An Evaluation of the In Vitro Roles and Mechanisms of Silibinin in Reducing Pyrazinamide- and Isoniazid-Induced Hepatocellular Damage



Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
2.1. Silibinin Mitigated Hepatotoxicity Induced by INH When Administered as a Rescue Adjuvant
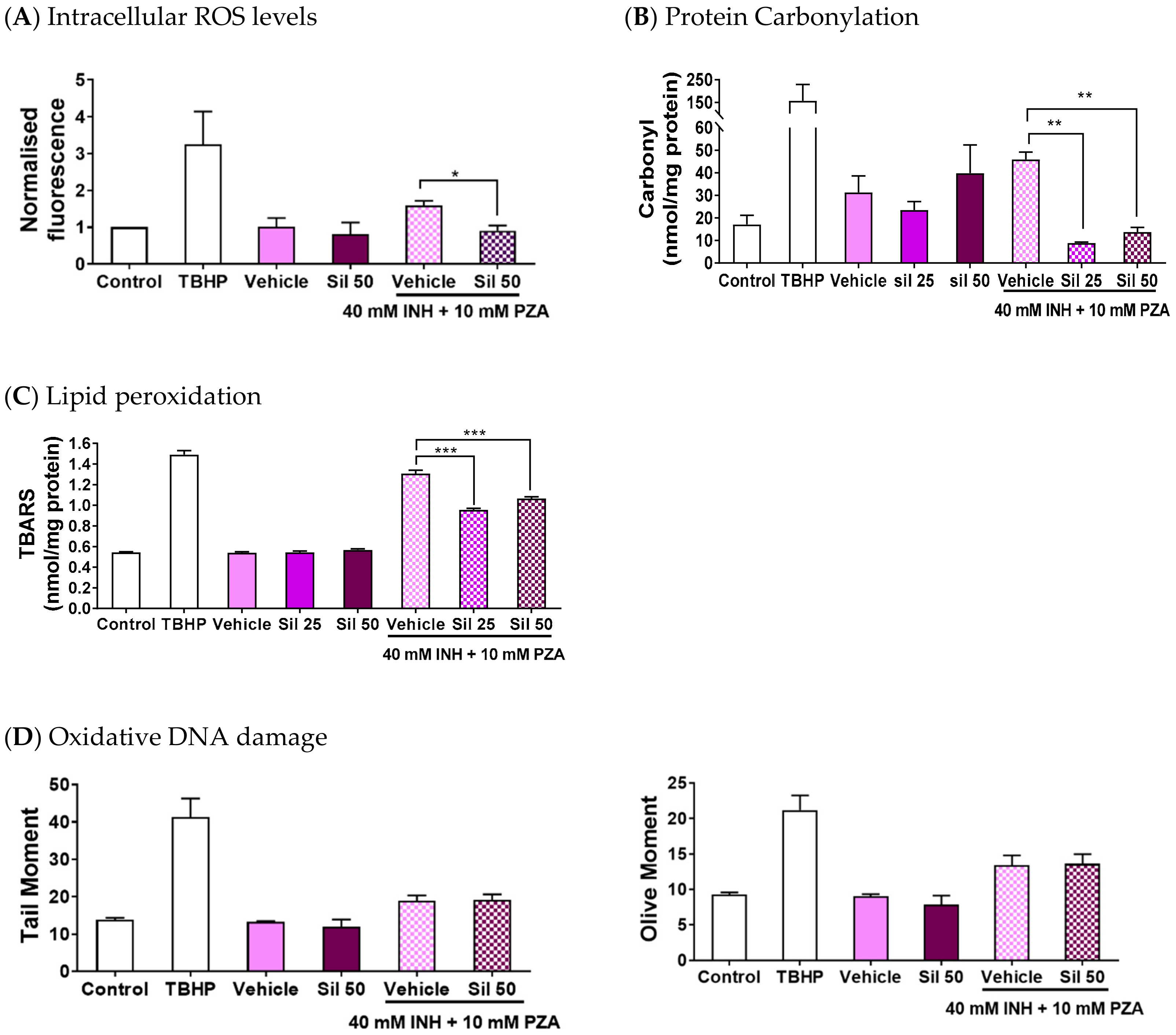
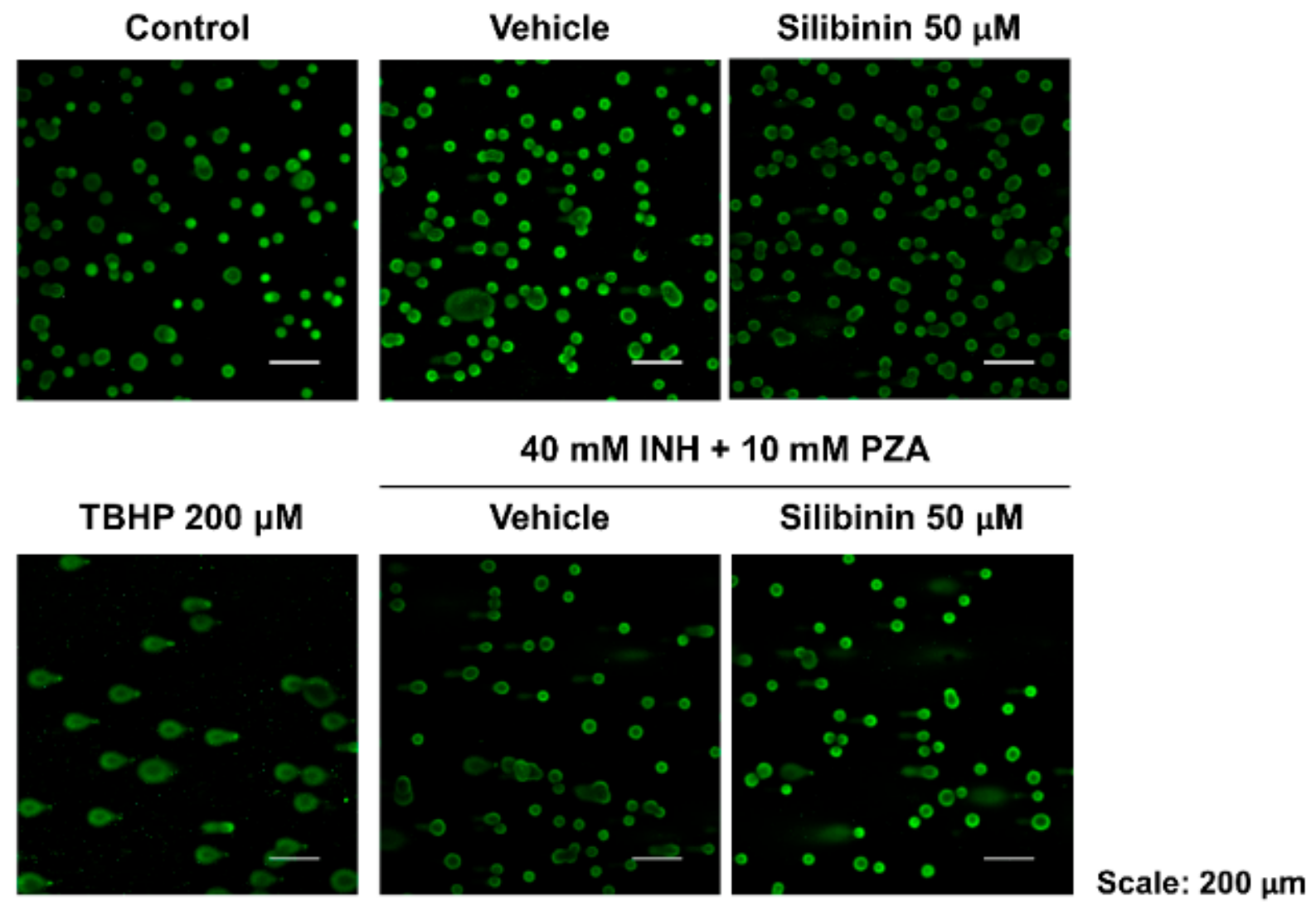
2.2. Silibinin Reduced Oxidative Damage of INH and PZA on Classical Intracellular Targets
2.3. Silibinin Protected against Apoptosis by Maintaining Mitochondrial Membrane Potential
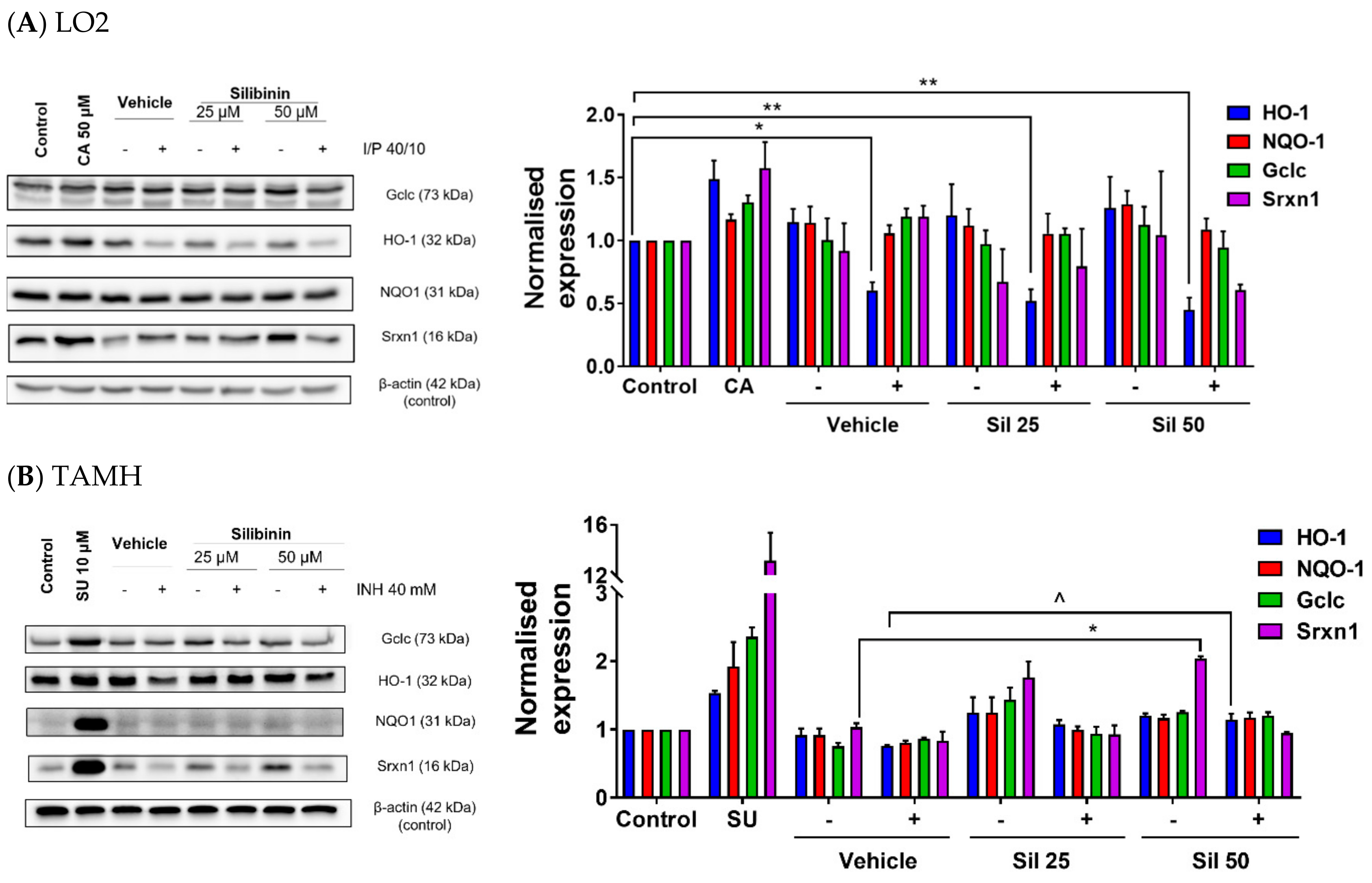
2.4. Silibinin Restored HO-1 Expression and Induced Srxn1 Expression in Transforming Growth Factor-α Transgenic Mouse Hepatocytes (TAMH)
3. Discussion
4. Materials and Methods
4.1. Cell Culture and Reagents
4.2. Cell Viability Assay
4.3. Direct ROS Quantitation
4.4. Lipid Peroxidation Quantitation
4.5. Protein Carbonylation Quantitation
4.6. Comet Assay
4.7. Mitochondrial Membrane Potential Measurement
4.8. Apoptosis Detection
4.9. Western Blot
4.10. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| ADP | Advanced digital processing |
| ARE | Antioxidant response element |
| ATT | Antitubercular therapy |
| BCA | Bicinchoninic acid |
| CA | Trans-cinnamaldehyde |
| CAT | Catalase |
| CPT | Camptothecin |
| DCFDA | 6-chloromethyl-2′,7′-dichlorodihydrofluorescein diacetate |
| DILI | Drug-induced liver injury |
| DMEM | Dulbecco’s minimum essential medium |
| DNA | Deoxyribonucleic acid |
| DTT | Dithiothreitol |
| EMB | Ethambutol |
| HEPES | 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid |
| IC50 | Half maximal inhibitory concentration |
| I/P | A combination of isoniazid and pyrazinamide |
| INH | Isoniazid |
| MAPK | Mitogen-activated protein kinase |
| MTT | 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide |
| NLRP3 | NOD-like receptor pyrin domain-containing-3 |
| Nrf2 | Nuclear factor (erythroid-derived 2)-like 2 |
| NQO1 | NAD(P)H quinone dehydrogenase 1 |
| PAGE | Polyacrylamide gel |
| PZA | Pyrazinamide |
| ROS | Reactive oxygen species |
| SDS | Sodium dodecyl sulfate |
| SOD | Superoxide dismutase |
| SU | Sulphoraphane |
| TAMH | Transforming growth factor-α transgenic mouse hepatocytes |
| TBARS | Thiobarbituric acid reactive substances |
| TBHP | Tert-butyl hydroperoxide |
| TCA | Trichloroacetic acid |
| TMRM | Tetramethylrhodamine-methyl-ester |
References
- Global Tuberculosis Report 2019; World Health Organization: Geneva, Switzerland, 2019.
- Kwon, Y.S.; Jeong, B.H.; Koh, W.J. Tuberculosis: Clinical trials and new drug regimens. Curr. Opin. Pulm. Med. 2014, 20, 280–286. [Google Scholar] [CrossRef] [PubMed]
- Cano-Paniagua, A.; Amariles, P.; Angulo, N.; Restrepo-Garay, M. Epidemiology of drug-induced liver injury in a University Hospital from Colombia: Updated RUCAM being used for prospective causality assessment. Ann. Hepatol. 2019, 18, 501–507. [Google Scholar] [CrossRef] [PubMed]
