Role of the Ang2–Tie2 Axis in Vascular Damage Driven by High Glucose or Nucleoside Diphosphate Kinase B Deficiency


 ,
,  and
and 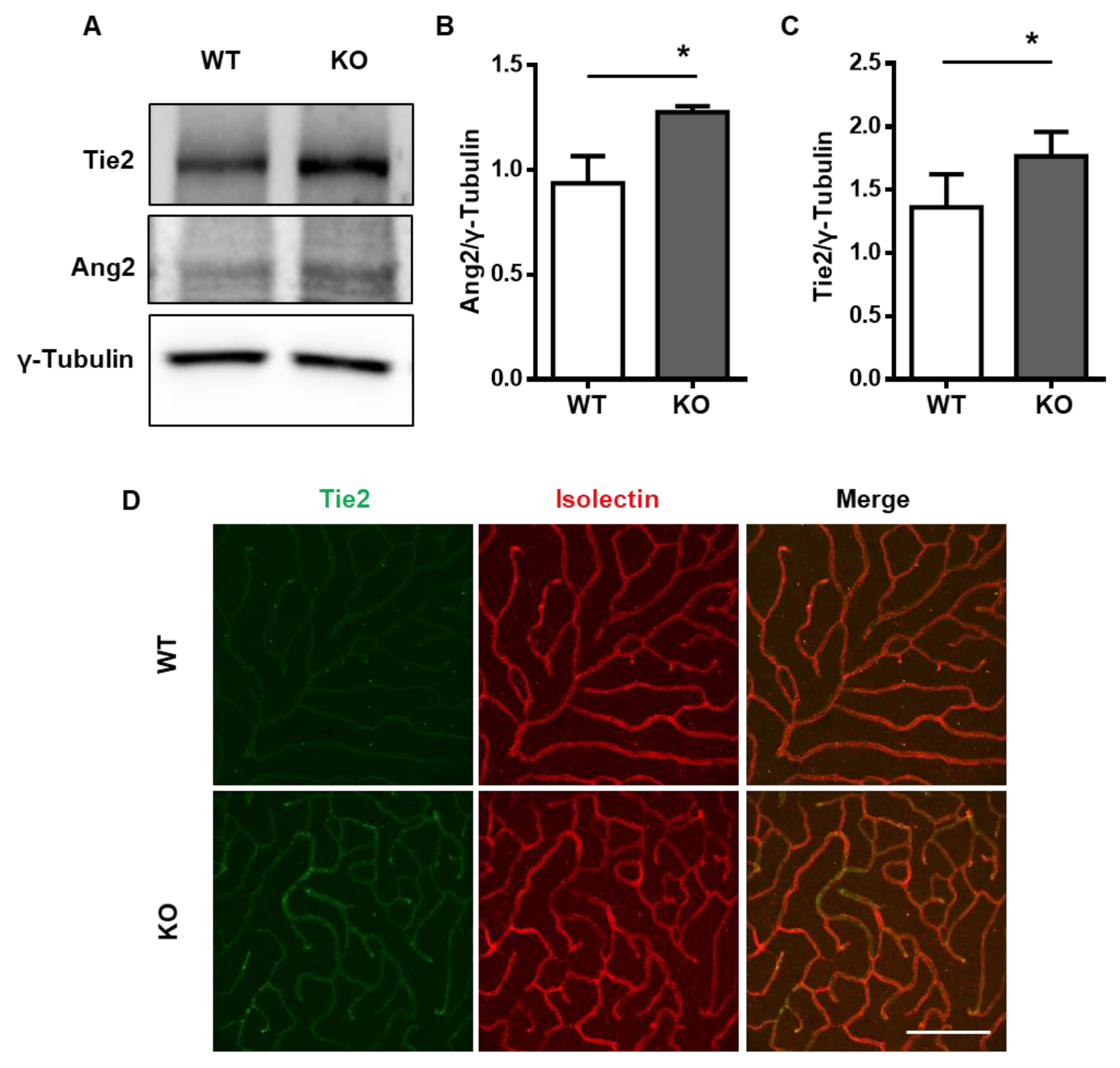{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
2.1. Tie2 Is Upregulated in the NDPK-B-Deficient Retina
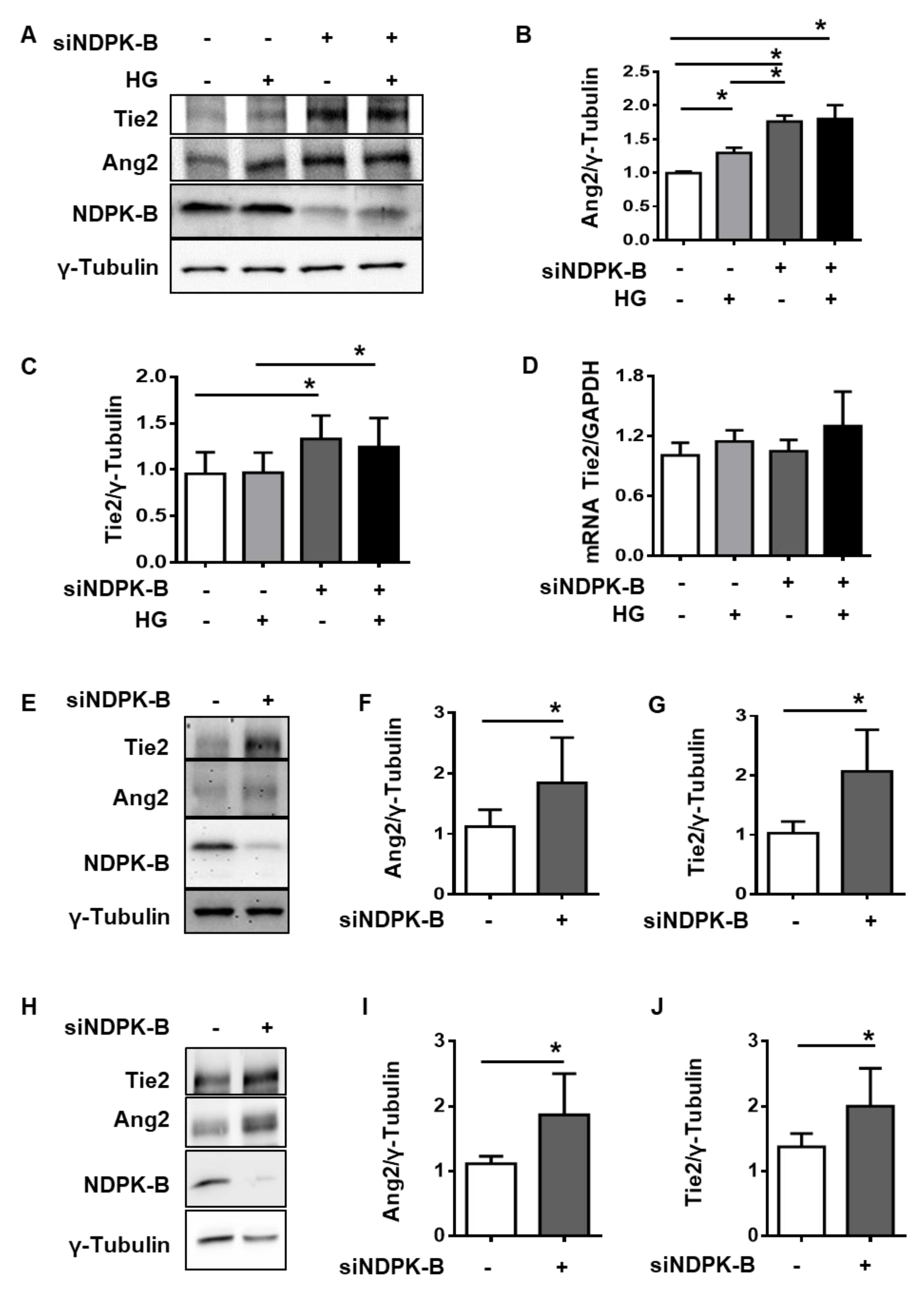
2.2. NDPK-B Depletion Upregulates Ang2 and Tie2 in Micro- and Macrovascular Endothelial Cells
2.3. NDPK-B Depletion Enhances Tie2 Levels at the Cell Membrane
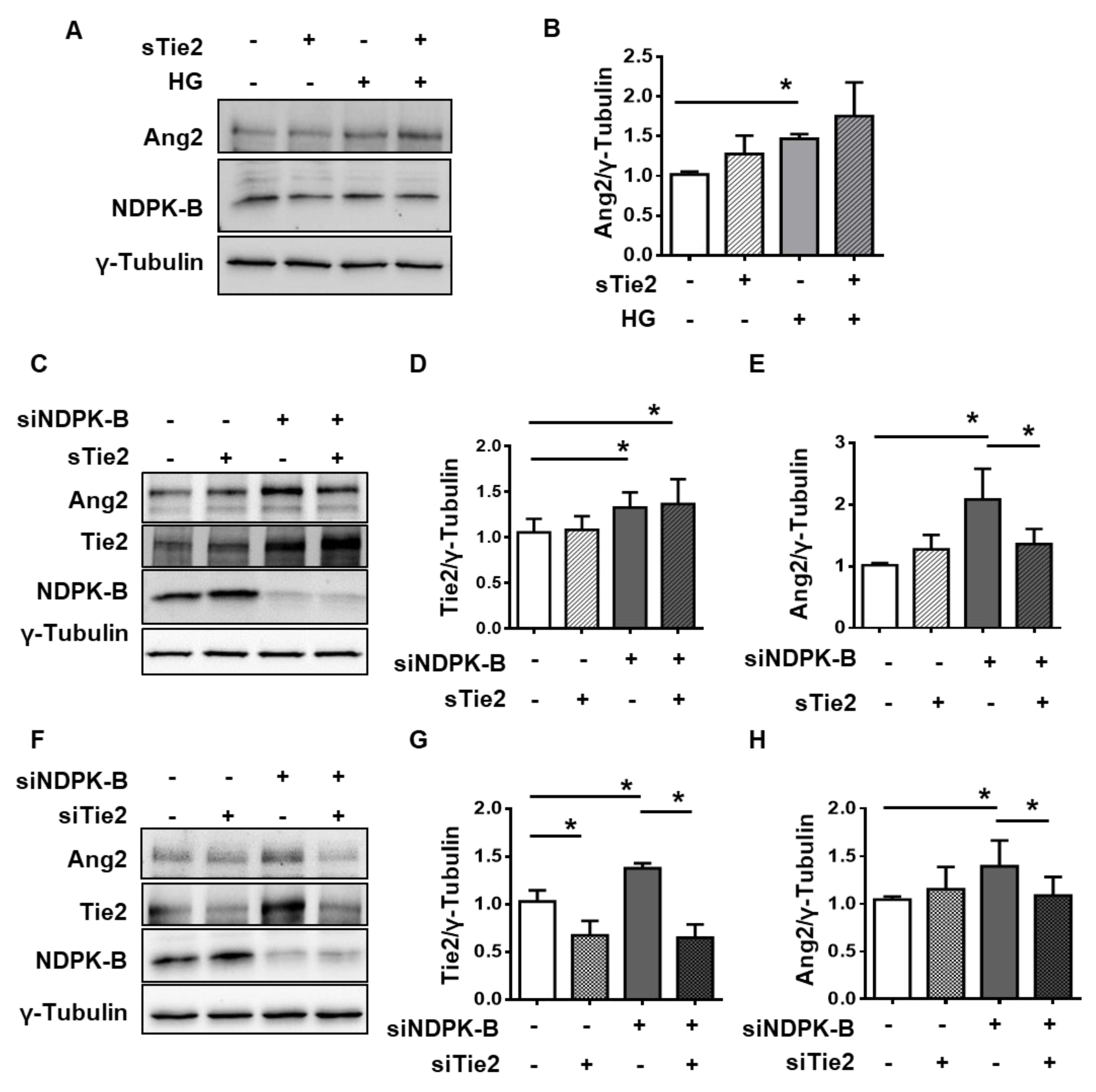
2.4. NDPK-B Depletion and High Glucose Reduce Tie2 Phosphorylation due to Elevated Ang2 Secretion
2.5. Tie2 Is Required for NDPK-B Depletion-Induced Ang2 Upregulation
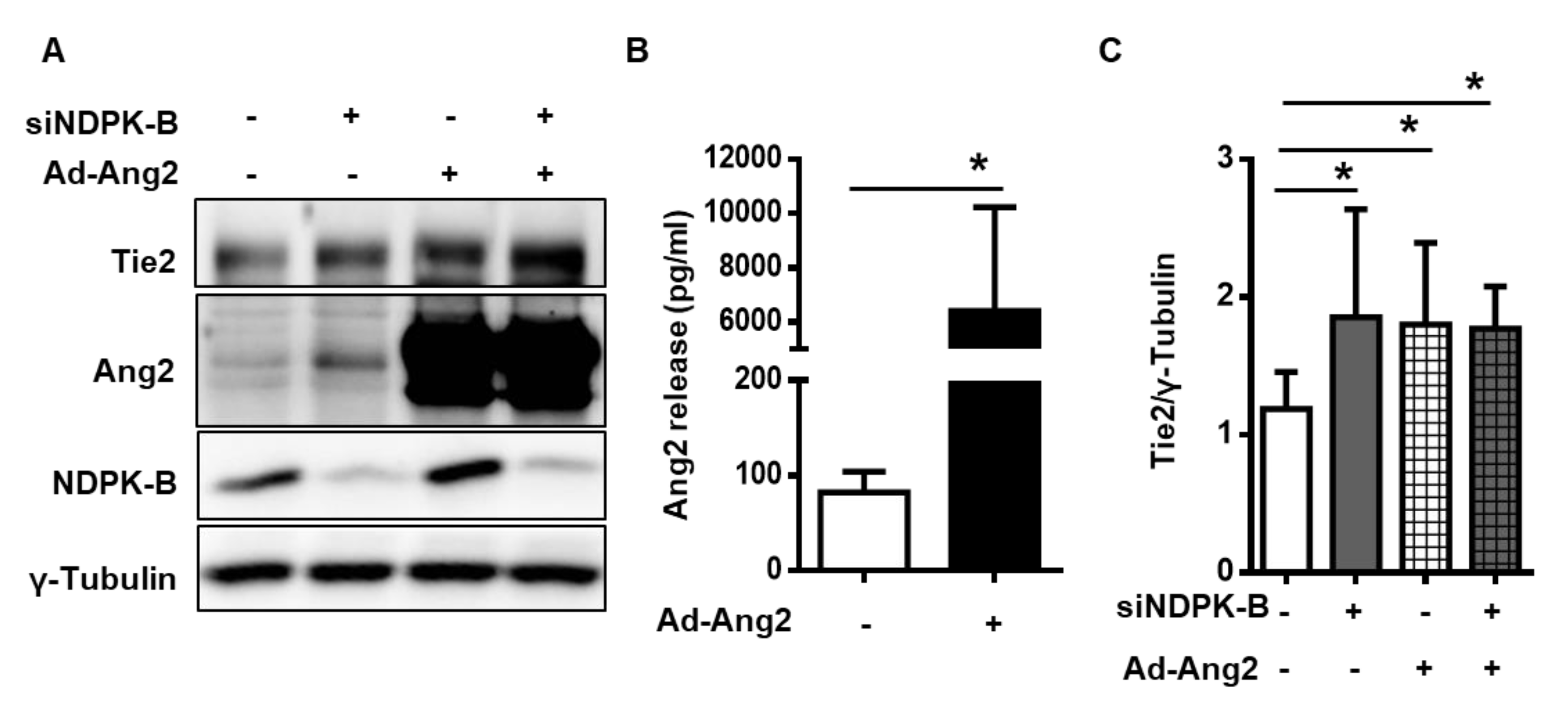
2.6. Excess Secreted Ang2 Promotes Upregulation of Tie2
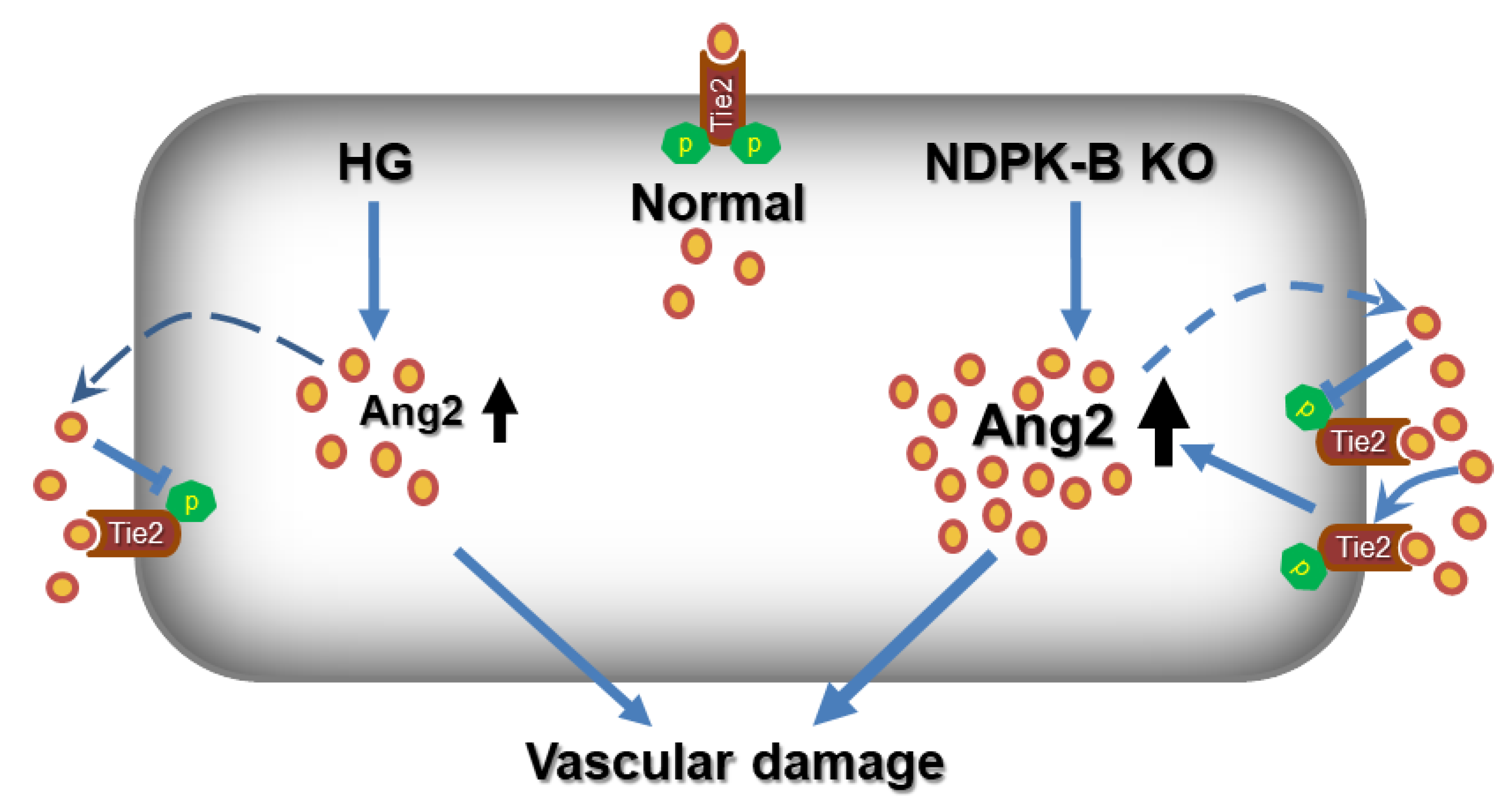
3. Discussion
4. Materials and Methods
4.1. Materials and Reagents
4.2. Animals
4.3. Retinal Whole-Mount Immunofluorescence Staining
4.4. Endothelial Immunofluorescence Staining
4.5. Isolation and Culture of Endothelial Cells
4.6. Cell Transfection
4.7. Subcellular Fractionation
4.8. Immunoprecipitation
4.9. Immunoblotting
4.10. ELISA
4.11. Adenovirus-Mediated Overexpression
4.12. Statistical Analysis
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| NDPK-B | Nucleoside diphosphate kinase B |
| ECs | Endothelial cells |
| Ang2 | Angiopoietin 2 |
| HG | High glucose |
| NDPKs | Nucleoside diphosphate kinases |
| DR | Diabetic retinopathy |
| NVU | Neurovascular unit |
| PC | Pericytes |
| AC | Acellular capillaries |
| Ang1 | Angiopoietin 1 |
| VEGF | Vascular endothelial growth factor |
