DNA Damage Regulates Senescence-Associated Extracellular Vesicle Release via the Ceramide Pathway to Prevent Excessive Inflammatory Responses

 , and
, and
Abstract
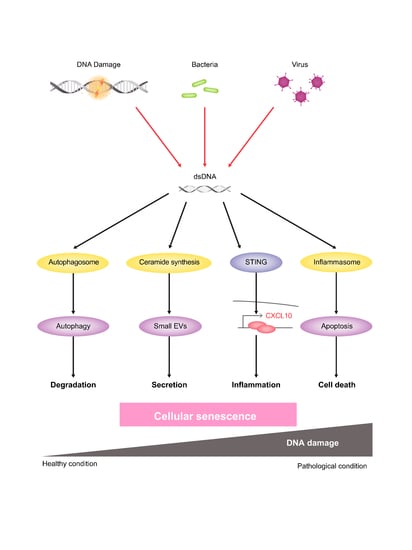
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
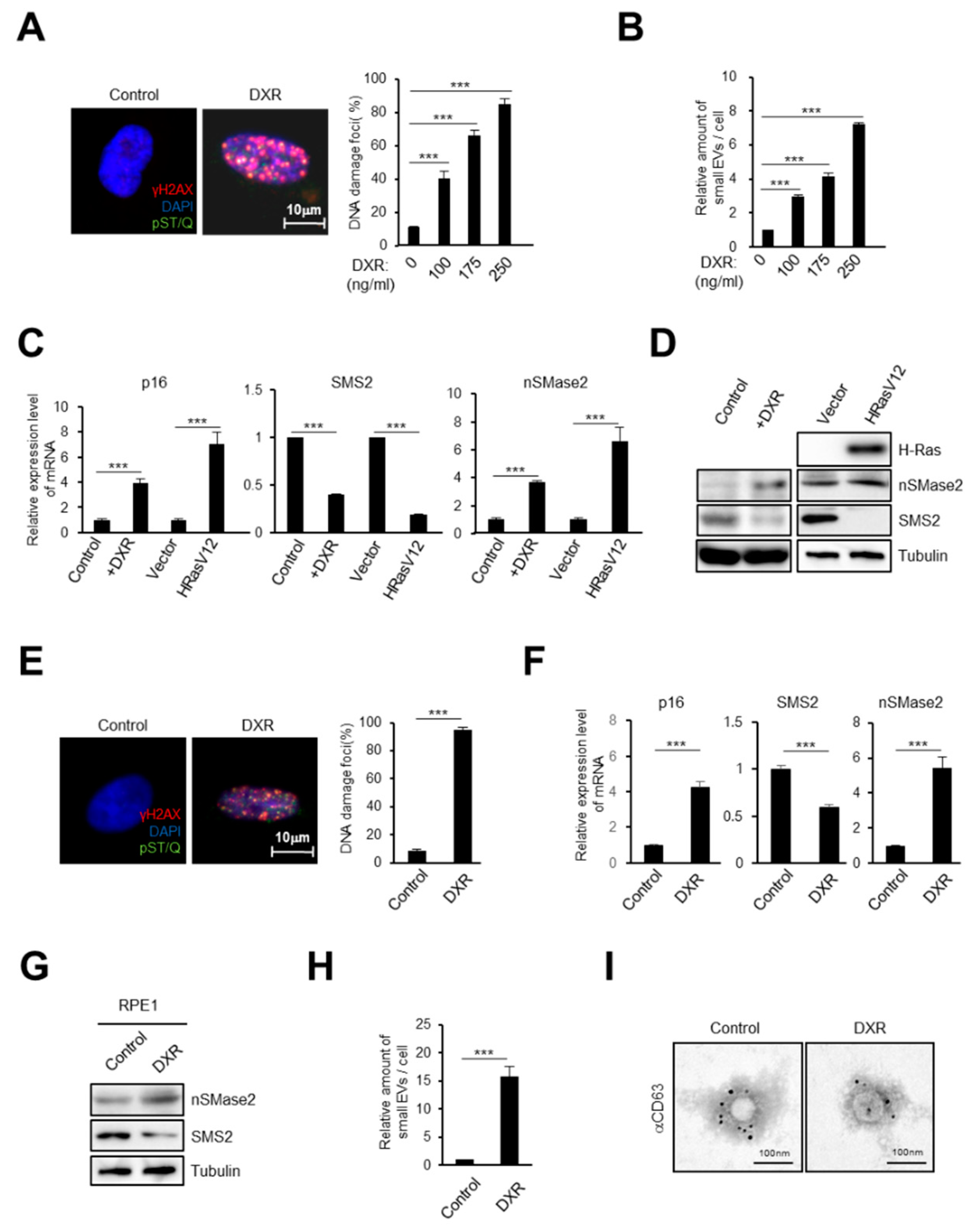
2.1. DNA Damage Induces Small EV Secretion from Normal Human Fibroblasts and Epithelial Cells
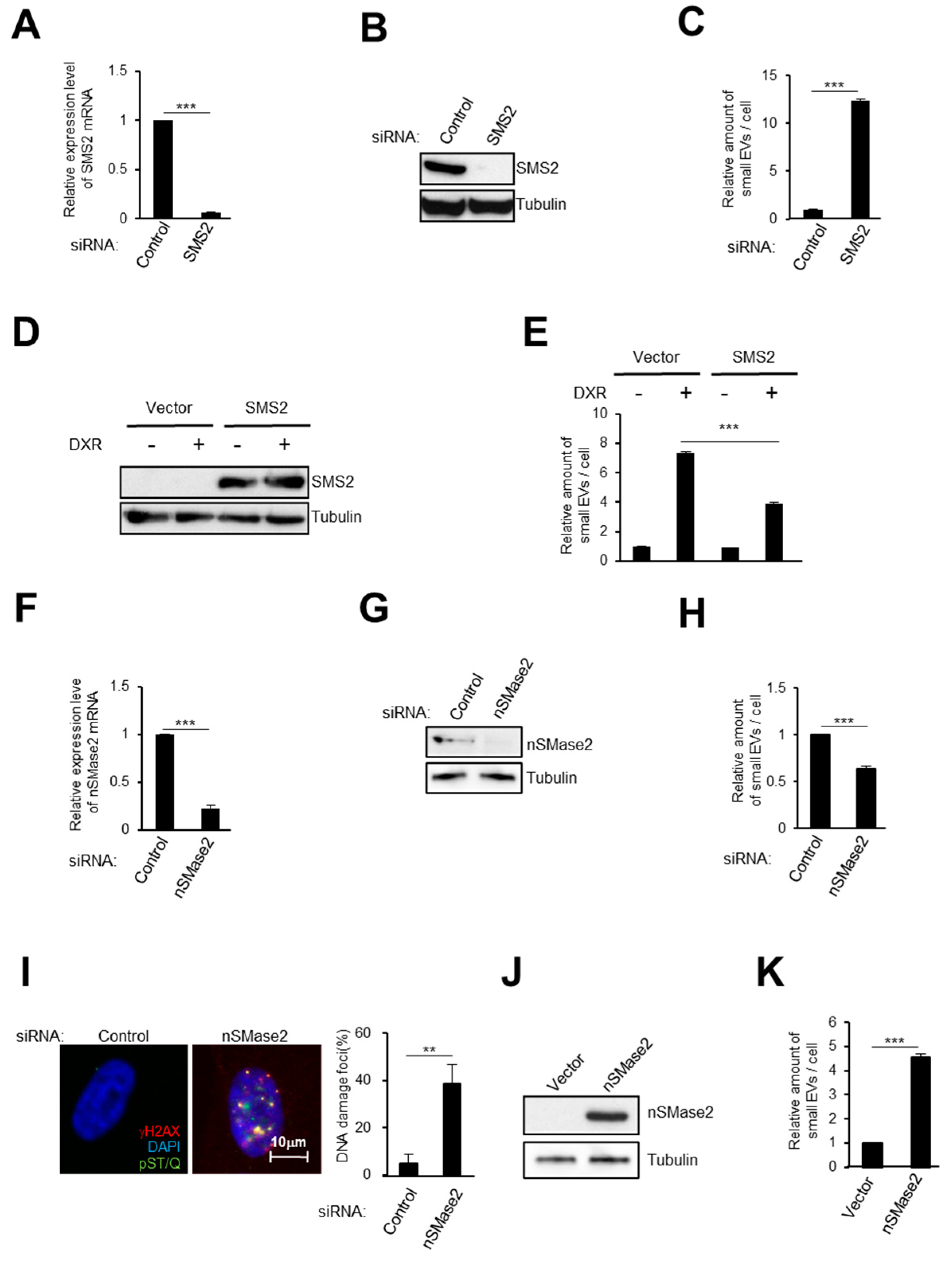
