IV or moderate/severe cGvHD; 5/5 GvHD in patients with nivo + DLI In 6/20 GvHD onset after 1 dose of CI 4 GvHD related deaths | [67] |
| 11 | r/r AML r/r (N)HL | phase I prospective single-center study AML/MDS, n = 8, lymphoma (DLBCL, HL), n = 3 no prior aGvHD > I or cGvHD | pembrolizumab (200 mg q3w) first application after allo-HCT N/A | ORR/CR/PR/SD/PD: 28/28/0/28/43% OS/PFS: N/A | 7/11 (any grade) pneumonitis (III-IV, n = 2), hyperthyrodism (III, n = 1), rash (II, n = 1) onset after 1-2 cycles of CI | none | [69] |
| 29 | r/r hematologic and solid malignancies | phase I multi-center study HL, n = 14; Myeloma, n = 6; AML, n = 2; CML, n = 2; CLL, n = 2; NHL, n = 1; breast cancer, n = 1; renal cell cancer, n = 1 no ongoing GvHD, no prior III/IV GvHD | ipilimumab (+ DLI if progressive after CI) single dose 0.1 mg/kg, n = 4 single dose 0.33 mg/kg, n = 3 single dose 0.66 mg/kg, n = 4 single dose 1 mg/kg, n = 3 single dose 3 mg/kg, n = 15 application after allo-HCT N/A | CR: 2/29 (cHL) PR: 1/29 (NHL) OS/PFS: N/A | polyarthropathy (III, n = 1); hyperthyrodism (I-II, n = 1); dyspnea on exertion (N/A); pneumonitis (IV, n = 1) | no grade III-IV time of GvHD onset after CI N/A GvHD related deaths N/A | [70] |
| 28 | r/r HM | phase I/Ib multicenter study AML, n = 12; HL, n = 7; NHL, n = 4; MDS, n = 2; MM, n = 1; MPN, n = 1; ALL, n = 1 72% received ≥ 1 salvage therapy post allo-HCT before anti-PD-1 mAb no ongoing GvHD, no prior III/IV aGvHD | ipilimumab (q3w, 4 courses) 3 mg/kg; n = 6 3 mg escalated to 10 mg/kg; n = 7 10 mg/kg; n = 15 first application 56 mo. after allo-HCT (median) | ORR/CR/PR/SD/PD 23/9/27/41% (10 mg/kg) no response with 3 mg/kg OS/PFS: N/A | 6/28 (any grade) pneumonitis (II-IV, n = 3) colitis (III, n = 1) ITP (II, n = 1) diarrhea (II, n = 1) death (n = 1) | with 10 mg/kg ipilimumab aGvHD gut (N/A, n = 1) cGvHD liver (N/A, n = 3) time of GvHD onset after CI N/A GvHD related deaths N/A | [71] |
| 28 | r/r HM | phase I/Ib multi-center study AML, n = 11; MDS n = 7; HL, n = 5; NHL, n = 3; MPN, n = 1; CLL, n = 1 64% received ≥ 1 salvage therapy post allo-HCT | nivolumab (q2w) 1 mg/kg (initial dose), n = 6 0.5 mg/kg (dose de-escalation), n = 8 0.5 mg/kg (initial dose), n = 14 3 mg/kg (dose escalation not realized due to toxicity) first application 21 mo. after allo-HCT (median) | with 1 mg/kg nivolumab CR/PR: 17/33% with 0.5 mg/kg nivolumab ORR/CR/PR/SD/PD 16/8/8/47/37% 6 mo. PFS/OS: 39/61% | with 1 mg/kg nivolumab sepsis/fatal ARDS (n = 1) fatal APS (n = 1) pneumonitis (III, n = 1) transaminitis (III, n = 1) bilirubinemia (III, n = 1) | with 1 mg/kg nivolumab aGvHD liver, gut (III, n = 2) with 0.5 mg/kg nivolumab new onset/worsening of GvHD (n = 10) aGvHD (N/A, n = 1) cGvHD (N/A, n = 7) aGvHD+ cGvHD (N/A, n = 2) time of GvHD onset after CI N/A GvHD related deaths N/A | [68] |
| 10 | r/r lymphoma | phase II multi-center study FL, n = 2; MCL, n = 3; THL, n = 1; DLBCL, n = 1; CLL, n = 2; ALCL, n = 1 no ongoing GvHD at study entry 44% had prior extensive cGvHD | lenalidomide (10 mg/day x 21d) + ipilimumab (3 mg/kg single-dose) 1st anti-CTLA-4 3d after completed 1st cycle of len repetition of len-cycle after 30d 2nd anti-CTLA-4 dose as before first application 29 mo. after allo- HCT (median) | ORR/CR/PR/SD 77/44/33/22% OS/PFS: N/A 4 mo RFS: 100% 12 mo RFS: 56% | hypothyrodism (II, n = 1) | cGvHD liver, mouth (N/A, n = 1) time of GvHD onset after CI N/A GvHD related deaths N/A | [72] |