| sTie2 | Soluble Tie2 |
| HUVECs | Human umbilical vein endothelial cells |
| ECGM | Endothelial cell growth medium |
| HRMVECs | Human retinal microvascular endothelial cells |
| HMECs | Human microvascular endothelial cells |
References
- Morera, S.; Chiadmi, M.; LeBras, G.; Lascu, I.; Janin, J. Mechanism of phosphate transfer by nucleoside diphosphate kinase: X-ray structures of the phosphohistidine intermediate of the enzymes from Drosophila and Dictyostelium. Biochemistry 1995, 34, 11062–11070. [Google Scholar] [CrossRef] [PubMed]
- Fan, Z.; Beresford, P.J.; Oh, D.Y.; Zhang, D.; Lieberman, J. Tumor suppressor NM23-H1 is a granzyme A-activated DNase during CTL-mediated apoptosis, and the nucleosome assembly protein SET is its inhibitor. Cell 2003, 112, 659–672. [Google Scholar] [CrossRef] [Green Version]
- Hippe, H.J.; Lutz, S.; Cuello, F.; Knorr, K.; Vogt, A.; Jakobs, K.H.; Wieland, T.; Niroomand, F. Activation of heterotrimeric G proteins by a high energy phosphate transfer via nucleoside diphosphate kinase (NDPK) B and Gbeta subunits. Specific activation of Gsalpha by an NDPK B.Gbetagamma complex in H10 cells. J. Biol. Chem. 2003, 278, 7227–7233. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Postel, E.H.; Berberich, S.J.; Flint, S.J.; Ferrone, C.A. Human c-myc transcription factor PuF identified as nm23-H2 nucleoside diphosphate kinase, a candidate suppressor of tumor metastasis. Science 1993, 261, 478–480. [Google Scholar] [CrossRef] [PubMed]
