Intrinsic Disorder in Tetratricopeptide Repeat Proteins




Abstract
:1. Introduction
2. Results and Discussion
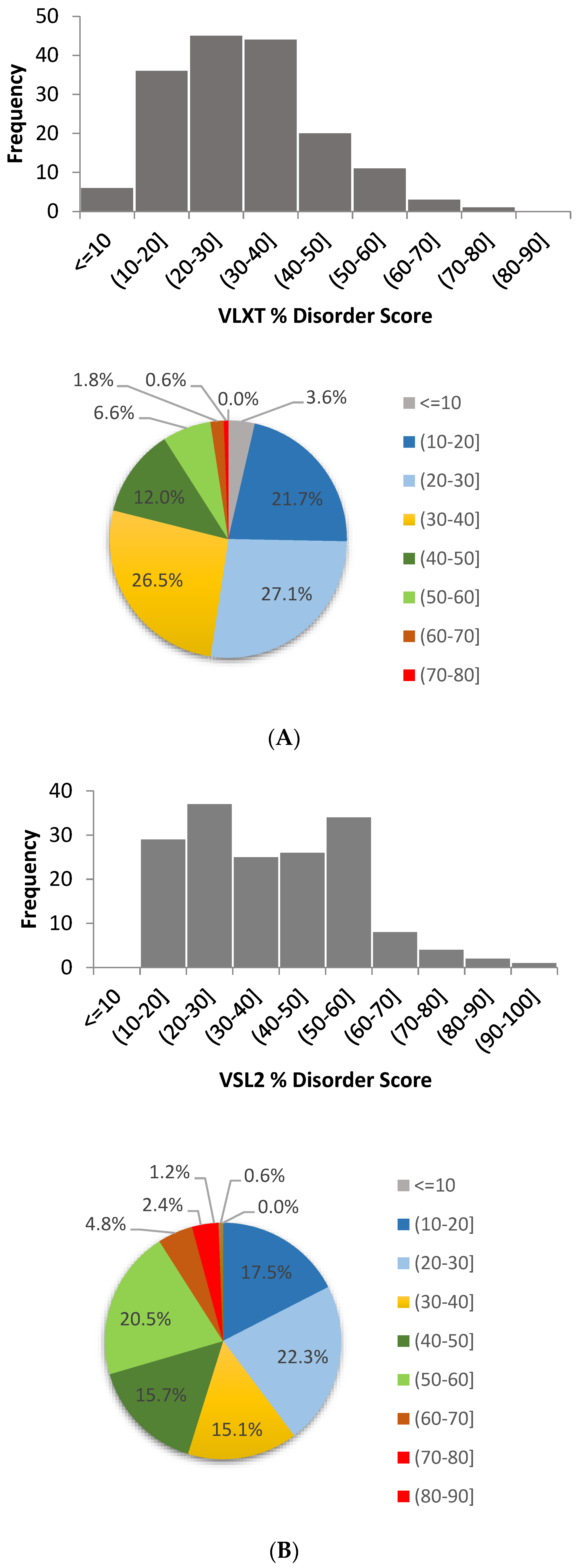
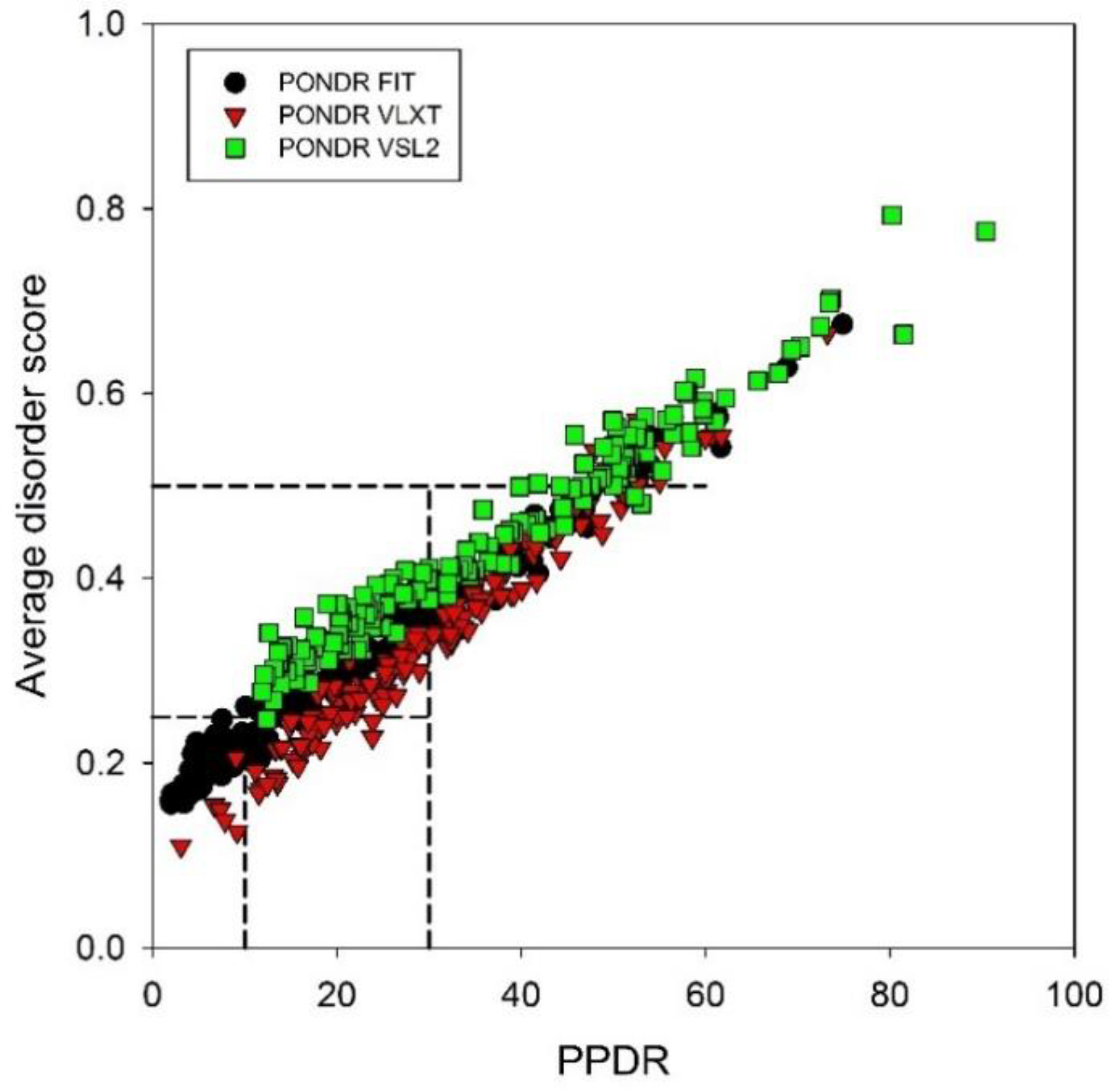
2.1. Per-Residue Intrinsic Disorder Predisposition of Human TPR Proteins
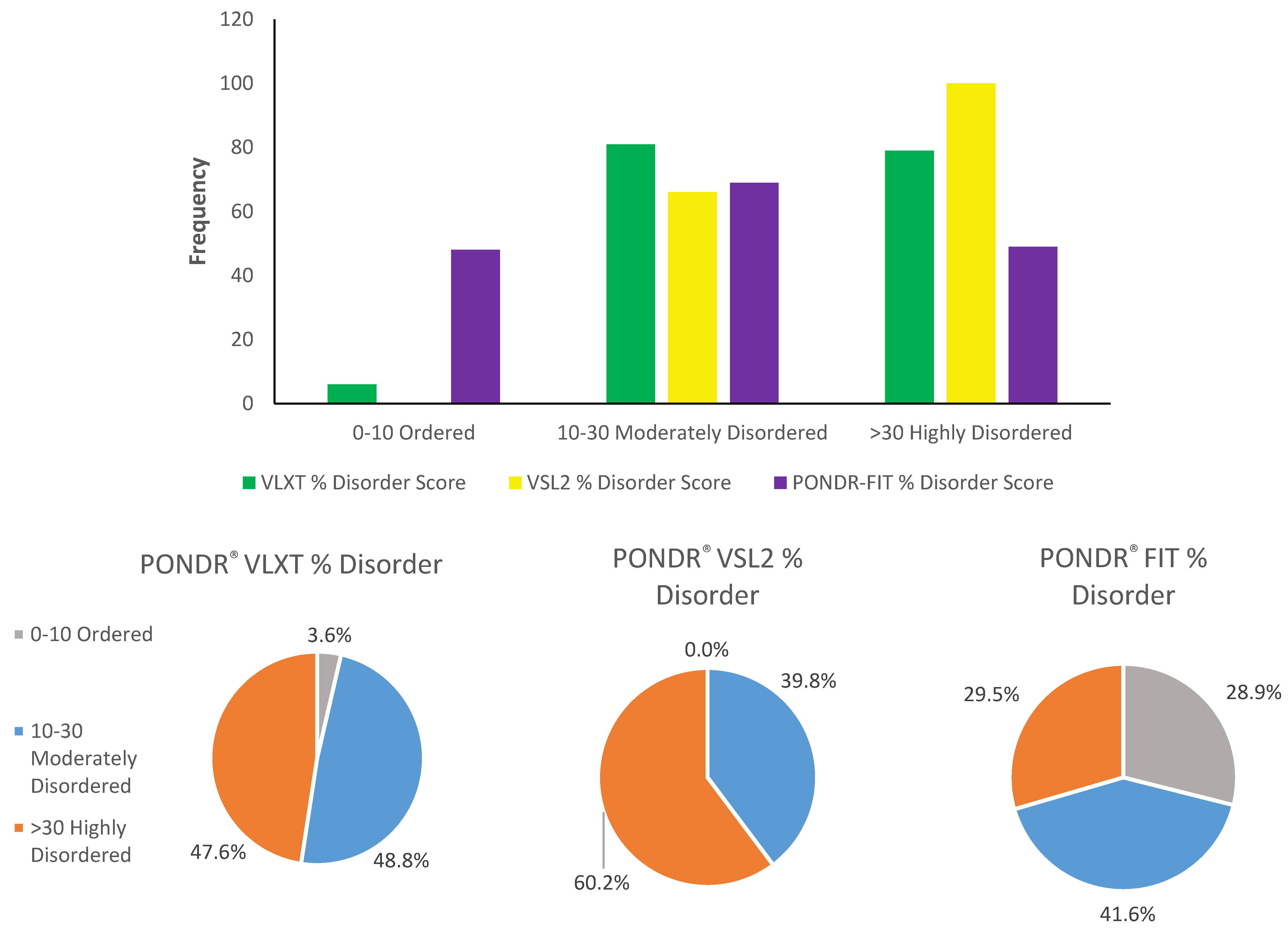
2.2. Binary Classification of the Intrinsic Disorder Status of Human TPR Proteins
2.3. Analysis of the Intrinsic Disorder-Based Functionality of Human TPR Proteins
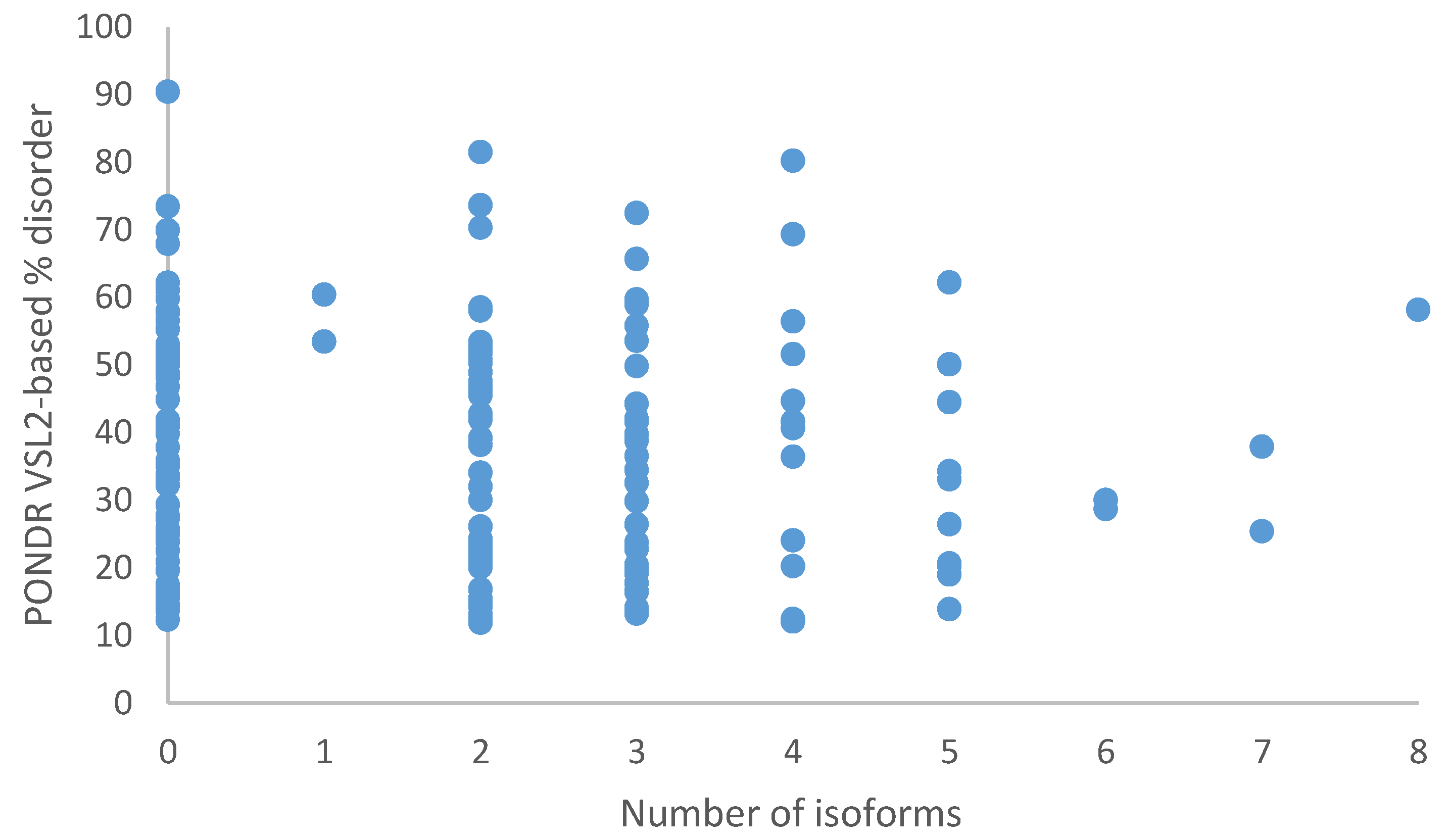
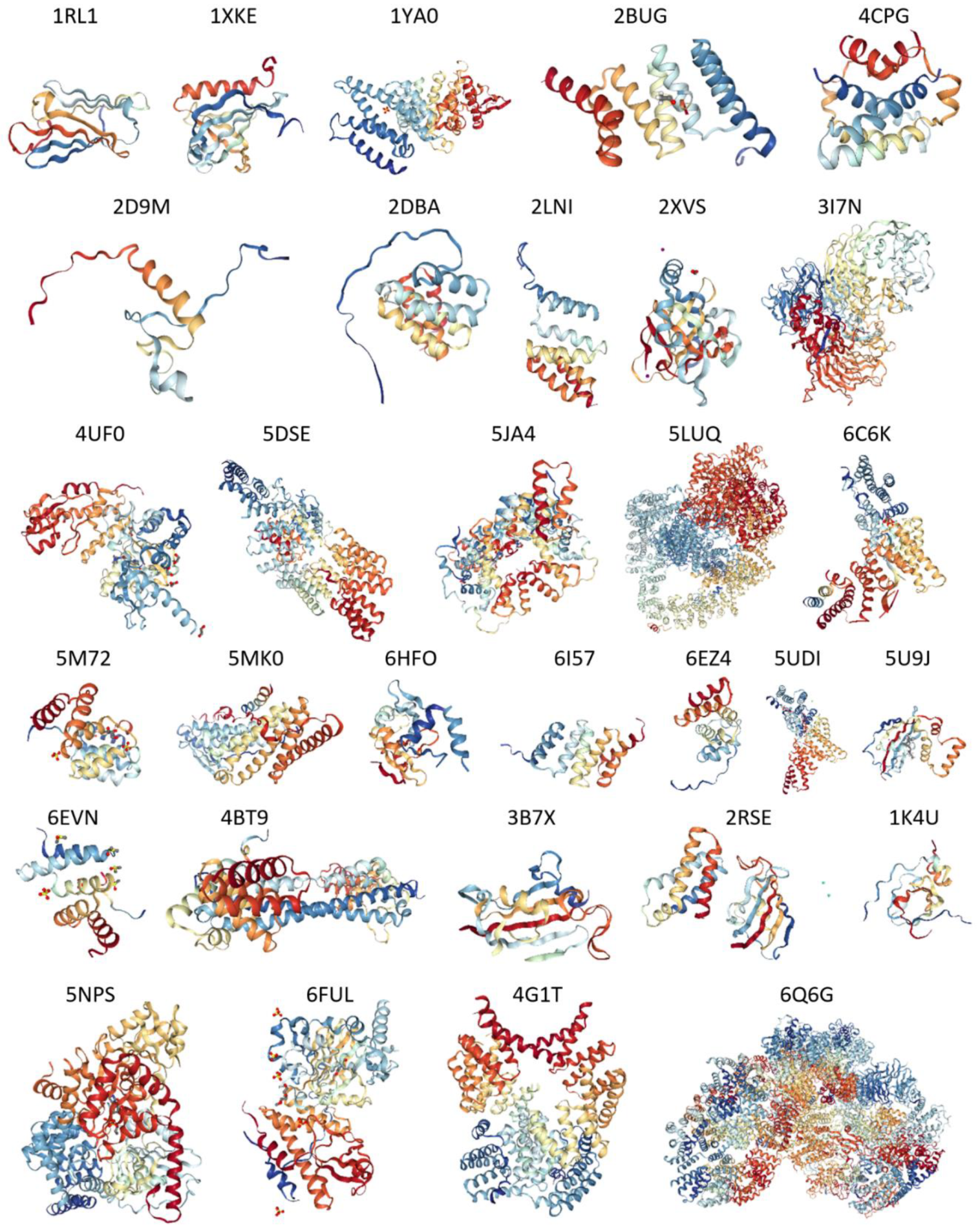
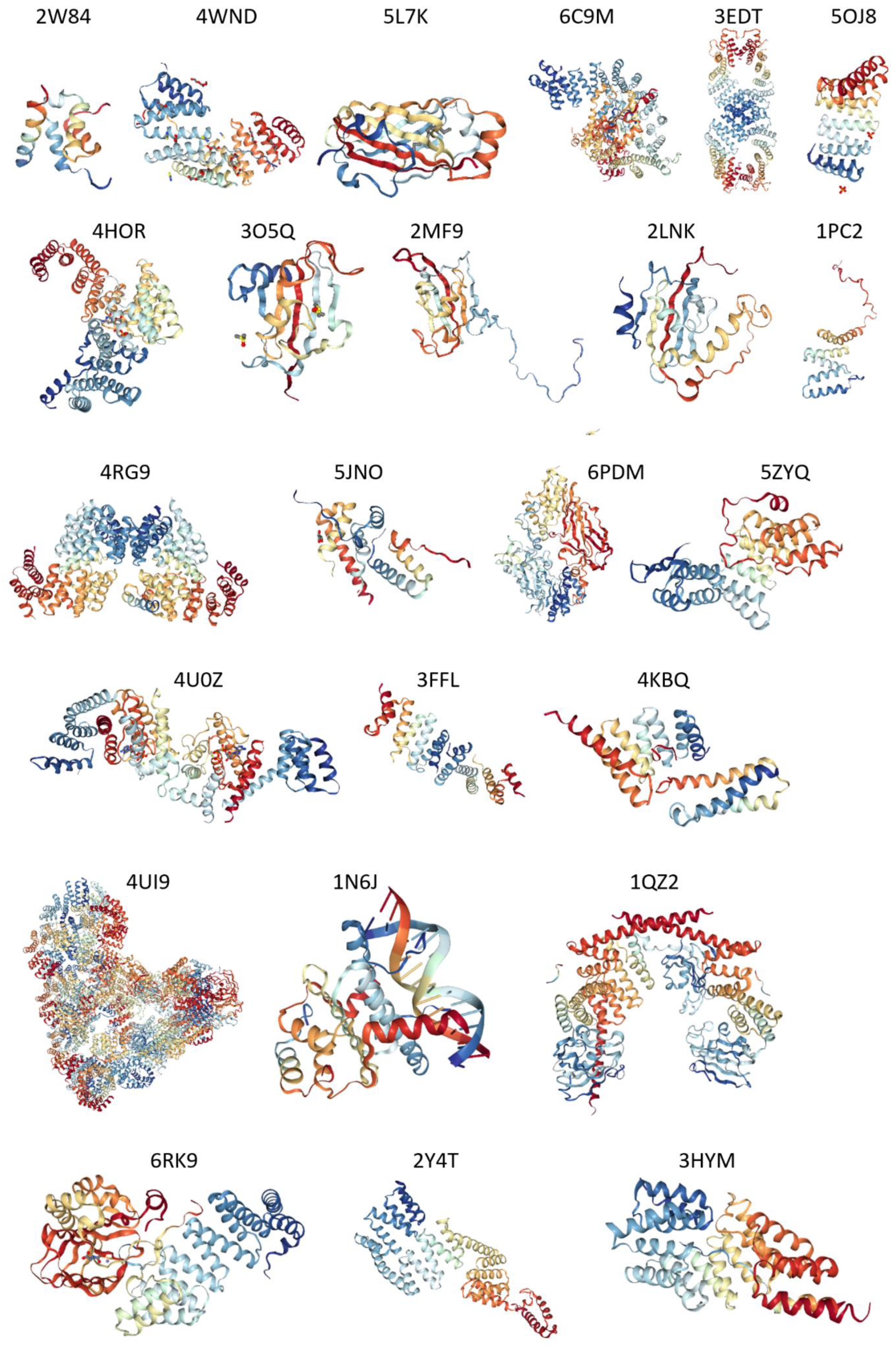
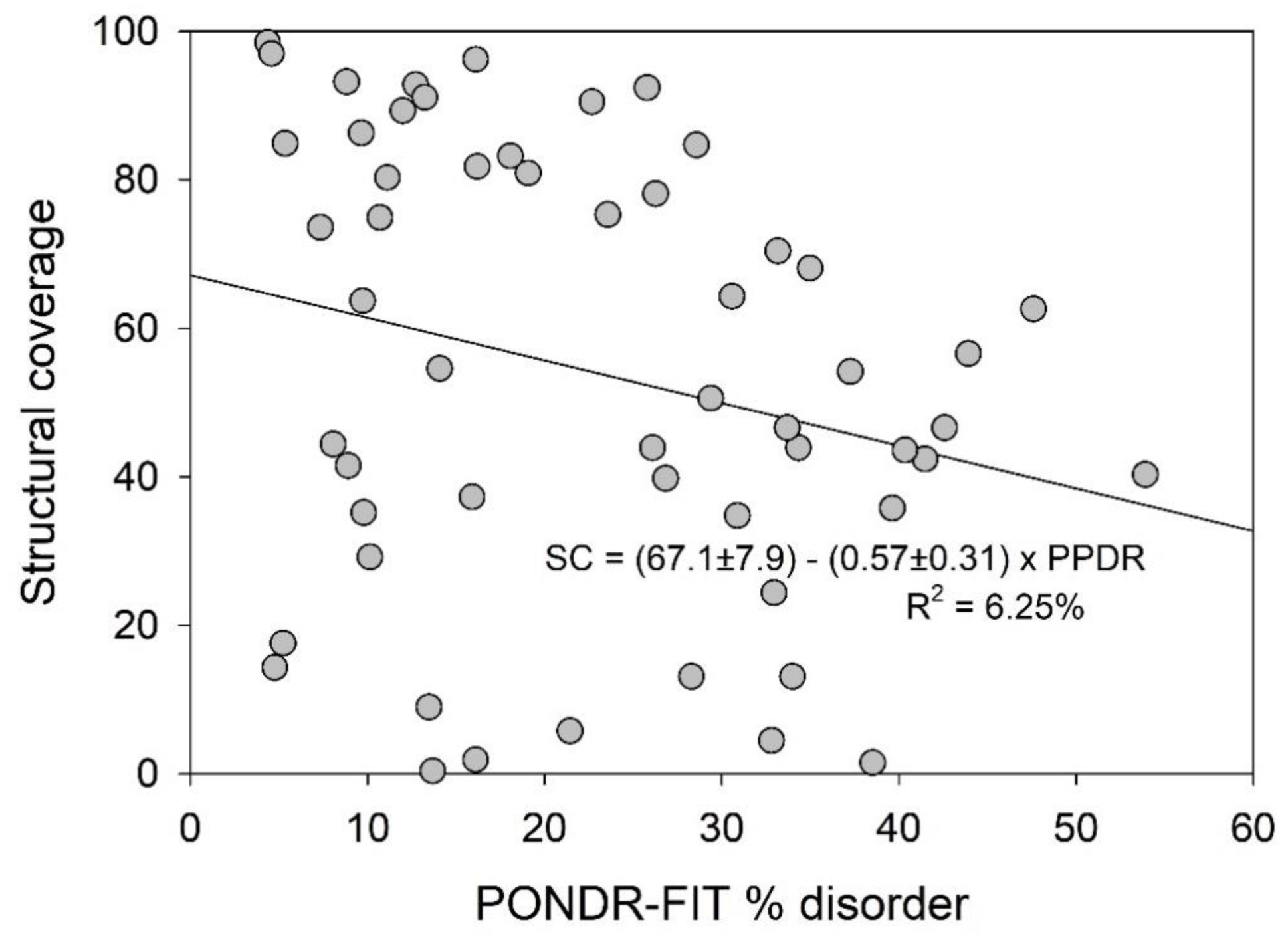
2.4. Structural Properties of Human TPR Proteins
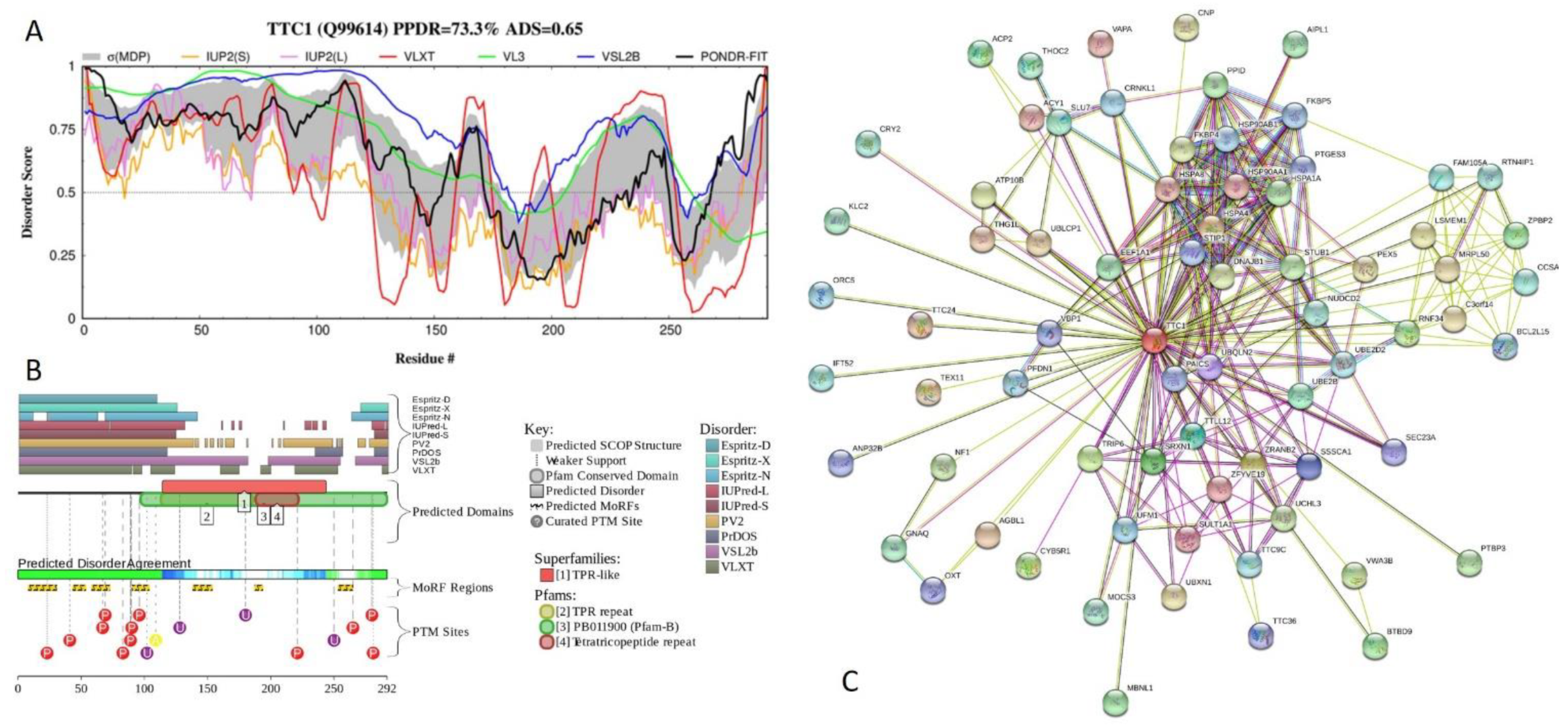
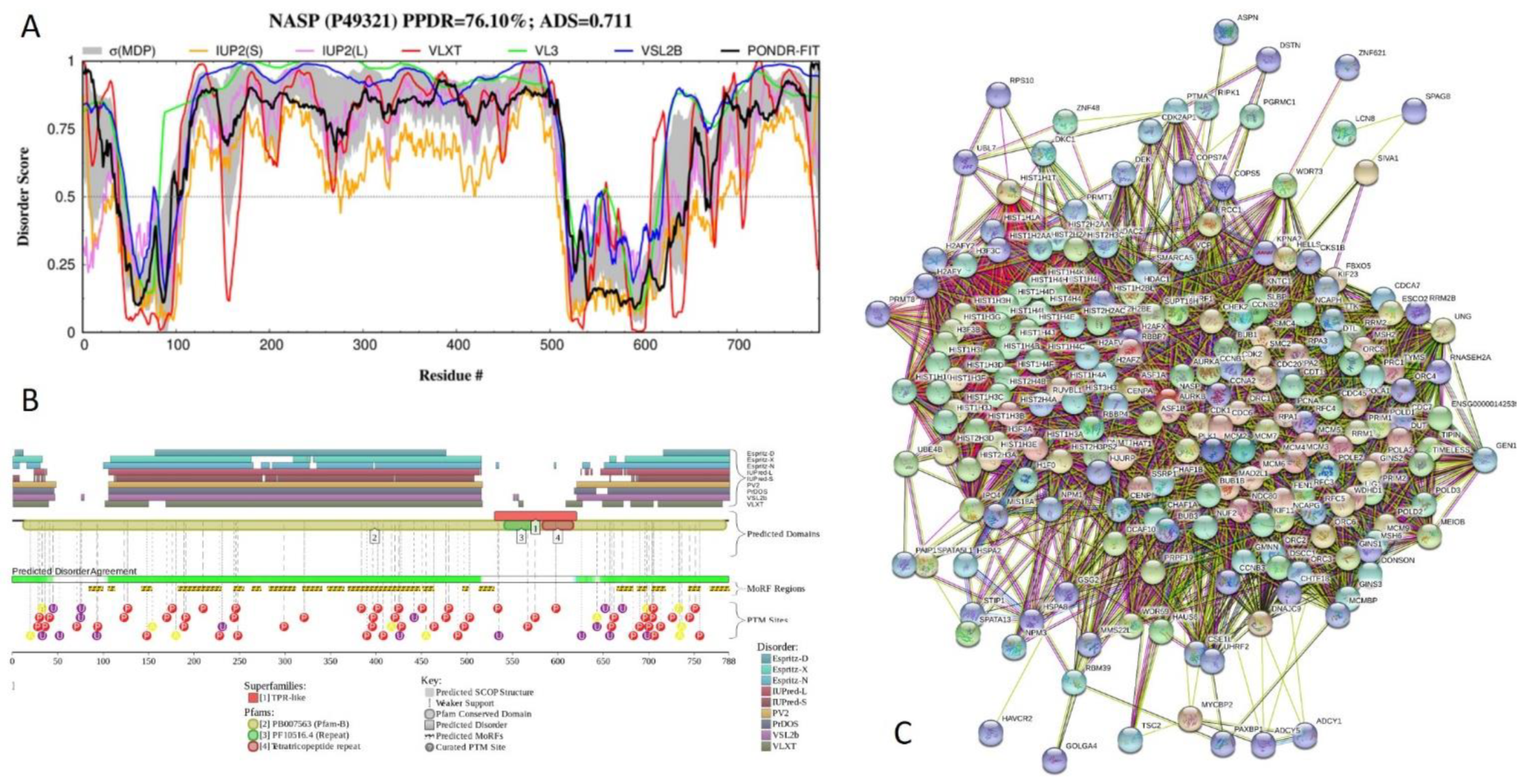
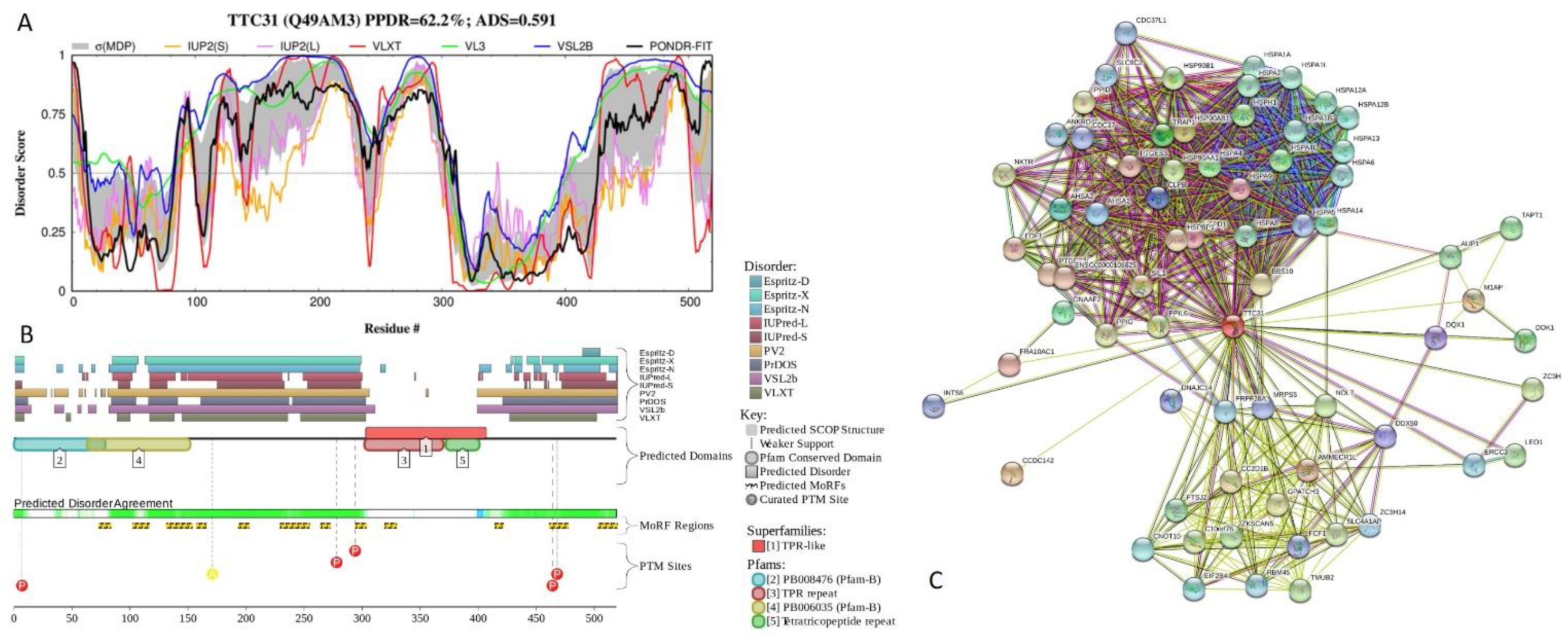
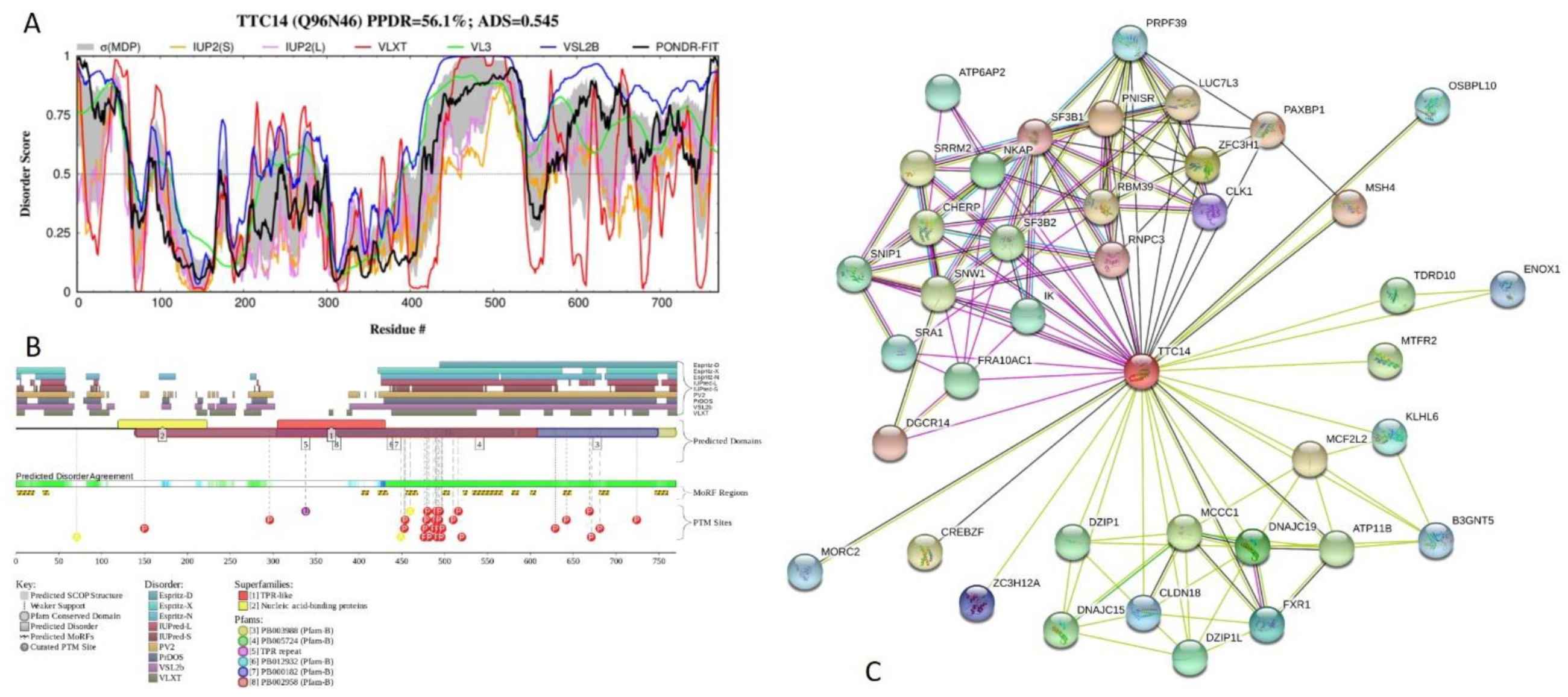
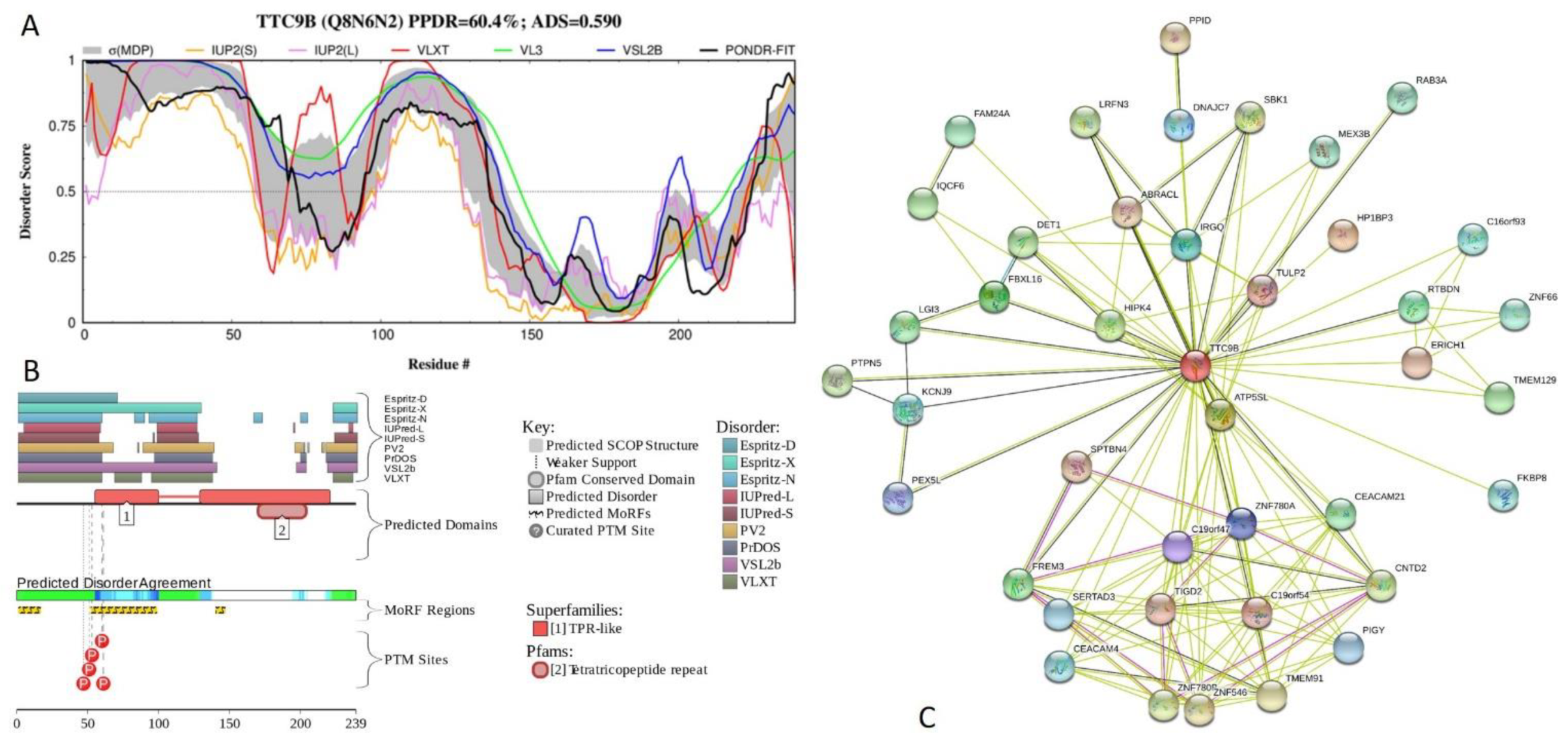
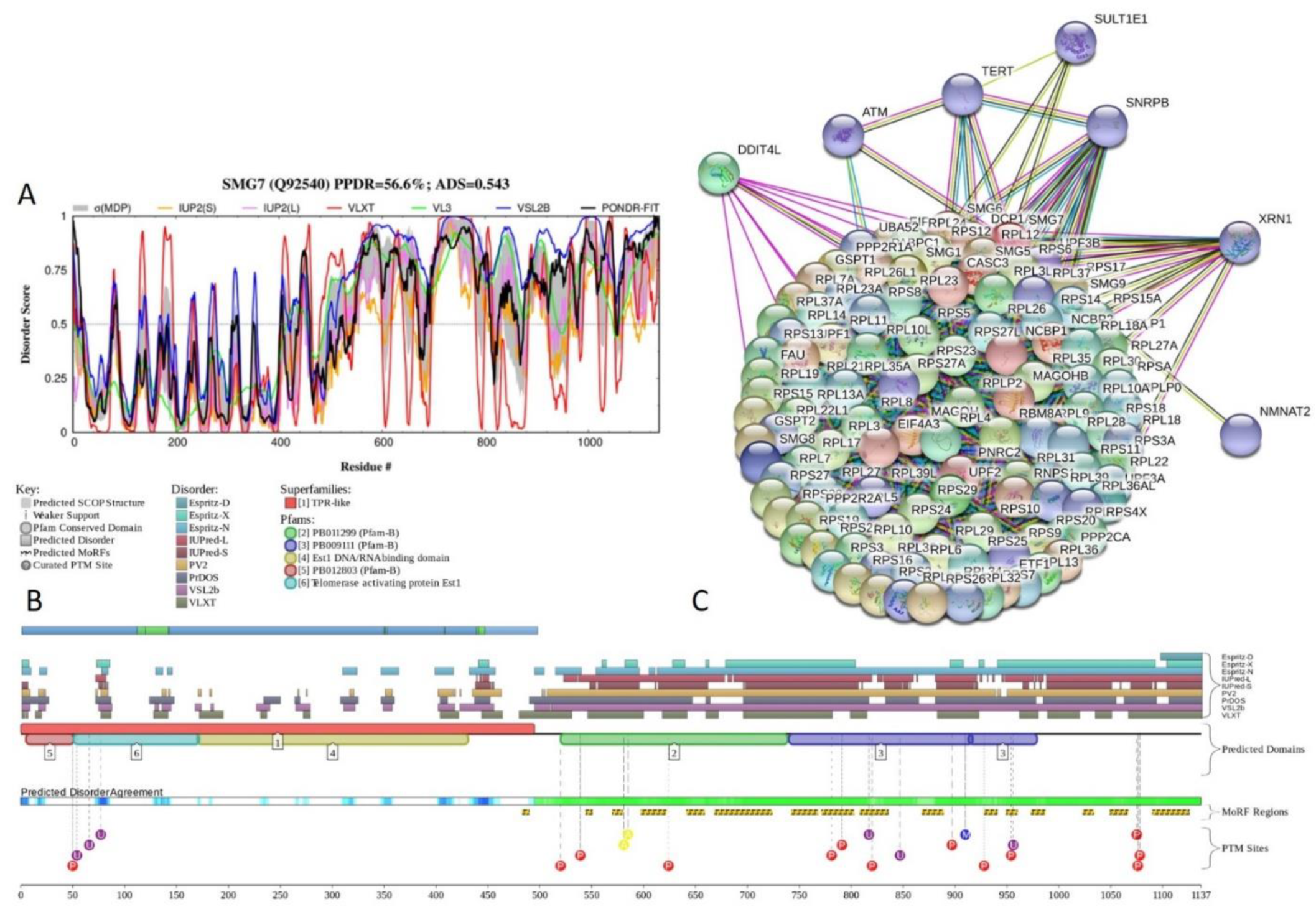
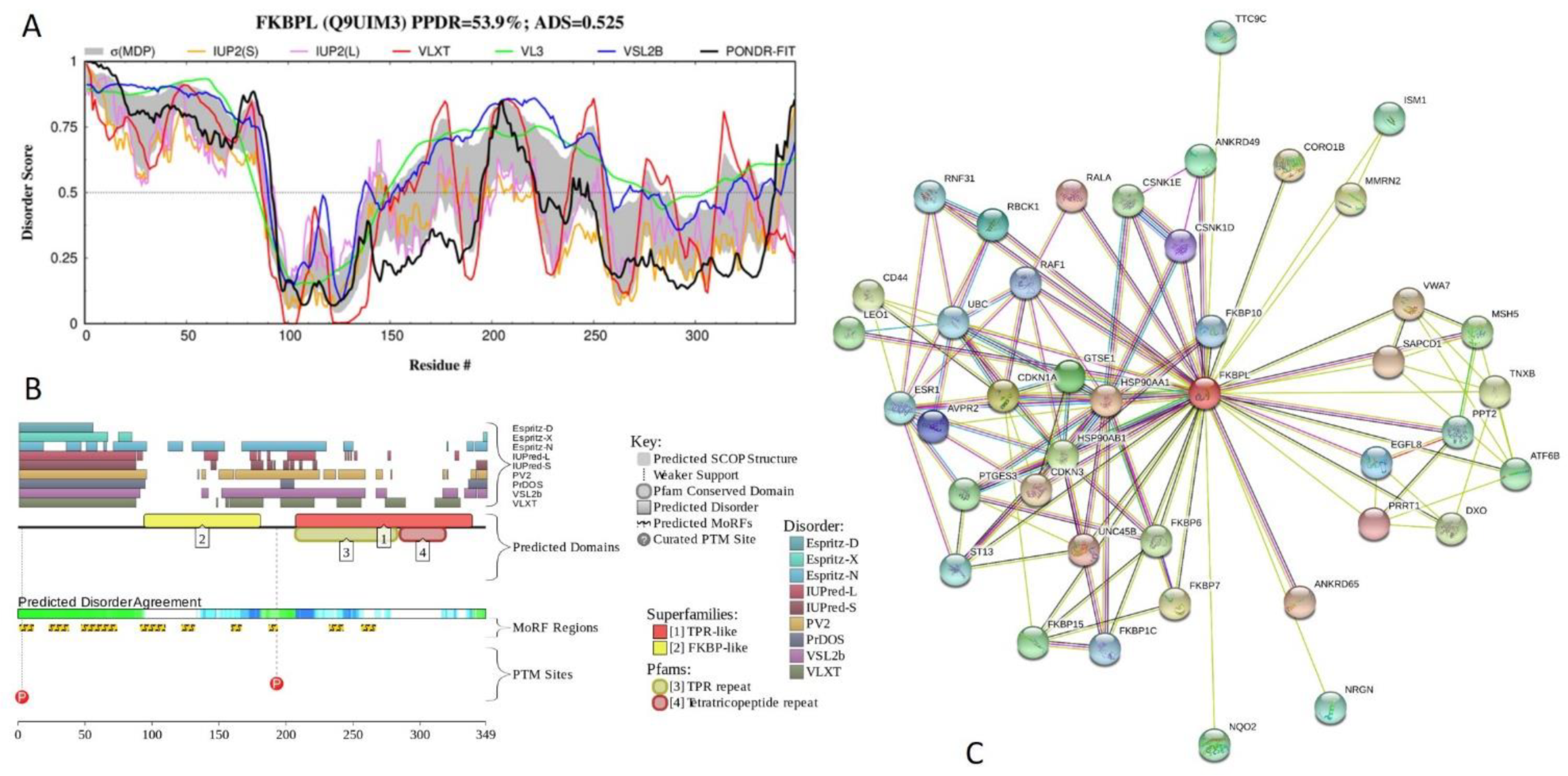
2.5. Functional Analysis of Most Disordered Human TPR Proteins
3. Materials and Methods
3.1. Datasets
3.2. Computational Characterization of Intrinsic Disorder in Human TPR Proteins
3.3. Computational Evaluation of Interactability of the Human TPR Proteins
4. Conclusions
Supplementary Materials
Author Contributions
Acknowledgments
Conflicts of Interest
References
- Fischer, E. Einfluss der configuration auf die wirkung der enzyme. Ber. Dt. Chem. Ges. 1894, 27, 2985–2993. [Google Scholar] [CrossRef] [Green Version]
- Landsteiner, K. The Specificity of Serological Reactions; C. C. Thomas: Baltimore, MD, USA, 1936. [Google Scholar]
- Wright, P.E.; Dyson, H.J. Intrinsically unstructured proteins: Re-assessing the protein structure-function paradigm. J. Mol. Biol. 1999, 293, 321–331. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Uversky, V.N.; Gillespie, J.R.; Fink, A.L. Why are “natively unfolded” proteins unstructured under physiologic conditions? Proteins 2000, 41, 415–427. [Google Scholar] [CrossRef]
- Dunker, A.K.; Lawson, J.D.; Brown, C.J.; Williams, R.M.; Romero, P.; Oh, J.S.; Oldfield, C.J.; Campen, A.M.; Ratliff, C.M.; Hipps, K.W.; et al. Intrinsically disordered protein. J. Mol. Graph. Model. 2001, 19, 26–59. [Google Scholar] [CrossRef] [Green Version]
- Tompa, P. Intrinsically unstructured proteins. Trends Biochem. Sci. 2002, 27, 527–533. [Google Scholar] [CrossRef]
- Andreeva, A.; Howorth, D.; Chothia, C.; Kulesha, E.; Murzin, A.G. SCOP2 prototype: A new approach to protein structure mining. Nucleic Acids Res. 2014, 42, D310–D314. [Google Scholar] [CrossRef]
- Dunker, A.K.; Obradovic, Z.; Romero, P.; Garner, E.C.; Brown, C.J. Intrinsic protein disorder in complete genomes. Genome Inf. Ser Workshop Genome Inf. 2000, 11, 161–171. [Google Scholar]
- Uversky, V.N. The mysterious unfoldome: Structureless, underappreciated, yet vital part of any given proteome. J. Biomed. Biotechnol. 2010, 2010, 568068. [Google Scholar] [CrossRef]
- Ward, J.J.; Sodhi, J.S.; McGuffin, L.J.; Buxton, B.F.; Jones, D.T. Prediction and functional analysis of native disorder in proteins from the three kingdoms of life. J. Mol. Biol. 2004, 337, 635–645. [Google Scholar] [CrossRef]
- Uversky, V.N.; Dunker, A.K. Understanding protein non-folding. Biochim. Biophys. Acta 2010, 1804, 1231–1264. [Google Scholar] [CrossRef] [Green Version]
- Xue, B.; Dunker, A.K.; Uversky, V.N. Orderly order in protein intrinsic disorder distribution: Disorder in 3500 proteomes from viruses and the three domains of life. J. Biomol. Struct. Dyn. 2012, 30, 137–149. [Google Scholar] [CrossRef]
- Peng, Z.; Yan, J.; Fan, X.; Mizianty, M.J.; Xue, B.; Wang, K.; Hu, G.; Uversky, V.N.; Kurgan, L. Exceptionally abundant exceptions: Comprehensive characterization of intrinsic disorder in all domains of life. Cell. Mol. Life Sci. 2015, 72, 137–151. [Google Scholar] [CrossRef]
- Oldfield, C.J.; Cheng, Y.; Cortese, M.S.; Brown, C.J.; Uversky, V.N.; Dunker, A.K. Comparing and combining predictors of mostly disordered proteins. Biochemistry 2005, 44, 1989–2000. [Google Scholar] [CrossRef] [PubMed]
- Dunker, A.K.; Brown, C.J.; Lawson, J.D.; Iakoucheva, L.M.; Obradovic, Z. Intrinsic disorder and protein function. Biochemistry 2002, 41, 6573–6582. [Google Scholar] [CrossRef] [Green Version]