- Abbara, A.; Chitty, S.; Roe, J.K.; Ghani, R.; Collin, S.M.; Ritchie, A.; Kon, O.M.; Dzvova, J.; Davidson, H.; Edwards, T.E.; et al. Drug-induced liver injury from antituberculous treatment: A retrospective study from a large TB centre in the UK. BMC Infect. Dis. 2017, 17, 231. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ramappa, V.; Aithal, G.P. Hepatotoxicity Related to Anti-tuberculosis Drugs: Mechanisms and Management. J. Clin. Exp. Hepatol. 2013, 3, 37–49. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chang, K.C.; Leung, C.C.; Yew, W.W.; Lau, T.Y.; Tam, C.M. Hepatotoxicity of pyrazinamide: Cohort and case-control analyses. Am. J. Respir. Crit. Care. Med. 2008, 177, 1391–1396. [Google Scholar] [CrossRef] [PubMed]
- Singh, M.; Sasi, P.; Gupta, V.H.; Rai, G.; Amarapurkar, D.N.; Wangikar, P.P. Protective effect of curcumin, silymarin and N-acetylcysteine on antitubercular drug-induced hepatotoxicity assessed in an in vitro model. Hum. Exp. Toxicol. 2012, 31, 788–797. [Google Scholar] [CrossRef]
- Wang, P.; Pradhan, K.; Zhong, X.B.; Ma, X. Isoniazid metabolism and hepatotoxicity. Acta Pharm Sin. B 2016, 6, 384–392. [Google Scholar] [CrossRef] [Green Version]
- Harrington, T.; Manangan, L.; Jereb, J.; Navin, T.; Powell, K. Severe isoniazid-associated liver injuries among persons being treated for latent tuberculosis infection—United States, 2004–2008. Morb. Mortal. Wkly. Rep. 2010, 59, 224–229. [Google Scholar]
- Eminzade, S.; Uraz, F.; Izzettin, F.V. Silymarin protects liver against toxic effects of anti-tuberculosis drugs in experimental animals. Nutr. Metab. 2008, 5, 18. [Google Scholar] [CrossRef] [Green Version]
- Srivastava, R.K.; Sharma, S.; Verma, S.; Arora, B.; Lal, H. Influence of diabetes on liver injury induced by antitubercular drugs and on silymarin hepatoprotection in rats. Methods Find Exp. Clin. Pharm. 2008, 30, 731–737. [Google Scholar] [CrossRef]
- Tasduq, S.A.; Peerzada, K.; Koul, S.; Bhat, R.; Johri, R.K. Biochemical manifestations of anti-tuberculosis drugs induced hepatotoxicity and the effect of silymarin. Hepatol. Res. 2005, 31, 132–135. [Google Scholar] [CrossRef] [PubMed]
- Victorrajmohan, C.; Pradeep, K.; Karthikeyan, S. Influence of silymarin administration on hepatic glutathione-conjugating enzyme system in rats treated with antitubercular drugs. Drugs R D 2005, 6, 395–400. [Google Scholar] [CrossRef] [PubMed]
- Jimenez-Arellanes, M.A.; Gutierrez-Rebolledo, G.A.; Meckes-Fischer, M.; Leon-Diaz, R. Medical plant extracts and natural compounds with a hepatoprotective effect against damage caused by antitubercular drugs: A review. Asian Pac. J. Trop. Med. 2016, 9, 1141–1149. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zumla, A.; Nahid, P.; Cole, S.T. Advances in the development of new tuberculosis drugs and treatment regimens. Nat. Rev. Drug. Discov. 2013, 12, 388–404. [Google Scholar] [CrossRef]
- Betteridge, D.J. What is oxidative stress? Metabolism 2000, 49, 3–8. [Google Scholar] [CrossRef]
- Pitre, D.; Facino, R.M.; Carini, M.; Carlo, A. In vitro biotransformation of pyrazinamide by rat liver: Identification of a new metabolite. Pharm. Res. Commun. 1981, 13, 351–362. [Google Scholar] [CrossRef]
- Shih, T.Y.; Pai, C.Y.; Yang, P.; Chang, W.L.; Wang, N.C.; Hu, O.Y. A novel mechanism underlies the hepatotoxicity of pyrazinamide. Antimicrob. Agents Chemother. 2013, 57, 1685–1690. [Google Scholar] [CrossRef] [Green Version]