- Di, L.; Srivastava, S.; Zhdanova, O.; Sun, Y.; Li, Z.; Skolnik, E.Y. Nucleoside diphosphate kinase B knock-out mice have impaired activation of the K+ channel KCa3.1, resulting in defective T cell activation. J. Biol. Chem. 2010, 285, 38765–38771. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gurnik, S.; Devraj, K.; Macas, J.; Yamaji, M.; Starke, J.; Scholz, A.; Sommer, K.; Di Tacchio, M.; Vutukuri, R.; Beck, H.; et al. Angiopoietin-2-induced blood-brain barrier compromise and increased stroke size are rescued by VE-PTP-dependent restoration of Tie2 signaling. Acta Neuropathol. 2016, 131, 753–773. [Google Scholar] [CrossRef] [Green Version]
- Hammes, H.P.; Feng, Y.; Pfister, F.; Brownlee, M. Diabetic retinopathy: Targeting vasoregression. Diabetes 2011, 60, 9–16. [Google Scholar] [CrossRef] [Green Version]
- Gross, S.; Devraj, K.; Feng, Y.; Macas, J.; Liebner, S.; Wieland, T. Nucleoside diphosphate kinase B regulates angiogenic responses in the endothelium via caveolae formation and c-Src-mediated caveolin-1 phosphorylation. J. Cereb Blood Flow Metab. 2017, 37, 2471–2484. [Google Scholar] [CrossRef] [Green Version]
- Feng, Y.; Gross, S.; Wolf, N.M.; Butenschon, V.M.; Qiu, Y.; Devraj, K.; Liebner, S.; Kroll, J.; Skolnik, E.Y.; Hammes, H.P.; et al. Nucleoside diphosphate kinase B regulates angiogenesis through modulation of vascular endothelial growth factor receptor type 2 and endothelial adherens junction proteins. Arterioscler. Thromb. Vasc. Biol. 2014, 34, 2292–2300. [Google Scholar] [CrossRef] [Green Version]
- Qiu, Y.; Zhao, D.; Butenschon, V.M.; Bauer, A.T.; Schneider, S.W.; Skolnik, E.Y.; Hammes, H.P.; Wieland, T.; Feng, Y. Nucleoside diphosphate kinase B deficiency causes a diabetes-like vascular pathology via up-regulation of endothelial angiopoietin-2 in the retina. Acta Diabetol. 2016, 53, 81–89. [Google Scholar] [CrossRef]