- Dunker, A.K.; Brown, C.J.; Obradovic, Z. Identification and functions of usefully disordered proteins. Adv. Protein. Chem. 2002, 62, 25–49. [Google Scholar] [PubMed]
- Dunker, A.K.; Obradovic, Z. The protein trinity--linking function and disorder. Nat. Biotechnol. 2001, 19, 805–806. [Google Scholar] [CrossRef] [PubMed]
- Dyson, H.J.; Wright, P.E. Coupling of folding and binding for unstructured proteins. Curr. Opin. Struct. Biol. 2002, 12, 54–60. [Google Scholar] [CrossRef]
- Dyson, H.J.; Wright, P.E. Intrinsically unstructured proteins and their functions. Nat. Rev. Mol. Cell Biol. 2005, 6, 197–208. [Google Scholar] [CrossRef] [PubMed]
- Van der Lee, R.; Buljan, M.; Lang, B.; Weatheritt, R.J.; Daughdrill, G.W.; Dunker, A.K.; Fuxreiter, M.; Gough, J.; Gsponer, J.; Jones, D.T.; et al. Classification of intrinsically disordered regions and proteins. Chem. Rev. 2014, 114, 6589–6631. [Google Scholar] [CrossRef]
- Uversky, V.N. Unusual biophysics of intrinsically disordered proteins. Biochim Biophys Acta 2013, 1834, 932–951. [Google Scholar] [CrossRef]
- Uversky, V.N. A decade and a half of protein intrinsic disorder: Biology still waits for physics. Protein Sci. 2013, 22, 693–724. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Uversky, V.N. Intrinsic disorder-based protein interactions and their modulators. Curr. Pharm. Des. 2013, 19, 4191–4213. [Google Scholar] [CrossRef] [PubMed]
- Jakob, U.; Kriwacki, R.; Uversky, V.N. Conditionally and transiently disordered proteins: Awakening cryptic disorder to regulate protein function. Chem. Rev. 2014, 114, 6779–6805. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dunker, A.K.; Silman, I.; Uversky, V.N.; Sussman, J.L. Function and structure of inherently disordered proteins. Curr. Opin. Struct. Biol. 2008, 18, 756–764. [Google Scholar] [CrossRef]
- Tokuriki, N.; Oldfield, C.J.; Uversky, V.N.; Berezovsky, I.N.; Tawfik, D.S. Do viral proteins possess unique biophysical features? Trends Biochem. Sci. 2009, 34, 53–59. [Google Scholar] [CrossRef]
- Xue, B.; Williams, R.W.; Oldfield, C.J.; Dunker, A.K.; Uversky, V.N. Archaic chaos: Intrinsically disordered proteins in Archaea. BMC Syst. Biol. 2010, 4 (Suppl. 1), S1. [Google Scholar] [CrossRef] [Green Version]
- Tompa, P.; Dosztanyi, Z.; Simon, I. Prevalent structural disorder in E. coli and S. cerevisiae proteomes. J. Proteome Res. 2006, 5, 1996–2000. [Google Scholar] [CrossRef]
- Krasowski, M.D.; Reschly, E.J.; Ekins, S. Intrinsic disorder in nuclear hormone receptors. J. Proteome Res. 2008, 7, 4359–4372. [Google Scholar] [CrossRef] [Green Version]
- Shimizu, K.; Toh, H. Interaction between intrinsically disordered proteins frequently occurs in a human protein-protein interaction network. J. Mol. Biol. 2009, 392, 1253–1265. [Google Scholar] [CrossRef]
- Pentony, M.M.; Jones, D.T. Modularity of intrinsic disorder in the human proteome. Proteins 2010, 78, 212–221. [Google Scholar] [CrossRef]
- Tompa, P.; Kalmar, L. Power law distribution defines structural disorder as a structural element directly linked with function. J. Mol. Biol. 2010, 403, 346–350. [Google Scholar] [CrossRef] [PubMed]
- Schad, E.; Tompa, P.; Hegyi, H. The relationship between proteome size, structural disorder and organism complexity. Genome Biol. 2011, 12, R120. [Google Scholar] [CrossRef] [Green Version]
- Dyson, H.J. Expanding the proteome: Disordered and alternatively folded proteins. Q. Rev. Biophys. 2011, 44, 467–518. [Google Scholar] [CrossRef] [Green Version]
- Pancsa, R.; Tompa, P. Structural disorder in eukaryotes. PLoS ONE 2012, 7, e34687. [Google Scholar] [CrossRef] [PubMed]
- Midic, U.; Obradovic, Z. Intrinsic disorder in putative protein sequences. Proteome Sci. 2012, 10 (Suppl. 1), S19. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hegyi, H.; Tompa, P. Increased structural disorder of proteins encoded on human sex chromosomes. Mol. Biosyst. 2012, 8, 229–236. [Google Scholar] [CrossRef]
- Korneta, I.; Bujnicki, J.M. Intrinsic disorder in the human spliceosomal proteome. PLoS Comput. Biol. 2012, 8, e1002641. [Google Scholar] [CrossRef] [Green Version]
- Kahali, B.; Ghosh, T.C. Disorderness in Escherichia coli proteome: Perception of folding fidelity and protein-protein interactions. J. Biomol. Struct. Dyn. 2013, 31, 472–476. [Google Scholar] [CrossRef]