| n | Disease | Characteristics | Intervention | Response | irAEs (Grade) | CRS/ICANS (Grade) | Ref. |
|---|---|---|---|---|---|---|---|
11 | r/r B-NHL | retrospective single-center study DLBCL (Stage III-IV), n = 10 Burkitt’s lymphoma, n = 1 | CD19 CAR + nivolumab (3 mg/kg single-dose) anti-PD-1 applied on d3 after CAR infusion | ORR/CR/PR/NR: 82/46/36/18% PFS: 6 (1–14 months) | no grade III-IV toxicity | CRS (I, n = 3; II, n = 6) ICANS (N/A, n = 1) | [103] |
| 14 | r/r B-ALL r/r B-NHL | retrospective single-center study B-ALL, n = 13 B-lymphoblastic lymphoma, n = 1 | CD19 CAR + pembrolizumab (200 mg, q3w) CD19 CAR + nivolumab (3 mg/kg, q3w) anti-PD-1 application ≧ 14d after CAR T cell infusion, median time after CAR T cell infusion N/A | ORR/CR/PR/PD: 43/14/29/7% PFS: N/A | pancreatitis (N/A, n = 1) hypothyroidism (N/A, n = 1) urticaria (N/A, n = 1) arthropathy (N/A, n = 1) no grade V toxicities | CRS (N/A, n = 3) | [100] |
| 4 | r/r DLBCL | phase 1b prospective multi-center study (PORTIA) DLBCL, n = 4 | CD19 CAR + pembrolizumab (200 mg, q3w, 6 courses) first anti-PD-1 application on d15 after CAR T cell infusion | N/A | none | CRS (N/A, n = 1) | [104] |
| 12 | r/r DLBCL | phase 1 prospective multi-center study (ZUMA-6) DLBCL, n = 12 | CD19 CAR + atezolizumab (1200 mg, q3w, 4 courses) first anti-PD-L1 application on d21 (cohort 1, n = 3), d14 (cohort 2, n = 3), d1 (cohort 3, n = 6) after CAR infusion | ORR/CR/PR: 90/60/30% PFS: N/A | N/A | CRS (≥ III, n = 3) ICANS (≥ III, n = 6) | [105] |
| 11 | r/r DLBCL | phase 1/2 prospective multi-center study (ALEXANDER) DLBCL NOS, n = 4 DLBCL transformed from FL/MZL, n = 7 | AUTO-3 CD19/CD22 CAR mono (n = 4) AUTO-3 CAR + pembrolizumab (200 mg, q3w) n = 7 first anti-PD-1 application on d14 Cohort 1: 50 × 106 AUTO3, n = 7 Cohort 2: 150 × 106 AUTO3, n = 4 | Cohort 1: 50 × 106 AUTO3 ORR/CR/PR: 57/29/28% PFS: N/A Cohort 2: 150 × 106 AUTO3 N/A | N/A | CRS (I, n = 3) ICANS (III, n = 1) | [106] |
| Clinical Trial | Phase | Disease | Intervention | Sponsor |
|---|---|---|---|---|
NCT02981914 | I | r/r HL, B-NHL, AML, MDS after allo-HCT | pembrolizumab (q3w, 200 mg) | University of Chicago |
| NCT03286114 | IB | r/r MDS, AML, ALL after allo-HCT | pembrolizumab (q3w, 200 mg) | University of Michigan Rogel Cancer Center |
| NCT04361058 | I | r/r high risk AML, MDS after allo-HCT | nivolumab (q2w, 0.25 mg/kg, 4 courses) | SCRI Development Innovations, LLC |
| NCT02890329 | I | r/r AML(+MRC), MDS after allo-HCT | decitabine + ipilimumab (q4w, dose N/A) priming: decitabine (d1-5 of 28 days) induction: decitabine (d1-5) + ipilimumab (d1); 4 courses maintenance: decitabine (d1-5) + ipilimumab (d1); 4 courses | National Cancer Institute (NCI) |
| NCT03588936 | I | r/r AL, CL, MDS, lymphoma after allo-HCT | tocilizumab (8 mg/kg on day 0 and 29) + nivolumab (q2w, 0.25 or 0.5 mg/kg on day 1; up to 4 courses) | Medical College of Wisconsin |