- Liu, X.; Zhao, M.; Mi, J.; Chen, H.; Sheng, L.; Li, Y. Protective Effect of Bicyclol on Anti-Tuberculosis Drug Induced Liver Injury in Rats. Molecules 2017, 22, 524. [Google Scholar] [CrossRef] [Green Version]
- Tafazoli, S.; Mashregi, M.; O’Brien, P.J. Role of hydrazine in isoniazid-induced hepatotoxicity in a hepatocyte inflammation model. Toxicol. Appl. Pharm. 2008, 229, 94–101. [Google Scholar] [CrossRef]
- Hassan, H.M.; Yousef, B.A.; Guo, H.; Xiaoxin, L.; Zhang, L.; Jiang, Z. Investigating the CYP2E1 Potential Role in the Mechanisms Behind INH/LPS-Induced Hepatotoxicity. Front. Pharm. 2018, 9, 198. [Google Scholar] [CrossRef]
- Shen, L.; Zhang, B.; Sun, S.; Feng, F. Methylation of cytochrome p450 2E1 promoter induced by low dosage of isoniazid. Environ. Toxicol. Pharmacol. 2013, 36, 149–151. [Google Scholar] [CrossRef] [PubMed]
- Ahadpour, M.; Eskandari, M.R.; Mashayekhi, V.; Haj Mohammad Ebrahim Tehrani, K.; Jafarian, I.; Naserzadeh, P.; Hosseini, M.J. Mitochondrial oxidative stress and dysfunction induced by isoniazid: Study on isolated rat liver and brain mitochondria. Drug Chem. Toxicol. 2016, 39, 224–232. [Google Scholar] [CrossRef] [PubMed]
- Sharma, R.; Battu, P.; Singla, M.; Goyal, N.; Sharma, V.L. Expression Profile of Markers of Oxidative Stress, Injury and Apoptosis in anti-tuberculosis drugs induced nephrotoxicity. Nephrology 2018. [Google Scholar] [CrossRef] [PubMed]
- Cao, J.; Mi, Y.; Shi, C.; Bian, Y.; Huang, C.; Ye, Z.; Liu, L.; Miao, L. First-line anti-tuberculosis drugs induce hepatotoxicity: A novel mechanism based on a urinary metabolomics platform. Biochem. Biophys. Res. Commun. 2018, 497, 485–491. [Google Scholar] [CrossRef] [PubMed]
- Rawat, A.; Chaturvedi, S.; Singh, A.K.; Guleria, A.; Dubey, D.; Keshari, A.K.; Raj, V.; Rai, A.; Prakash, A.; Kumar, U.; et al. Metabolomics approach discriminates toxicity index of pyrazinamide and its metabolic products, pyrazinoic acid and 5-hydroxy pyrazinoic acid. Hum. Exp. Toxicol. 2018, 37, 373–389. [Google Scholar] [CrossRef]
- Chen, Y.; Xue, P.; Hou, Y.; Zhang, H.; Zheng, H.; Zhou, T.; Qu, W.; Teng, W.; Zhang, Q.; Andersen, M.E.; et al. Isoniazid suppresses antioxidant response element activities and impairs adipogenesis in mouse and human preadipocytes. Toxicol. Appl. Pharm. 2013, 273, 435–441. [Google Scholar] [CrossRef] [PubMed]
- Peng, H.; Wang, H.; Xue, P.; Hou, Y.; Dong, J.; Zhou, T.; Qu, W.; Peng, S.; Li, J.; Carmichael, P.L.; et al. Suppression of NRF2-ARE activity sensitizes chemotherapeutic agent-induced cytotoxicity in human acute monocytic leukemia cells. Toxicol. Appl. Pharm. 2016, 292, 1–7. [Google Scholar] [CrossRef]
- Singh, M.; Sasi, P.; Rai, G.; Gupta, V.H.; Amarapurkar, D.; Wangikar, P.P. Studies on toxicity of antitubercular drugs namely isoniazid, rifampicin, and pyrazinamide in an in vitro model of HepG2 cell line. Med. Chem. Res. 2010, 20, 1611–1615. [Google Scholar] [CrossRef]
- Li, S.; Tan, H.Y.; Wang, N.; Zhang, Z.J.; Lao, L.; Wong, C.W.; Feng, Y. The Role of Oxidative Stress and Antioxidants in Liver Diseases. Int. J. Mol. Sci. 2015, 16, 26087–26124. [Google Scholar] [CrossRef] [Green Version]
- Surai, P. Silymarin as a Natural Antioxidant: An Overview of the Current Evidence and Perspectives. Antioxidants 2015, 4, 204. [Google Scholar] [CrossRef] [Green Version]