- Di Domenico, T.; Walsh, I.; Tosatto, S.C. Analysis and consensus of currently available intrinsic protein disorder annotation sources in the MobiDB database. BMC Bioinform. 2013, 14 (Suppl. 7), S3. [Google Scholar] [CrossRef] [Green Version]
- Oldfield, C.J.; Dunker, A.K. Intrinsically disordered proteins and intrinsically disordered protein regions. Annu. Rev. Biochem. 2014, 83, 553–584. [Google Scholar] [CrossRef]
- Uversky, V.N. Multitude of binding modes attainable by intrinsically disordered proteins: A portrait gallery of disorder-based complexes. Chem. Soc. Rev. 2011, 40, 1623–1634. [Google Scholar] [CrossRef]
- Uversky, V.N. Functional roles of transiently and intrinsically disordered regions within proteins. FEBS J. 2015, 282, 1182–1189. [Google Scholar] [CrossRef] [PubMed]
- Uversky, V.N. Intrinsically disordered proteins and their ‘mysterious’ (meta)physics. Front. Phys. 2019, 7. [Google Scholar] [CrossRef] [Green Version]
- Uversky, V.N.; Oldfield, C.J.; Dunker, A.K. Intrinsically disordered proteins in human diseases: Introducing the D2 concept. Annu. Rev. Biophys. 2008, 37, 215–246. [Google Scholar] [CrossRef] [PubMed]
- Uversky, V.N.; Dave, V.; Iakoucheva, L.M.; Malaney, P.; Metallo, S.J.; Pathak, R.R.; Joerger, A.C. Pathological unfoldomics of uncontrolled chaos: Intrinsically disordered proteins and human diseases. Chem. Rev. 2014, 114, 6844–6879. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Iakoucheva, L.M.; Brown, C.J.; Lawson, J.D.; Obradovic, Z.; Dunker, A.K. Intrinsic disorder in cell-signaling and cancer-associated proteins. J. Mol. Biol. 2002, 323, 573–584. [Google Scholar] [CrossRef] [Green Version]
- Uversky, V.N. Amyloidogenesis of natively unfolded proteins. Curr. Alzheimer Res. 2008, 5, 260–287. [Google Scholar] [CrossRef]
- Cheng, Y.; LeGall, T.; Oldfield, C.J.; Dunker, A.K.; Uversky, V.N. Abundance of intrinsic disorder in protein associated with cardiovascular disease. Biochemistry 2006, 45, 10448–10460. [Google Scholar] [CrossRef] [PubMed]
- Du, Z.; Uversky, V.N. A Comprehensive Survey of the Roles of Highly Disordered Proteins in Type 2 Diabetes. Int. J. Mol. Sci. 2017, 18, 2010. [Google Scholar] [CrossRef] [PubMed]
- Uversky, V.N. The triple power of D(3): Protein intrinsic disorder in degenerative diseases. Front. Biosci. 2014, 19, 181–258. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Uversky, V.N. Intrinsic disorder in proteins associated with neurodegenerative diseases. Front. Biosci. 2009, 14, 5188–5238. [Google Scholar] [CrossRef] [PubMed]
- Tewari, R.; Bailes, E.; Bunting, K.A.; Coates, J.C. Armadillo-repeat protein functions: Questions for little creatures. Trends Cell Biol. 2010, 20, 470–481. [Google Scholar] [CrossRef]
- Parra, R.G.; Espada, R.; Verstraete, N.; Ferreiro, D.U. Structural and Energetic Characterization of the Ankyrin Repeat Protein Family. PLoS Comput. Biol. 2015, 11, e1004659. [Google Scholar] [CrossRef]
- Yoshimura, S.H.; Hirano, T. HEAT repeats—Versatile arrays of amphiphilic helices working in crowded environments? J. Cell Sci. 2016, 129, 3963–3970. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Matsushima, N.; Takatsuka, S.; Miyashita, H.; Kretsinger, R.H. Leucine Rich Repeat Proteins: Sequences, Mutations, Structures and Diseases. Protein Pept. Lett. 2019, 26, 108–131. [Google Scholar] [CrossRef] [PubMed]
- Adams, J.; Kelso, R.; Cooley, L. The kelch repeat superfamily of proteins: Propellers of cell function. Trends Cell Biol. 2000, 10, 17–24. [Google Scholar] [CrossRef]
- Neer, E.J.; Schmidt, C.J.; Nambudripad, R.; Smith, T.F. The ancient regulatory-protein family of WD-repeat proteins. Nature 1994, 371, 297–300. [Google Scholar] [CrossRef]
- Vetting, M.W.; Hegde, S.S.; Fajardo, J.E.; Fiser, A.; Roderick, S.L.; Takiff, H.E.; Blanchard, J.S. Pentapeptide repeat proteins. Biochemistry 2006, 45, 1–10. [Google Scholar] [CrossRef] [Green Version]
- Saravanan, K.M.; Ponnuraj, K. Sequence and structural analysis of fibronectin-binding protein reveals importance of multiple intrinsic disordered tandem repeats. J. Mol. Recognit. 2019, 32, e2768. [Google Scholar] [CrossRef]
- Li, X.; Tao, Y.; Murphy, J.W.; Scherer, A.N.; Lam, T.T.; Marshall, A.G.; Koleske, A.J.; Boggon, T.J. The repeat region of cortactin is intrinsically disordered in solution. Sci. Rep. 2017, 7, 16696. [Google Scholar] [CrossRef] [Green Version]