- Shan, S.; Chatterjee, A.; Qiu, Y.; Hammes, H.P.; Wieland, T.; Feng, Y. O-GlcNAcylation of FoxO1 mediates nucleoside diphosphate kinase B deficiency induced endothelial damage. Sci. Rep. 2018, 8, 10581. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hammes, H.-P.; Porta, M. Experimental Approaches to Diabetic Retinopathy; Karger Medical and Scientific Publishers: Basel, Switzerland, 2010; Volume 20. [Google Scholar]
- Jeon, B.H.; Khanday, F.; Deshpande, S.; Haile, A.; Ozaki, M.; Irani, K. Tie-ing the antiinflammatory effect of angiopoietin-1 to inhibition of NF-kappaB. Circ. Res. 2003, 92, 586–588. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ramsauer, M.; D’Amore, P.A. Getting Tie(2)d up in angiogenesis. J. Clin. Investig. 2002, 110, 1615–1617. [Google Scholar] [CrossRef] [PubMed]
- Wong, A.L.; Haroon, Z.A.; Werner, S.; Dewhirst, M.W.; Greenberg, C.S.; Peters, K.G. Tie2 expression and phosphorylation in angiogenic and quiescent adult tissues. Circ. Res. 1997, 81, 567–574. [Google Scholar] [CrossRef] [PubMed]
- Davis, S.; Aldrich, T.H.; Jones, P.F.; Acheson, A.; Compton, D.L.; Jain, V.; Ryan, T.E.; Bruno, J.; Radziejewski, C.; Maisonpierre, P.C.; et al. Isolation of angiopoietin-1, a ligand for the TIE2 receptor, by secretion-trap expression cloning. Cell 1996, 87, 1161–1169. [Google Scholar] [CrossRef] [Green Version]