| NCT03146468 | II | r/r hematologic disease after allo-HCT | nivolumab (q2w, 3 mg/kg) | Melbourne Health |
| NCT01822509 | I/IB | r/r AML, MDS, MPN, ALL, CLL, CML, (N)HL, MM after allo-HCT | nivolumab or ipilimumab induction: nivolumab (q2w, dose N/A, 8 courses) or ipilimumab (q3w, dose N/A; 4 courses) maintenance: nivolumab (q2w, dose N/A, up to a total of 60 weeks) or ipilimumab (q12w, dose N/A; 4 courses) | National Cancer Institute (NCI) |
| NCT03600155 | IB | r/r high risk AML, MDS after allo-HCT | nivolumab or ipilimumab or nivolumab + ipilimumab Arm A: nivolumab (q4w, d1+15, dose N/A 6 courses) ≥ six weeks post allo-HCT Arm B: ipilimumab (q3w, d1, dose N/A, 6 courses) ≥ six weeks post allo-HCT Arm C; nivolumab (q6w, d1,14,28, dose N/A 6 courses) + ipilimumab (q6w, d1, dose N/A, 6 courses) ≥ six weeks post allo-HCT | M.D. Anderson Cancer Center |
| NCT00586391 | I | B-NHL, CLL, ALL | CD19CAR-28-zeta T cells Dose Level 1: 2 × 107 T cells/m2 Dose Level 2: 1 × 108 T cells/m2 Dose Level 3: 2 × 108 T cells/m2 ± ipilimumab (once in week 2 after CAR infusion, dose N/A, only in patients with low/intermediate grade leukemia/ lymphoma) | Baylor College of Medicine |
| NCT03630159 | IB | r/r DLBCL | tisagenlecleucel + pembrolizumab timing and dose N/A | Novartis Pharmaceuticals |
| NCT03630159 | I/II | r/r DLBCL | axicabtagene ciloleucel + atezolizumab timing and dose N/A | Kite, A Gilead Company |
| NCT03287817 | I/II | r/r DLBCL | AUTO-3 (50 × 106 to 900 × 106 CD19/CD22 CAR T cells) ± pembrolizumab timing and dose N/A | Autolus Limited |
| NCT04134325 | I | r/r HL after CAR T cell therapy | pembrolizumab (q3w, 200 mg) or nivolumab (q2w, 240 mg or q4w, 480 mg) | UNC Lineberger Comprehensive Cancer Center |
| NCT02650999 | I/II | r/r DLBCL, FL, MCL after CAR T cell therapy | pembrolizumab (timing N/A, 200 mg) | Abramson Cancer Center of the University of Pennsylvania |
| NCT04205409 | II | r/r CLL, DLBCL, FL, MZL, NHL, MM after CAR T cell therapy | nivolumab (q4w, dose N/A) | University of Washington |
| NCT04337606 | I/II | r/r NHL after CAR T cell therapy | cohort 1: chidamide (q3w, 10 mg on d1-5 and 20 mg on d8,11,15,18) + decitabine (q3w, 10 mg on d1-5) cohort 2: decitabine (q3w, 10 mg on d1-5) + camrelizumab (q3w, 200 mg on d6) | Chinese PLA General Hospital |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Schmitz, F.; Wolf, D.; Holderried, T.A.W. The Role of Immune Checkpoints after Cellular Therapy. Int. J. Mol. Sci. 2020, 21, 3650. https://doi.org/10.3390/ijms21103650
Schmitz F, Wolf D, Holderried TAW. The Role of Immune Checkpoints after Cellular Therapy. International Journal of Molecular Sciences. 2020; 21(10):3650. https://doi.org/10.3390/ijms21103650
Chicago/Turabian StyleSchmitz, Friederike, Dominik Wolf, and Tobias A.W. Holderried. 2020. "The Role of Immune Checkpoints after Cellular Therapy" International Journal of Molecular Sciences 21, no. 10: 3650. https://doi.org/10.3390/ijms21103650
APA StyleSchmitz, F., Wolf, D., & Holderried, T. A. W. (2020). The Role of Immune Checkpoints after Cellular Therapy. International Journal of Molecular Sciences, 21(10), 3650. https://doi.org/10.3390/ijms21103650