- Darling, A.L.; Uversky, V.N. Intrinsic Disorder in Proteins with Pathogenic Repeat Expansions. Molecules 2017, 22, 2027. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roberts, S.; Dzuricky, M.; Chilkoti, A. Elastin-like polypeptides as models of intrinsically disordered proteins. FEBS Lett. 2015, 589, 2477–2486. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jorda, J.; Xue, B.; Uversky, V.N.; Kajava, A.V. Protein tandem repeats—The more perfect, the less structured. Febs J. 2010, 277, 2673–2682. [Google Scholar] [CrossRef] [PubMed]
- Denning, D.P.; Patel, S.S.; Uversky, V.; Fink, A.L.; Rexach, M. Disorder in the nuclear pore complex: The FG repeat regions of nucleoporins are natively unfolded. Proc. Natl. Acad. Sci. USA 2003, 100, 2450–2455. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- D’Andrea, L.D.; Regan, L. TPR proteins: The versatile helix. Trends Biochem. Sci. 2003, 28, 655–662. [Google Scholar] [CrossRef]
- Zeytuni, N.; Zarivach, R. Structural and functional discussion of the tetra-trico-peptide repeat, a protein interaction module. Structure 2012, 20, 397–405. [Google Scholar] [CrossRef] [Green Version]
- Alva, V.; Nam, S.Z.; Soding, J.; Lupas, A.N. The MPI bioinformatics Toolkit as an integrative platform for advanced protein sequence and structure analysis. Nucleic Acids Res. 2016, 44, W410–W415. [Google Scholar] [CrossRef]
- Biegert, A.; Mayer, C.; Remmert, M.; Soding, J.; Lupas, A.N. The MPI Bioinformatics Toolkit for protein sequence analysis. Nucleic Acids. Res. 2006, 34, W335–W339. [Google Scholar] [CrossRef] [Green Version]
- Das, A.K.; Cohen, P.W.; Barford, D. The structure of the tetratricopeptide repeats of protein phosphatase 5: Implications for TPR-mediated protein-protein interactions. EMBO J. 1998, 17, 1192–1199. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Perez-Riba, A.; Itzhaki, L.S. The tetratricopeptide-repeat motif is a versatile platform that enables diverse modes of molecular recognition. Curr. Opin. Struct. Biol. 2019, 54, 43–49. [Google Scholar] [CrossRef] [PubMed]
- Main, E.R.; Xiong, Y.; Cocco, M.J.; D’Andrea, L.; Regan, L. Design of stable alpha-helical arrays from an idealized TPR motif. Structure 2003, 11, 497–508. [Google Scholar] [CrossRef] [Green Version]
- Kajander, T.; Cortajarena, A.L.; Mochrie, S.; Regan, L. Structure and stability of designed TPR protein superhelices: Unusual crystal packing and implications for natural TPR proteins. Acta Cryst. D Biol. Cryst. 2007, 63, 800–811. [Google Scholar] [CrossRef] [PubMed]
- Mears, H.V.; Sweeney, T.R. Better together: The role of IFIT protein-protein interactions in the antiviral response. J. Gen. Virol. 2018, 99, 1463–1477. [Google Scholar] [CrossRef] [PubMed]
- Pidugu, V.K.; Pidugu, H.B.; Wu, M.M.; Liu, C.J.; Lee, T.C. Emerging Functions of Human IFIT Proteins in Cancer. Front. Mol. Biosci. 2019, 6, 148. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Burchfield, J.S.; Li, Q.; Wang, H.Y.; Wang, R.F. JMJD3 as an epigenetic regulator in development and disease. Int. J. Biochem. Cell Biol. 2015, 67, 148–157. [Google Scholar] [CrossRef]
- Wang, L.; Shilatifard, A. UTX Mutations in Human Cancer. Cancer Cell 2019, 35, 168–176. [Google Scholar] [CrossRef] [Green Version]
- Edkins, A.L. CHIP: A co-chaperone for degradation by the proteasome. Subcell Biochem. 2015, 78, 219–242. [Google Scholar] [CrossRef]
- Ni, W.; Odunuga, O.O. UCS proteins: Chaperones for myosin and co-chaperones for Hsp90. Subcell Biochem. 2015, 78, 133–152. [Google Scholar] [CrossRef]
- Graham, J.B.; Canniff, N.P.; Hebert, D.N. TPR-containing proteins control protein organization and homeostasis for the endoplasmic reticulum. Crit. Rev. Biochem. Mol. Biol. 2019, 54, 103–118. [Google Scholar] [CrossRef]
- Bohne, A.V.; Schwenkert, S.; Grimm, B.; Nickelsen, J. Roles of Tetratricopeptide Repeat Proteins in Biogenesis of the Photosynthetic Apparatus. Int. Rev. Cell Mol. Biol. 2016, 324, 187–227. [Google Scholar] [CrossRef]
- Lu, Y. Identification and Roles of Photosystem II Assembly, Stability, and Repair Factors in Arabidopsis. Front. Plant Sci 2016, 7, 168. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roberts, J.D.; Thapaliya, A.; Martinez-Lumbreras, S.; Krysztofinska, E.M.; Isaacson, R.L. Structural and Functional Insights into Small, Glutamine-Rich, Tetratricopeptide Repeat Protein Alpha. Front. Mol. Biosci. 2015, 2, 71. [Google Scholar] [CrossRef] [PubMed]