- Daly, C.; Wong, V.; Burova, E.; Wei, Y.; Zabski, S.; Griffiths, J.; Lai, K.M.; Lin, H.C.; Ioffe, E.; Yancopoulos, G.D.; et al. Angiopoietin-1 modulates endothelial cell function and gene expression via the transcription factor FKHR (FOXO1). Genes Dev. 2004, 18, 1060–1071. [Google Scholar] [CrossRef] [Green Version]
- Hanahan, D. Signaling vascular morphogenesis and maintenance. Science 1997, 277, 48–50. [Google Scholar] [CrossRef]
- Maisonpierre, P.C.; Suri, C.; Jones, P.F.; Bartunkova, S.; Wiegand, S.J.; Radziejewski, C.; Compton, D.; McClain, J.; Aldrich, T.H.; Papadopoulos, N.; et al. Angiopoietin-2, a natural antagonist for Tie2 that disrupts in vivo angiogenesis. Science 1997, 277, 55–60. [Google Scholar] [CrossRef]
- Akwii, R.G.; Sajib, M.S.; Zahra, F.T.; Mikelis, C.M. Role of Angiopoietin-2 in Vascular Physiology and Pathophysiology. Cells 2019, 8. [Google Scholar] [CrossRef] [Green Version]
- Kim, M.; Allen, B.; Korhonen, E.A.; Nitschke, M.; Yang, H.W.; Baluk, P.; Saharinen, P.; Alitalo, K.; Daly, C.; Thurston, G.; et al. Opposing actions of angiopoietin-2 on Tie2 signaling and FOXO1 activation. J. Clin. Investig. 2016, 126, 3511–3525. [Google Scholar] [CrossRef]
- Yao, D.; Taguchi, T.; Matsumura, T.; Pestell, R.; Edelstein, D.; Giardino, I.; Suske, G.; Rabbani, N.; Thornalley, P.J.; Sarthy, V.P.; et al. High glucose increases angiopoietin-2 transcription in microvascular endothelial cells through methylglyoxal modification of mSin3A. J. Biol. Chem. 2007, 282, 31038–31045. [Google Scholar] [CrossRef] [Green Version]
- Fiedler, U.; Scharpfenecker, M.; Koidl, S.; Hegen, A.; Grunow, V.; Schmidt, J.M.; Kriz, W.; Thurston, G.; Augustin, H.G. The Tie-2 ligand angiopoietin-2 is stored in and rapidly released upon stimulation from endothelial cell Weibel-Palade bodies. Blood 2004, 103, 4150–4156. [Google Scholar] [CrossRef]