- Patel, N.; Joseph, C.; Corcoran, G.B.; Ray, S.D. Silymarin modulates doxorubicin-induced oxidative stress, Bcl-xL and p53 expression while preventing apoptotic and necrotic cell death in the liver. Toxicol. Appl. Pharm. 2010, 245, 143–152. [Google Scholar] [CrossRef] [PubMed]
- Fanoudi, S.; Alavi, M.S.; Karimi, G.; Hosseinzadeh, H. Milk thistle (Silybum Marianum) as an antidote or a protective agent against natural or chemical toxicities: A review. Drug Chem. Toxicol. 2018, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Ma, Q.; Wang, L.H.; Jiang, J.G. Hepatoprotective effect of flavonoids from Cirsium japonicum DC on hepatotoxicity in comparison with silymarin. Food Funct. 2016, 7, 2179–2184. [Google Scholar] [CrossRef] [PubMed]
- Tran, V.N.; Viktorova, J.; Augustynkova, K.; Jelenova, N.; Dobiasova, S.; Rehorova, K.; Fenclova, M.; Stranska-Zachariasova, M.; Vitek, L.; Hajslova, J. In Silico and In Vitro Studies of Mycotoxins and Their Cocktails; Their Toxicity and Its Mitigation by Silibinin Pre-Treatment. Toxins 2020, 12, 148. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Flora, K.; Hahn, M.; Rosen, H.; Benner, K. Milk thistle (Silybum marianum) for the therapy of liver disease. Am. J. Gastroenterol. 1998, 93, 139–143. [Google Scholar] [CrossRef] [PubMed]
- Polachi, N.; Bai, G.; Li, T.; Chu, Y.; Wang, X.; Li, S.; Gu, N.; Wu, J.; Li, W.; Zhang, Y.; et al. Modulatory effects of silibinin in various cell signaling pathways against liver disorders and cancer—A comprehensive review. Eur. J. Med. Chem. 2016, 123, 577–595. [Google Scholar] [CrossRef]
- Rodriguez-Garcia, A.; Hevia, D.; Mayo, J.C.; Gonzalez-Menendez, P.; Coppo, L.; Lu, J.; Holmgren, A.; Sainz, R.M. Thioredoxin 1 modulates apoptosis induced by bioactive compounds in prostate cancer cells. Redox. Biol. 2017, 12, 634–647. [Google Scholar] [CrossRef]
- Vargas-Mendoza, N.; Madrigal-Santillan, E.; Morales-Gonzalez, A.; Esquivel-Soto, J.; Esquivel-Chirino, C.; Garcia-Luna, Y.G.-R.M.; Gayosso-de-Lucio, J.A.; Morales-Gonzalez, J.A. Hepatoprotective effect of silymarin. World J. Hepatol. 2014, 6, 144–149. [Google Scholar] [CrossRef]
- Essid, E.; Dernawi, Y.; Petzinger, E. Apoptosis Induction by OTA and TNF-α in Cultured Primary Rat Hepatocytes and Prevention by Silibinin. Toxins 2012, 4, 1139–1156. [Google Scholar] [CrossRef] [Green Version]
- Essid, E.; Petzinger, E. Silibinin pretreatment protects against Ochratoxin A-mediated apoptosis in primary rat hepatocytes. Mycotoxin Res. 2011, 27, 167–176. [Google Scholar] [CrossRef]
- Abenavoli, L.; Izzo, A.A.; Milic, N.; Cicala, C.; Santini, A.; Capasso, R. Milk thistle (Silybum marianum): A concise overview on its chemistry, pharmacological, and nutraceutical uses in liver diseases. Phytother. Res. 2018, 32, 2202–2213. [Google Scholar] [CrossRef]
- Abdel-Moneim, A.M.; Al-Kahtani, M.A.; El-Kersh, M.A.; Al-Omair, M.A. Free Radical-Scavenging, Anti-Inflammatory/Anti-Fibrotic and Hepatoprotective Actions of Taurine and Silymarin against CCl4 Induced Rat Liver Damage. PLoS ONE 2015, 10, e0144509. [Google Scholar] [CrossRef] [Green Version]
- Kim, E.J.; Lee, M.Y.; Jeon, Y.J. Silymarin Inhibits Morphological Changes in LPS-Stimulated Macrophages by Blocking NF-kappaB Pathway. Korean J. Physiol. Pharm. 2015, 19, 211–218. [Google Scholar] [CrossRef] [Green Version]