- Bando, T.; Mito, T.; Hamada, Y.; Ishimaru, Y.; Noji, S.; Ohuchi, H. Molecular mechanisms of limb regeneration: Insights from regenerating legs of the cricket Gryllus bimaculatus. Int. J. Dev. Biol. 2018, 62, 559–569. [Google Scholar] [CrossRef] [PubMed]
- Schwenkert, S.; Dittmer, S.; Soll, J. Structural components involved in plastid protein import. Essays Biochem. 2018, 62, 65–75. [Google Scholar] [CrossRef] [PubMed]
- Speltz, E.B.; Brown, R.S.; Hajare, H.S.; Schlieker, C.; Regan, L. A designed repeat protein as an affinity capture reagent. Biochem. Soc. Trans. 2015, 43, 874–880. [Google Scholar] [CrossRef]
- Romera, D.; Couleaud, P.; Mejias, S.H.; Aires, A.; Cortajarena, A.L. Biomolecular templating of functional hybrid nanostructures using repeat protein scaffolds. Biochem. Soc. Trans. 2015, 43, 825–831. [Google Scholar] [CrossRef]
- Rajagopalan, K.; Mooney, S.M.; Parekh, N.; Getzenberg, R.H.; Kulkarni, P. A majority of the cancer/testis antigens are intrinsically disordered proteins. J. Cell. Biochem. 2011, 112, 3256–3267. [Google Scholar] [CrossRef] [Green Version]
- Xue, B.; Dunbrack, R.L.; Williams, R.W.; Dunker, A.K.; Uversky, V.N. PONDR-FIT: A meta-predictor of intrinsically disordered amino acids. Biochim. Biophys. Acta 2010, 1804, 996–1010. [Google Scholar] [CrossRef] [Green Version]
- Xue, B.; Oldfield, C.J.; Dunker, A.K.; Uversky, V.N. CDF it all: Consensus prediction of intrinsically disordered proteins based on various cumulative distribution functions. FEBS Lett. 2009, 583, 1469–1474. [Google Scholar] [CrossRef] [Green Version]
- Huang, F.; Oldfield, C.J.; Xue, B.; Hsu, W.L.; Meng, J.; Liu, X.; Shen, L.; Romero, P.; Uversky, V.N.; Dunker, A. Improving protein order-disorder classification using charge-hydropathy plots. BMC Bioinform. 2014, 15 (Suppl. 17), S4. [Google Scholar] [CrossRef] [Green Version]
- Mohan, A.; Sullivan, W.J., Jr.; Radivojac, P.; Dunker, A.K.; Uversky, V.N. Intrinsic disorder in pathogenic and non-pathogenic microbes: Discovering and analyzing the unfoldomes of early-branching eukaryotes. Mol. Biosyst. 2008, 4, 328–340. [Google Scholar] [CrossRef] [PubMed]
- Huang, F.; Oldfield, C.; Meng, J.; Hsu, W.L.; Xue, B.; Uversky, V.N.; Romero, P.; Dunker, A.K. Subclassifying disordered proteins by the CH-CDF plot method. Pac. Symp. Biocomput. 2012, 128–139. [Google Scholar]
- Kriventseva, E.V.; Koch, I.; Apweiler, R.; Vingron, M.; Bork, P.; Gelfand, M.S.; Sunyaev, S. Increase of functional diversity by alternative splicing. Trends Genet. 2003, 19, 124–128. [Google Scholar] [CrossRef]
- Romero, P.R.; Zaidi, S.; Fang, Y.Y.; Uversky, V.N.; Radivojac, P.; Oldfield, C.J.; Cortese, M.S.; Sickmeier, M.; LeGall, T.; Obradovic, Z.; et al. Alternative splicing in concert with protein intrinsic disorder enables increased functional diversity in multicellular organisms. Proc. Natl. Acad. Sci. USA 2006, 103, 8390–8395. [Google Scholar] [CrossRef] [Green Version]
- Hegyi, H.; Kalmar, L.; Horvath, T.; Tompa, P. Verification of alternative splicing variants based on domain integrity, truncation length and intrinsic protein disorder. Nucleic Acids Res. 2011, 39, 1208–1219. [Google Scholar] [CrossRef] [Green Version]
- Buljan, M.; Chalancon, G.; Eustermann, S.; Wagner, G.P.; Fuxreiter, M.; Bateman, A.; Babu, M.M. Tissue-specific splicing of disordered segments that embed binding motifs rewires protein interaction networks. Mol. Cell 2012, 46, 871–883. [Google Scholar] [CrossRef] [Green Version]
- Uversky, V.N. The multifaceted roles of intrinsic disorder in protein complexes. FEBS Lett. 2015. [Google Scholar] [CrossRef] [Green Version]
- Mammen, M.; Choi, S.K.; Whitesides, G.M. Polyvalent interactions in biological systems: Implications for design and use of multivalent ligands and inhibitors. Angew. Chem. Int. Ed. 1998, 37, 2755–2794. [Google Scholar] [CrossRef]
- Schulz, G.E. Nucleotide Binding Proteins. In Molecular Mechanism of Biological Recognition; Balaban, M., Ed.; Elsevier/North-Holland Biomedical Press: New York, NY, USA, 1979; pp. 79–94. [Google Scholar]
- Wright, P.E.; Dyson, H.J. Linking folding and binding. Curr. Opin. Struct. Biol. 2009, 19, 31–38. [Google Scholar] [CrossRef] [Green Version]