- Findley, C.M.; Cudmore, M.J.; Ahmed, A.; Kontos, C.D. VEGF induces Tie2 shedding via a phosphoinositide 3-kinase/Akt dependent pathway to modulate Tie2 signaling. Arterioscler. Thromb. Vasc. Biol. 2007, 27, 2619–2626. [Google Scholar] [CrossRef] [Green Version]
- Rangasamy, S.; Srinivasan, R.; Maestas, J.; McGuire, P.G.; Das, A. A potential role for angiopoietin 2 in the regulation of the blood-retinal barrier in diabetic retinopathy. Investig. Ophthalmol. Vis. Sci. 2011, 52, 3784–3791. [Google Scholar] [CrossRef] [Green Version]
- Singh, H.; Brindle, N.P.; Zammit, V.A. High glucose and elevated fatty acids suppress signaling by the endothelium protective ligand angiopoietin-1. Microvasc. Res. 2010, 79, 121–127. [Google Scholar] [CrossRef]
- Yoon, M.J.; Cho, C.H.; Lee, C.S.; Jang, I.H.; Ryu, S.H.; Koh, G.Y. Localization of Tie2 and phospholipase D in endothelial caveolae is involved in angiopoietin-1-induced MEK/ERK phosphorylation and migration in endothelial cells. Biochem. Biophys. Res. Commun. 2003, 308, 101–105. [Google Scholar] [CrossRef]
- Daly, C.; Pasnikowski, E.; Burova, E.; Wong, V.; Aldrich, T.H.; Griffiths, J.; Ioffe, E.; Daly, T.J.; Fandl, J.P.; Papadopoulos, N.; et al. Angiopoietin-2 functions as an autocrine protective factor in stressed endothelial cells. Proc. Natl. Acad. Sci. USA 2006, 103, 15491–15496. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hansen, T.M.; Singh, H.; Tahir, T.A.; Brindle, N.P. Effects of angiopoietins-1 and -2 on the receptor tyrosine kinase Tie2 are differentially regulated at the endothelial cell surface. Cell Signal. 2010, 22, 527–532. [Google Scholar] [CrossRef] [PubMed]