- Zhang, B.; Xu, D.; She, L.; Wang, Z.; Yang, N.; Sun, R.; Zhang, Y.; Yan, C.; Wei, Q.; Aa, J.; et al. Silybin inhibits NLRP3 inflammasome assembly through the NAD(+)/SIRT2 pathway in mice with nonalcoholic fatty liver disease. FASEB J. 2018, 32, 757–767. [Google Scholar] [CrossRef] [Green Version]
- Roubalova, L.; Dinkova-Kostova, A.T.; Biedermann, D.; Kren, V.; Ulrichova, J.; Vrba, J. Flavonolignan 2,3-dehydrosilydianin activates Nrf2 and upregulates NAD(P)H:quinone oxidoreductase 1 in Hepa1c1c7 cells. Fitoterapia 2017, 119, 115–120. [Google Scholar] [CrossRef] [Green Version]
- Mann, A.; Pelz, T.; Rennert, K.; Mosig, A.; Decker, M.; Lupp, A. Evaluation of HepaRG cells for the assessment of indirect drug-induced hepatotoxicity using INH as a model substance. Hum. Cell 2017, 1–12. [Google Scholar] [CrossRef]
- Raghu, R.; Karthikeyan, S. Zidovudine and isoniazid induced liver toxicity and oxidative stress: Evaluation of mitigating properties of silibinin. Environ. Toxicol Pharm. 2016, 46, 217–226. [Google Scholar] [CrossRef]
- Luangchosiri, C.; Thakkinstian, A.; Chitphuk, S.; Stitchantrakul, W.; Petraksa, S.; Sobhonslidsuk, A. A double-blinded randomized controlled trial of silymarin for the prevention of antituberculosis drug-induced liver injury. BMC Complement. Altern. Med. 2015, 15, 334. [Google Scholar] [CrossRef] [Green Version]
- Marjani, M.; Baghaei, P.; Kazempour Dizaji, M.; Gorji Bayani, P.; Fahimi, F.; Tabarsi, P.; Velayati, A.A. Evaluation of Hepatoprotective Effect of Silymarin Among Under Treatment Tuberculosis Patients: A Randomized Clinical Trial. Iran. J. Pharm. Res. 2016, 15, 247–252. [Google Scholar]
- Gu, J.; Tang, S.-J.; Tan, S.-Y.; Wu, Q.; Zhang, X.; Liu, C.-X.; Gao, X.-S.; Yuan, B.-D.; Han, L.-J.; Gao, A.-P.; et al. An open-label, randomized and multi-center clinical trial to evaluate the efficacy of Silibinin in preventing drug-induced liver injury. Int. J. Clin. Exp. Med. 2015, 8, 4320–4327. [Google Scholar]
- Bokemeyer, C.; Lentzen, H.; Stolte, H.; Schmoll, H.; Gaedeke, J.; Fels, L.; Dunn, T.; Voigt, W. Silibinin protects against cisplatin-induced nephrotoxicity without compromising cisplatin or ifosfamide anti-tumour activity. Br. J. Cancer 1996, 74, 2036. [Google Scholar] [CrossRef] [PubMed]
- Elhag, R.; Mazzio, E.A.; Soliman, K.F. The effect of silibinin in enhancing toxicity of temozolomide and etoposide in p53 and PTEN-mutated resistant glioma cell lines. Anticancer Res. 2015, 35, 1263–1269. [Google Scholar] [PubMed]
- Tiwari, P.; Mishra, K. Silibinin in cancer therapy: A promising prospect. Cancer Res. Front. 2015, 1, 303–318. [Google Scholar] [CrossRef]
- Shen, C.; Meng, Q.; Zhang, G.; Hu, W. Rifampicin exacerbates isoniazid-induced toxicity in human but not in rat hepatocytes in tissue-like cultures. Br. J. Pharmacol. 2008, 153, 784–791. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Z.-H.; Tang, J.-H.; Zhan, Z.-L.; Zhang, X.-L.; Wu, H.-H.; Hou, Y.-N. Cellular toxicity of isoniazid together with rifampicin and the metabolites of isoniazid on QSG-7701 hepatocytes. Asian Pac. J. Trop. Med. 2012, 5, 306–309. [Google Scholar] [CrossRef] [Green Version]