- Meador, W.E.; Means, A.R.; Quiocho, F.A. Modulation of calmodulin plasticity in molecular recognition on the basis of x-ray structures. Science 1993, 262, 1718–1721. [Google Scholar] [CrossRef]
- Kriwacki, R.W.; Hengst, L.; Tennant, L.; Reed, S.I.; Wright, P.E. Structural studies of p21Waf1/Cip1/Sdi1 in the free and Cdk2-bound state: Conformational disorder mediates binding diversity. Proc. Natl. Acad. Sci. USA 1996, 93, 11504–11509. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dunker, A.K.; Garner, E.; Guilliot, S.; Romero, P.; Albrecht, K.; Hart, J.; Obradovic, Z.; Kissinger, C.; Villafranca, J.E. Protein disorder and the evolution of molecular recognition: Theory, predictions and observations. Pac. Symp. Biocomput. 1998, 473–484. [Google Scholar]
- Uversky, V.N. Protein folding revisited. A polypeptide chain at the folding-misfolding-nonfolding cross-roads: Which way to go? Cell. Mol. Life. Sci. 2003, 60, 1852–1871. [Google Scholar] [CrossRef] [PubMed]
- Dunker, A.K.; Cortese, M.S.; Romero, P.; Iakoucheva, L.M.; Uversky, V.N. Flexible nets: The roles of intrinsic disorder in protein interaction networks. FEBS J. 2005, 272, 5129–5148. [Google Scholar] [CrossRef] [PubMed]
- Dajani, R.; Fraser, E.; Roe, S.M.; Yeo, M.; Good, V.M.; Thompson, V.; Dale, T.C.; Pearl, L.H. Structural basis for recruitment of glycogen synthase kinase 3beta to the axin-APC scaffold complex. EMBO J. 2003, 22, 494–501. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hsu, W.L.; Oldfield, C.J.; Xue, B.; Meng, J.; Huang, F.; Romero, P.; Uversky, V.N.; Dunker, A.K. Exploring the binding diversity of intrinsically disordered proteins involved in one-to-many binding. Protein Sci. 2013, 22, 258–273. [Google Scholar] [CrossRef] [Green Version]
- Oldfield, C.J.; Meng, J.; Yang, J.Y.; Yang, M.Q.; Uversky, V.N.; Dunker, A.K. Flexible nets: Disorder and induced fit in the associations of p53 and 14-3-3 with their partners. Bmc Genom. 2008, 9 (Suppl. 1), S1. [Google Scholar] [CrossRef] [Green Version]
- Tompa, P.; Fuxreiter, M. Fuzzy complexes: Polymorphism and structural disorder in protein-protein interactions. Trends Biochem. Sci. 2008, 33, 2–8. [Google Scholar] [CrossRef]
- Hazy, E.; Tompa, P. Limitations of induced folding in molecular recognition by intrinsically disordered proteins. Chemphyschem 2009, 10, 1415–1419. [Google Scholar] [CrossRef]
- Sigalov, A.; Aivazian, D.; Stern, L. Homooligomerization of the cytoplasmic domain of the T cell receptor zeta chain and of other proteins containing the immunoreceptor tyrosine-based activation motif. Biochemistry 2004, 43, 2049–2061. [Google Scholar] [CrossRef]
- Sigalov, A.B.; Zhuravleva, A.V.; Orekhov, V.Y. Binding of intrinsically disordered proteins is not necessarily accompanied by a structural transition to a folded form. Biochimie 2007, 89, 419–421. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Permyakov, S.E.; Millett, I.S.; Doniach, S.; Permyakov, E.A.; Uversky, V.N. Natively unfolded C-terminal domain of caldesmon remains substantially unstructured after the effective binding to calmodulin. Proteins 2003, 53, 855–862. [Google Scholar] [CrossRef] [PubMed]
- Fuxreiter, M. Fuzziness: Linking regulation to protein dynamics. Mol. Biosyst. 2012, 8, 168–177. [Google Scholar] [CrossRef] [PubMed]
- Fuxreiter, M.; Tompa, P. Fuzzy complexes: A more stochastic view of protein function. Adv. Exp. Med. Biol. 2012, 725, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Sharma, R.; Raduly, Z.; Miskei, M.; Fuxreiter, M. Fuzzy complexes: Specific binding without complete folding. FEBS Lett. 2015. [Google Scholar] [CrossRef] [Green Version]
- Patil, A.; Nakamura, H. Disordered domains and high surface charge confer hubs with the ability to interact with multiple proteins in interaction networks. FEBS Lett. 2006, 580, 2041–2045. [Google Scholar] [CrossRef] [Green Version]
- Ekman, D.; Light, S.; Bjorklund, A.K.; Elofsson, A. What properties characterize the hub proteins of the protein-protein interaction network of Saccharomyces cerevisiae? Genome Biol. 2006, 7, R45. [Google Scholar] [CrossRef] [Green Version]
- Haynes, C.; Oldfield, C.J.; Ji, F.; Klitgord, N.; Cusick, M.E.; Radivojac, P.; Uversky, V.N.; Vidal, M.; Iakoucheva, L.M. Intrinsic disorder is a common feature of hub proteins from four eukaryotic interactomes. PLoS Comput. Biol. 2006, 2, e100. [Google Scholar] [CrossRef]