- Tuo, Q.H.; Xiong, G.Z.; Zeng, H.; Yu, H.D.; Sun, S.W.; Ling, H.Y.; Zhu, B.Y.; Liao, D.F.; Chen, J.X. Angiopoietin-1 protects myocardial endothelial cell function blunted by angiopoietin-2 and high glucose condition. Acta Pharmacol. Sin. 2011, 32, 45–51. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Felcht, M.; Luck, R.; Schering, A.; Seidel, P.; Srivastava, K.; Hu, J.; Bartol, A.; Kienast, Y.; Vettel, C.; Loos, E.K.; et al. Angiopoietin-2 differentially regulates angiogenesis through TIE2 and integrin signaling. J. Clin. Investig. 2012, 122, 1991–2005. [Google Scholar] [CrossRef] [Green Version]
- Hakanpaa, L.; Sipila, T.; Leppanen, V.M.; Gautam, P.; Nurmi, H.; Jacquemet, G.; Eklund, L.; Ivaska, J.; Alitalo, K.; Saharinen, P. Endothelial destabilization by angiopoietin-2 via integrin beta1 activation. Nat. Commun. 2015, 6, 5962. [Google Scholar] [CrossRef] [PubMed]
- Park, S.W.; Yun, J.H.; Kim, J.H.; Kim, K.W.; Cho, C.H.; Kim, J.H. Angiopoietin 2 induces pericyte apoptosis via alpha3beta1 integrin signaling in diabetic retinopathy. Diabetes 2014, 63, 3057–3068. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Campochiaro, P.A.; Khanani, A.; Singer, M.; Patel, S.; Boyer, D.; Dugel, P.; Kherani, S.; Withers, B.; Gambino, L.; Peters, K.; et al. Enhanced Benefit in Diabetic Macular Edema from AKB-9778 Tie2 Activation Combined with Vascular Endothelial Growth Factor Suppression. Ophthalmology 2016, 123, 1722–1730. [Google Scholar] [CrossRef]