- Biswas, A.; Santra, S.; Bishnu, D.; Dhali, G.K.; Chowdhury, A.; Santra, A. Isoniazid and Rifampicin Produce Hepatic Fibrosis through an Oxidative Stress-Dependent Mechanism. Int. J. Hepatol. 2020, 2020, 6987295. [Google Scholar] [CrossRef] [Green Version]
- Chowdhury, A.; Santra, A.; Bhattacharjee, K.; Ghatak, S.; Saha, D.R.; Dhali, G.K. Mitochondrial oxidative stress and permeability transition in isoniazid and rifampicin induced liver injury in mice. J. Hepatol. 2006, 45, 117–126. [Google Scholar] [CrossRef]
- Steele, M.A.; Burk, R.F.; DesPrez, R.M. Toxic Hepatitis with Isoniazid and Rifampin: A Meta-analysis. Chest 1991, 99, 465–471. [Google Scholar] [CrossRef]
- Yew, W.W. Clinically Significant Interactions with Drugs Used in the Treatment of Tuberculosis. Drug Saf. 2002, 25, 111–113. [Google Scholar] [CrossRef]
- Halliwell, B.; Whiteman, M. Measuring reactive species and oxidative damage in vivo and in cell culture: How should you do it and what do the results mean? Br. J. Pharmacol. 2004, 142, 231–255. [Google Scholar] [CrossRef] [Green Version]
- Collins, A.R. Measuring oxidative damage to DNA and its repair with the comet assay. Biochim. Biophys. Acta 2014, 1840, 794–800. [Google Scholar] [CrossRef] [PubMed]
- Redza-Dutordoir, M.; Averill-Bates, D.A. Activation of apoptosis signalling pathways by reactive oxygen species. Biochim. Biophys. Acta 2016, 1863, 2977–2992. [Google Scholar] [CrossRef]
- Boelsterli, U.A.; Lee, K.K. Mechanisms of isoniazid-induced idiosyncratic liver injury: Emerging role of mitochondrial stress. J. Gastroenterol. Hepatol. 2014, 29, 678–687. [Google Scholar] [CrossRef]
- Kim, M.; Yang, S.-G.; Kim, J.M.; Lee, J.-W.; Kim, Y.S.; Lee, J.I. Silymarin suppresses hepatic stellate cell activation in a dietary rat model of non-alcoholic steatohepatitis: Analysis of isolated hepatic stellate cells. Int. J. Mol. Med. 2012, 30, 473–479. [Google Scholar] [CrossRef] [Green Version]
- Björnsson, E. Hepatotoxicity by drugs: The most common implicated agents. Int. J. Mol. Sci. 2016, 17, 224. [Google Scholar] [CrossRef] [Green Version]
- Esselun, C.; Bruns, B.; Hagl, S.; Grewal, R.; Eckert, G.P. Differential Effects of Silibinin A on Mitochondrial Function in Neuronal PC12 and HepG2 Liver Cells. Oxidative Med. Cell. Longev. 2019. [Google Scholar] [CrossRef]
- Sun, Q.; Sha, W.; Liu, H.-P.; Wang, P.; Liu, Z.-B.; Sun, W.-W.; Xiao, H.-P. Genetic Polymorphisms in Antioxidant Enzymes Modulate the Susceptibility of Idiosyncratic Antituberculous Drug-Induced Liver Injury and Treatment Outcomes in Patients with Tuberculosis. Pharmacother. J. Hum. Pharmacol. Drug Ther. 2020, 40, 4–16. [Google Scholar] [CrossRef]
- Chen, P.N.; Hsieh, Y.S.; Chiang, C.L.; Chiou, H.L.; Yang, S.F.; Chu, S.C. Silibinin inhibits invasion of oral cancer cells by suppressing the MAPK pathway. J. Dent. Res. 2006, 85, 220–225. [Google Scholar] [CrossRef]
- Hamza, R.Z.; Al-Harbi, M.S. Amelioration of paracetamol hepatotoxicity and oxidative stress on mice liver with silymarin and Nigella sativa extract supplements. Asian Pac. J. Trop. Biomed. 2015, 5, 521–531. [Google Scholar] [CrossRef] [Green Version]