- Dosztanyi, Z.; Chen, J.; Dunker, A.K.; Simon, I.; Tompa, P. Disorder and sequence repeats in hub proteins and their implications for network evolution. J. Proteome Res. 2006, 5, 2985–2995. [Google Scholar] [CrossRef]
- Singh, G.P.; Dash, D. Intrinsic disorder in yeast transcriptional regulatory network. Proteins 2007, 68, 602–605. [Google Scholar] [CrossRef]
- Singh, G.P.; Ganapathi, M.; Dash, D. Role of intrinsic disorder in transient interactions of hub proteins. Proteins 2007, 66, 761–765. [Google Scholar] [CrossRef] [PubMed]
- Szklarczyk, D.; Franceschini, A.; Kuhn, M.; Simonovic, M.; Roth, A.; Minguez, P.; Doerks, T.; Stark, M.; Muller, J.; Bork, P.; et al. The STRING database in 2011: Functional interaction networks of proteins, globally integrated and scored. Nucleic Acids Res. 2011, 39, D561–D568. [Google Scholar] [CrossRef]
- Batova, I.; O’Rand, M.G. Histone-binding domains in a human nuclear autoantigenic sperm protein. Biol. Reprod. 1996, 54, 1238–1244. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fukuhara, N.; Ebert, J.; Unterholzner, L.; Lindner, D.; Izaurralde, E.; Conti, E. SMG7 is a 14-3-3-like adaptor in the nonsense-mediated mRNA decay pathway. Mol. Cell 2005, 17, 537–547. [Google Scholar] [CrossRef] [PubMed]
- Unterholzner, L.; Izaurralde, E. SMG7 acts as a molecular link between mRNA surveillance and mRNA decay. Mol. Cell 2004, 16, 587–596. [Google Scholar] [CrossRef] [PubMed]
- Jascur, T.; Brickner, H.; Salles-Passador, I.; Barbier, V.; El Khissiin, A.; Smith, B.; Fotedar, R.; Fotedar, A. Regulation of p21(WAF1/CIP1) stability by WISp39, a Hsp90 binding TPR protein. Mol. Cell 2005, 17, 237–249. [Google Scholar] [CrossRef] [PubMed]
- UniProt, C. UniProt: A hub for protein information. Nucleic Acids Res. 2015, 43, D204–D212. [Google Scholar] [CrossRef]
- Berman, H.M.; Westbrook, J.; Feng, Z.; Gilliland, G.; Bhat, T.N.; Weissig, H.; Shindyalov, I.N.; Bourne, P.E. The Protein Data Bank. Nucleic Acids Res. 2000, 28, 235–242. [Google Scholar] [CrossRef] [Green Version]
- Romero, P.; Obradovic, Z.; Li, X.; Garner, E.C.; Brown, C.J.; Dunker, A.K. Sequence complexity of disordered protein. Proteins 2001, 42, 38–48. [Google Scholar] [CrossRef]
- Peng, K.; Vucetic, S.; Radivojac, P.; Brown, C.J.; Dunker, A.K.; Obradovic, Z. Optimizing long intrinsic disorder predictors with protein evolutionary information. J. Bioinform. Comput. Biol. 2005, 3, 35–60. [Google Scholar] [CrossRef]
- Dosztányi, Z.; Csizmok, V.; Tompa, P.; Simon, I. IUPred: Web server for the prediction of intrinsically unstructured regions of proteins based on estimated energy content. Bioinformatics 2005, 21, 3433–3434. [Google Scholar] [CrossRef] [Green Version]
- Peng, K.; Radivojac, P.; Vucetic, S.; Dunker, A.K.; Obradovic, Z. Length-dependent prediction of protein intrinsic disorder. BMC Bioinform. 2006, 7, 208. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dosztanyi, Z.; Csizmok, V.; Tompa, P.; Simon, I. The pairwise energy content estimated from amino acid composition discriminates between folded and intrinsically unstructured proteins. J. Mol. Biol. 2005, 347, 827–839. [Google Scholar] [CrossRef] [PubMed]
- He, B.; Wang, K.; Liu, Y.; Xue, B.; Uversky, V.N.; Dunker, A.K. Predicting intrinsic disorder in proteins: An overview. Cell Res. 2009, 19, 929–949. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oates, M.E.; Romero, P.; Ishida, T.; Ghalwash, M.; Mizianty, M.J.; Xue, B.; Dosztanyi, Z.; Uversky, V.N.; Obradovic, Z.; Kurgan, L.; et al. D(2)P(2): Database of disordered protein predictions. Nucleic Acids Res. 2013, 41, D508–D516. [Google Scholar] [CrossRef] [Green Version]
- Ishida, T.; Kinoshita, K. PrDOS: Prediction of disordered protein regions from amino acid sequence. Nucleic Acids Res. 2007, 35, W460–W464. [Google Scholar] [CrossRef]
- Obradovic, Z.; Peng, K.; Vucetic, S.; Radivojac, P.; Dunker, A.K. Exploiting heterogeneous sequence properties improves prediction of protein disorder. Proteins: Struct. Funct. Bioinform. 2005, 61, 176–182. [Google Scholar] [CrossRef]
- Walsh, I.; Martin, A.J.; Di Domenico, T.; Tosatto, S.C. ESpritz: Accurate and fast prediction of protein disorder. Bioinformatics 2012, 28, 503–509. [Google Scholar] [CrossRef] [Green Version]