- Shen, J.; Frye, M.; Lee, B.L.; Reinardy, J.L.; McClung, J.M.; Ding, K.; Kojima, M.; Xia, H.; Seidel, C.; Lima e Silva, R.; et al. Targeting VE-PTP activates TIE2 and stabilizes the ocular vasculature. J. Clin. Investig. 2014, 124, 4564–4576. [Google Scholar] [CrossRef] [PubMed] [Green Version]







© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chatterjee, A.; Eshwaran, R.; Huang, H.; Zhao, D.; Schmidt, M.; Wieland, T.; Feng, Y. Role of the Ang2–Tie2 Axis in Vascular Damage Driven by High Glucose or Nucleoside Diphosphate Kinase B Deficiency. Int. J. Mol. Sci. 2020, 21, 3713. https://doi.org/10.3390/ijms21103713
Chatterjee A, Eshwaran R, Huang H, Zhao D, Schmidt M, Wieland T, Feng Y. Role of the Ang2–Tie2 Axis in Vascular Damage Driven by High Glucose or Nucleoside Diphosphate Kinase B Deficiency. International Journal of Molecular Sciences. 2020; 21(10):3713. https://doi.org/10.3390/ijms21103713
Chicago/Turabian StyleChatterjee, Anupriya, Rachana Eshwaran, Hongpeng Huang, Di Zhao, Martina Schmidt, Thomas Wieland, and Yuxi Feng. 2020. "Role of the Ang2–Tie2 Axis in Vascular Damage Driven by High Glucose or Nucleoside Diphosphate Kinase B Deficiency" International Journal of Molecular Sciences 21, no. 10: 3713. https://doi.org/10.3390/ijms21103713
APA StyleChatterjee, A., Eshwaran, R., Huang, H., Zhao, D., Schmidt, M., Wieland, T., & Feng, Y. (2020). Role of the Ang2–Tie2 Axis in Vascular Damage Driven by High Glucose or Nucleoside Diphosphate Kinase B Deficiency. International Journal of Molecular Sciences, 21(10), 3713. https://doi.org/10.3390/ijms21103713