- Malekinejad, H.; Rahmani, F.; Valivande-Azar, S.; Taheri-Broujerdi, M.; Bazargani-Gilani, B. Long-term administration of Silymarin augments proinflammatory mediators in the hippocampus of rats: Evidence for antioxidant and pro-oxidant effects. Hum. Exp. Toxicol. 2012, 31, 921–930. [Google Scholar] [CrossRef]
- Prochazkova, D.; Bousova, I.; Wilhelmova, N. Antioxidant and prooxidant properties of flavonoids. Fitoterapia 2011, 82, 513–523. [Google Scholar] [CrossRef]
- Forbes, S.J.; Newsome, P.N. Liver regeneration—Mechanisms and models to clinical application. Nat. Rev. Gastroenterol. Hepatol. 2016, 13, 473–485. [Google Scholar] [CrossRef]
- Ezhilarasan, D.; Evraerts, J.; Brice, S.; Buc-Calderon, P.; Karthikeyan, S.; Sokal, E.; Najimi, M. Silibinin Inhibits Proliferation and Migration of Human Hepatic Stellate LX-2 Cells. J. Clin. Exp. Hepatol. 2016, 6, 167–174. [Google Scholar] [CrossRef] [Green Version]
- Chan, Y.-T.; Wang, N.; Tan, H.Y.; Li, S.; Feng, Y. Targeting Hepatic Stellate Cells for the Treatment of Liver Fibrosis by Natural Products: Is It the Dawning of a New Era? Front. Pharmacol. 2020, 11. [Google Scholar] [CrossRef]
- Andrade, R.J. Reducing Risk of Severe Liver Injury in Patients Treated With Isoniazid. Clin. Gastroenterol. Hepatol. 2015, 13, 1683–1685. [Google Scholar] [CrossRef]
- Song, Z.; Deaciuc, I.; Song, M.; Lee, D.Y.; Liu, Y.; Ji, X.; McClain, C. Silymarin protects against acute ethanol-induced hepatotoxicity in mice. Alcohol Clin. Exp. Res. 2006, 30, 407–413. [Google Scholar] [CrossRef]
- Soleimani, V.; Delghandi, P.S.; Moallem, S.A.; Karimi, G. Safety and toxicity of silymarin, the major constituent of milk thistle extract: An updated review. Phytother. Res. 2019, 33, 1627–1638. [Google Scholar] [CrossRef]
- Liu, Y.; Xu, W.; Zhai, T.; You, J.; Chen, Y. Silibinin ameliorates hepatic lipid accumulation and oxidative stress in mice with non-alcoholic steatohepatitis by regulating CFLAR-JNK pathway. Acta Pharm. Sin. B 2019, 9, 745–757. [Google Scholar] [CrossRef]
- Li, S.; Li, J.; Shen, C.; Zhang, X.; Sun, S.; Cho, M.; Sun, C.; Song, Z. tert-Butylhydroquinone (tBHQ) protects hepatocytes against lipotoxicity via inducing autophagy independently of Nrf2 activation. Biochim. Biophys. Acta 2014, 1841, 22–33. [Google Scholar] [CrossRef] [Green Version]
- Kang, J.; Jeong, M.G.; Oh, S.; Jang, E.J.; Kim, H.K.; Hwang, E.S. A FoxO1-dependent, but NRF2-independent induction of heme oxygenase-1 during muscle atrophy. FEBS Lett. 2014, 588, 79–85. [Google Scholar] [CrossRef] [Green Version]
- Li, D.; Gu, Z.; Zhang, J.; Ma, S. Protective effect of inducible aldo-keto reductases on 4-hydroxynonenal- induced hepatotoxicity. Chem. Biol. Interact. 2019, 304, 124–130. [Google Scholar] [CrossRef]
- Davis, M.; Stamper, B.D. TAMH: A Useful In Vitro Model for Assessing Hepatotoxic Mechanisms. Biomed. Res. Int. 2016, 2016, 4780872. [Google Scholar] [CrossRef]
- Wu, Y.; Geng, X.C.; Wang, J.F.; Miao, Y.F.; Lu, Y.L.; Li, B. The HepaRG cell line, a superior in vitro model to L-02, HepG2 and hiHeps cell lines for assessing drug-induced liver injury. Cell Biol. Toxicol. 2016, 32, 37–59. [Google Scholar] [CrossRef]
- Liang, Q.; Sheng, Y.; Ji, L.; Min, Y.; Xia, Y.; Wang, Z. Acetaminophen-induced cytotoxicity on human normal liver L-02 cells and the protection of antioxidants. Toxicol. Mech. Methods 2010, 20, 273–278. [Google Scholar] [CrossRef]