- Andreeva, A.; Howorth, D.; Brenner, S.E.; Hubbard, T.J.; Chothia, C.; Murzin, A.G. SCOP database in 2004: Refinements integrate structure and sequence family data. Nucleic Acids Res. 2004, 32, D226–D229. [Google Scholar] [CrossRef] [Green Version]
- Murzin, A.G.; Brenner, S.E.; Hubbard, T.; Chothia, C. SCOP: A structural classification of proteins database for the investigation of sequences and structures. J. Mol. Biol. 1995, 247, 536–540. [Google Scholar] [CrossRef]
- de Lima Morais, D.A.; Fang, H.; Rackham, O.J.; Wilson, D.; Pethica, R.; Chothia, C.; Gough, J. SUPERFAMILY 1.75 including a domain-centric gene ontology method. Nucleic Acids Res. 2011, 39, D427–D434. [Google Scholar] [CrossRef] [Green Version]
- Meszaros, B.; Simon, I.; Dosztanyi, Z. Prediction of protein binding regions in disordered proteins. PLoS Comput. Biol. 2009, 5, e1000376. [Google Scholar] [CrossRef] [Green Version]
- Hornbeck, P.V.; Kornhauser, J.M.; Tkachev, S.; Zhang, B.; Skrzypek, E.; Murray, B.; Latham, V.; Sullivan, M. PhosphoSitePlus: A comprehensive resource for investigating the structure and function of experimentally determined post-translational modifications in man and mouse. Nucleic Acids Res. 2012, 40, D261–D270. [Google Scholar] [CrossRef] [Green Version]
- Uversky, V.N. p53 Proteoforms and Intrinsic Disorder: An Illustration of the Protein Structure-Function Continuum Concept. Int. J. Mol. Sci. 2016, 17, 1874. [Google Scholar] [CrossRef] [PubMed]
- Smith, L.M.; Kelleher, N.L.; Consortium for Top Down, P. Proteoform: A single term describing protein complexity. Nat. Methods 2013, 10, 186–187. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Uversky, V.N. Protein intrinsic disorder and structure-function continuum. Prog. Mol. Biol. Transl. Sci. 2019, 166, 1–17. [Google Scholar] [CrossRef] [PubMed]
- Fonin, A.V.; Darling, A.L.; Kuznetsova, I.M.; Turoverov, K.K.; Uversky, V.N. Multi-functionality of proteins involved in GPCR and G protein signaling: Making sense of structure-function continuum with intrinsic disorder-based proteoforms. Cell. Mol. Life Sci. 2019. [Google Scholar] [CrossRef]
- Midic, U.; Oldfield, C.J.; Dunker, A.K.; Obradovic, Z.; Uversky, V.N. Unfoldomics of human genetic diseases: Illustrative examples of ordered and intrinsically disordered members of the human diseasome. Protein Pept. Lett. 2009, 16, 1533–1547. [Google Scholar] [CrossRef]
- Uversky, V.N.; Oldfield, C.J.; Midic, U.; Xie, H.; Xue, B.; Vucetic, S.; Iakoucheva, L.M.; Obradovic, Z.; Dunker, A.K. Unfoldomics of human diseases: Linking protein intrinsic disorder with diseases. BMC Genom. 2009, 10 (Suppl. 1), S7. [Google Scholar] [CrossRef] [Green Version]
- Uversky, V.N. Targeting intrinsically disordered proteins in neurodegenerative and protein dysfunction diseases: Another illustration of the D(2) concept. Expert Rev. Proteom. 2010, 7, 543–564. [Google Scholar] [CrossRef] [Green Version]
- Uversky, V.N. Wrecked regulation of intrinsically disordered proteins in diseases: Pathogenicity of deregulated regulators. Front. Mol. Biosci. 2014, 1, 6. [Google Scholar] [CrossRef] [PubMed]


























{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| PONDR® VLXT | PONDR® VSL2 | |||
|---|---|---|---|---|
| # DR | Longest DR | # DR | Longest DR | |
| Min | 3 | 10 | 3 | 11 |
| Average | 14.5 | 61.2 | 14.0 | 128.5 |
| Max | 61 | 348 | 68 | 818 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Van Bibber, N.W.; Haerle, C.; Khalife, R.; Xue, B.; Uversky, V.N. Intrinsic Disorder in Tetratricopeptide Repeat Proteins. Int. J. Mol. Sci. 2020, 21, 3709. https://doi.org/10.3390/ijms21103709
Van Bibber NW, Haerle C, Khalife R, Xue B, Uversky VN. Intrinsic Disorder in Tetratricopeptide Repeat Proteins. International Journal of Molecular Sciences. 2020; 21(10):3709. https://doi.org/10.3390/ijms21103709
Chicago/Turabian StyleVan Bibber, Nathan W., Cornelia Haerle, Roy Khalife, Bin Xue, and Vladimir N. Uversky. 2020. "Intrinsic Disorder in Tetratricopeptide Repeat Proteins" International Journal of Molecular Sciences 21, no. 10: 3709. https://doi.org/10.3390/ijms21103709
APA StyleVan Bibber, N. W., Haerle, C., Khalife, R., Xue, B., & Uversky, V. N. (2020). Intrinsic Disorder in Tetratricopeptide Repeat Proteins. International Journal of Molecular Sciences, 21(10), 3709. https://doi.org/10.3390/ijms21103709