- Du, K.; Ramachandran, A.; Jaeschke, H. Oxidative stress during acetaminophen hepatotoxicity: Sources, pathophysiological role and therapeutic potential. Redox. Biol. 2016, 10, 148–156. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.T.; Chee, C.B.; Hsu, L.Y.; Jagadesan, R.; Kaw, G.J.; Kong, P.M.; Lew, Y.J.; Lim, C.S.; Lim, T.T.; Lu, K.F.; et al. Ministry of Health Clinical Practice Guidelines: Prevention, Diagnosis and Management of Tuberculosis. Singap. Med. J. 2016, 57, 118–124, quiz 125. [Google Scholar] [CrossRef] [Green Version]
- Ramachandran, A.; Duan, L.; Akakpo, J.Y.; Jaeschke, H. Mitochondrial dysfunction as a mechanism of drug-induced hepatotoxicity: Current understanding and future perspectives. J. Clin. Transl. Res. 2018, 5. [Google Scholar] [CrossRef]
- Mitchell, J.R.; Thorgeirsson, U.P.; Black, M.; Timbrell, J.A.; Snodgrass, W.R.; Potter, W.Z.; Jollow, D.J.; Keiser, H.R. Increased incidence of isoniazid hepatitis in rapid acetylators: Possible relation to hydrazine metabolites. Clin. Pharmacol. Ther. 1975, 18, 70–79. [Google Scholar] [CrossRef]
- Varga, Z.; Seres, I.; Nagy, E.; Ujhelyi, L.; Balla, G.; Balla, J.; Antus, S. Structure prerequisite for antioxidant activity of silybin in different biochemical systems in vitro. Phytomedicine 2006, 13, 85–93. [Google Scholar] [CrossRef]
- Hu, X.; Yang, T.; Li, C.; Zhang, L.; Li, M.; Huang, W.; Zhou, P. Human fetal hepatocyte line, L-02, exhibits good liver function in vitro and in an acute liver failure model. Transpl. Proc. 2013, 45, 695–700. [Google Scholar] [CrossRef]
- Wu, J.C.; Merlino, G.; Cveklova, K.; Mosinger, B.; Fausto, N. Autonomous Growth in Serum-free Medium and Production of Hepatocellular Carcinomas by Differentiated Hepatocyte Lines that Overexpress Transforming Growth Factor α. Cancer Res. 1994, 54, 5964. [Google Scholar]
- Gyori, B.M.; Venkatachalam, G.; Thiagarajan, P.S.; Hsu, D.; Clement, M.V. OpenComet: An automated tool for comet assay image analysis. Redox Biol. 2014, 2, 457–465. [Google Scholar] [CrossRef] [Green Version]






© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Goh, Z.-H.; Tee, J.K.; Ho, H.K. An Evaluation of the In Vitro Roles and Mechanisms of Silibinin in Reducing Pyrazinamide- and Isoniazid-Induced Hepatocellular Damage. Int. J. Mol. Sci. 2020, 21, 3714. https://doi.org/10.3390/ijms21103714
Goh Z-H, Tee JK, Ho HK. An Evaluation of the In Vitro Roles and Mechanisms of Silibinin in Reducing Pyrazinamide- and Isoniazid-Induced Hepatocellular Damage. International Journal of Molecular Sciences. 2020; 21(10):3714. https://doi.org/10.3390/ijms21103714
Chicago/Turabian StyleGoh, Zhang-He, Jie Kai Tee, and Han Kiat Ho. 2020. "An Evaluation of the In Vitro Roles and Mechanisms of Silibinin in Reducing Pyrazinamide- and Isoniazid-Induced Hepatocellular Damage" International Journal of Molecular Sciences 21, no. 10: 3714. https://doi.org/10.3390/ijms21103714
APA StyleGoh, Z. -H., Tee, J. K., & Ho, H. K. (2020). An Evaluation of the In Vitro Roles and Mechanisms of Silibinin in Reducing Pyrazinamide- and Isoniazid-Induced Hepatocellular Damage. International Journal of Molecular Sciences, 21(10), 3714. https://doi.org/10.3390/ijms21103714