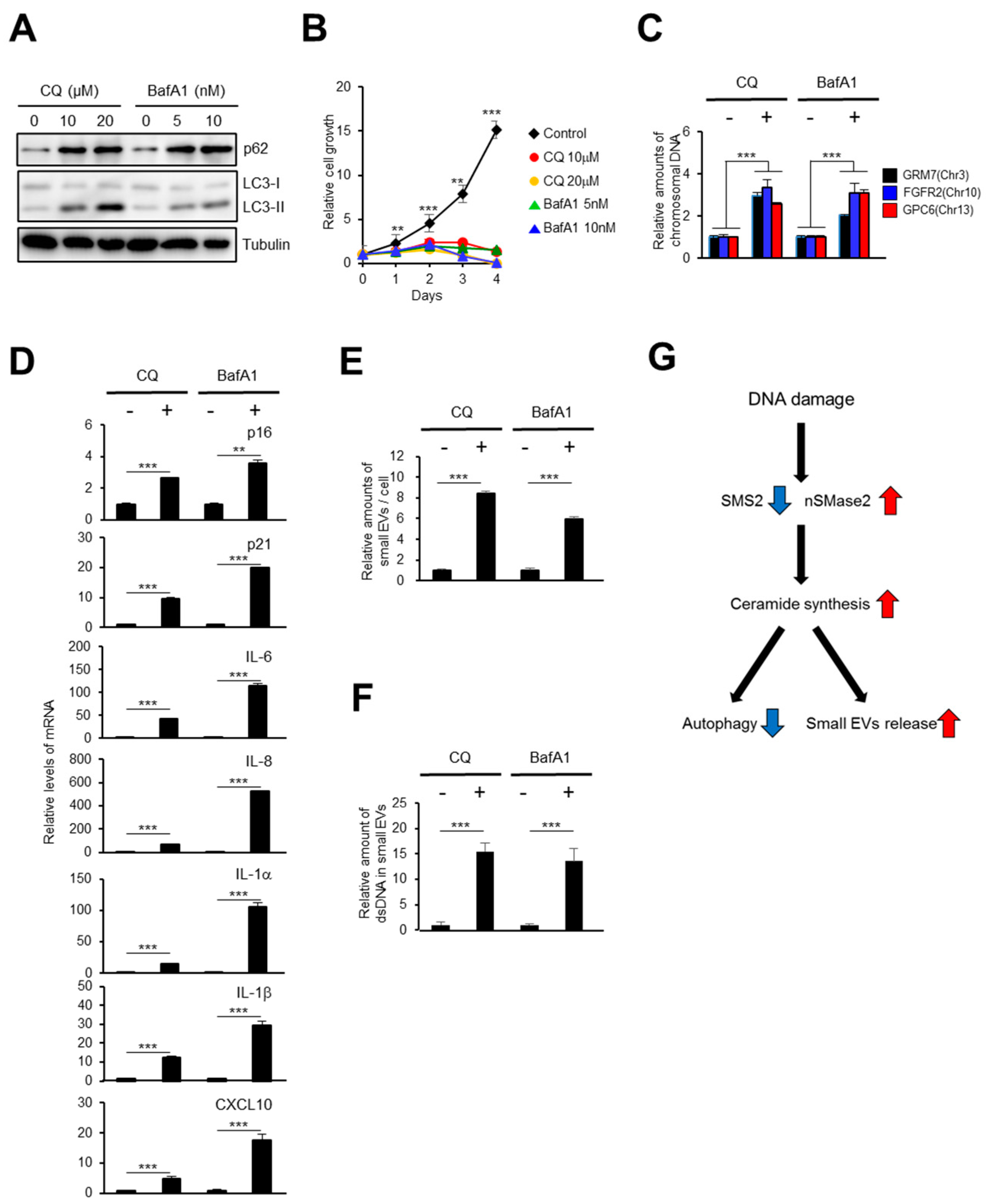
2.2. Activation of the Ceramide Synthetic Pathway Promotes Small EV Release from Cells
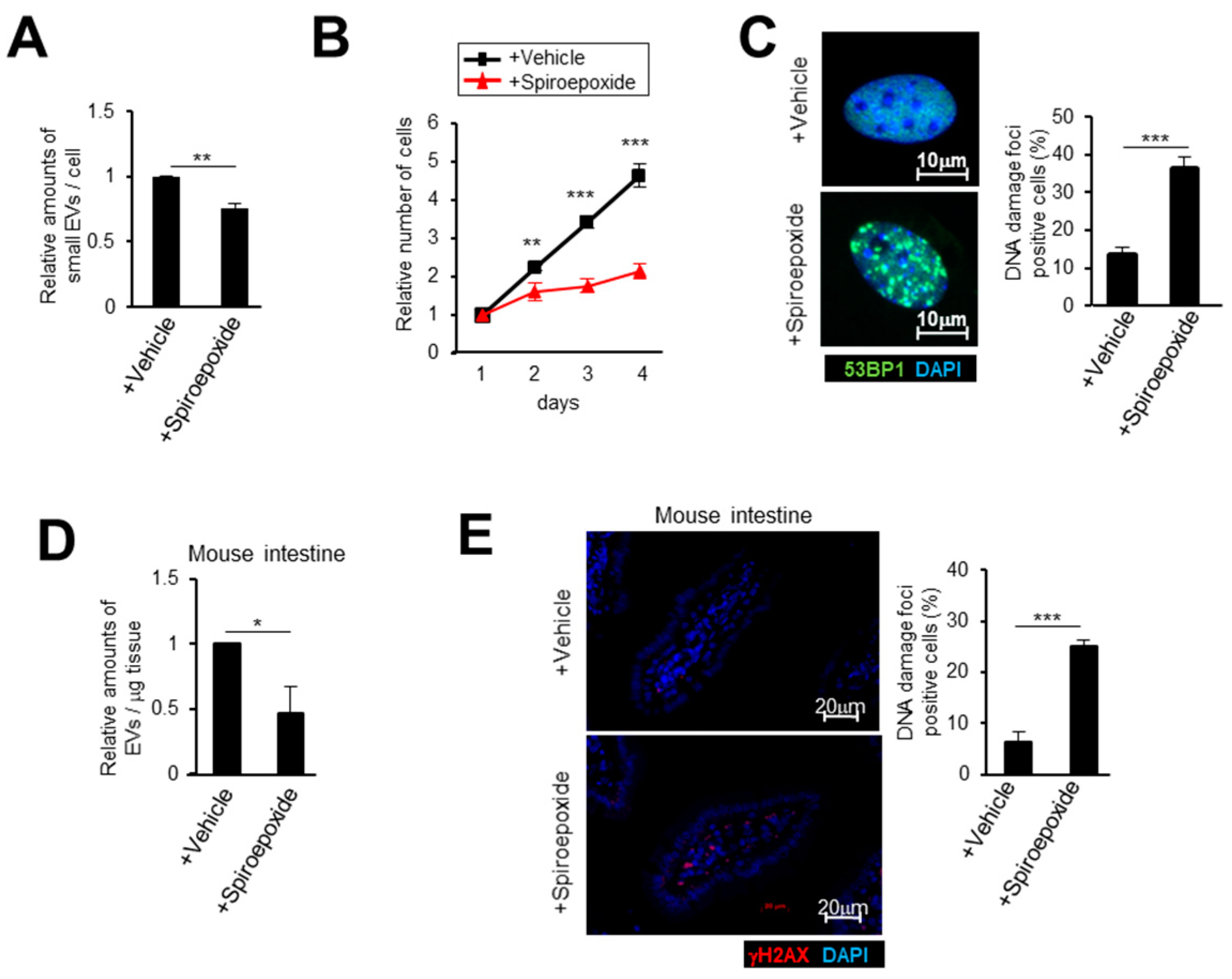
2.3. Small EV Release Via the Ceramide Pathway Prevents DNA Damage Accumulation in Mice
2.4. The Autophagy Pathway Prevents Accumulation of Chromosomal DNA Fragments in the Cytoplasm to Block SASP-Factor Gene Expression in Accompany with Small EV Release
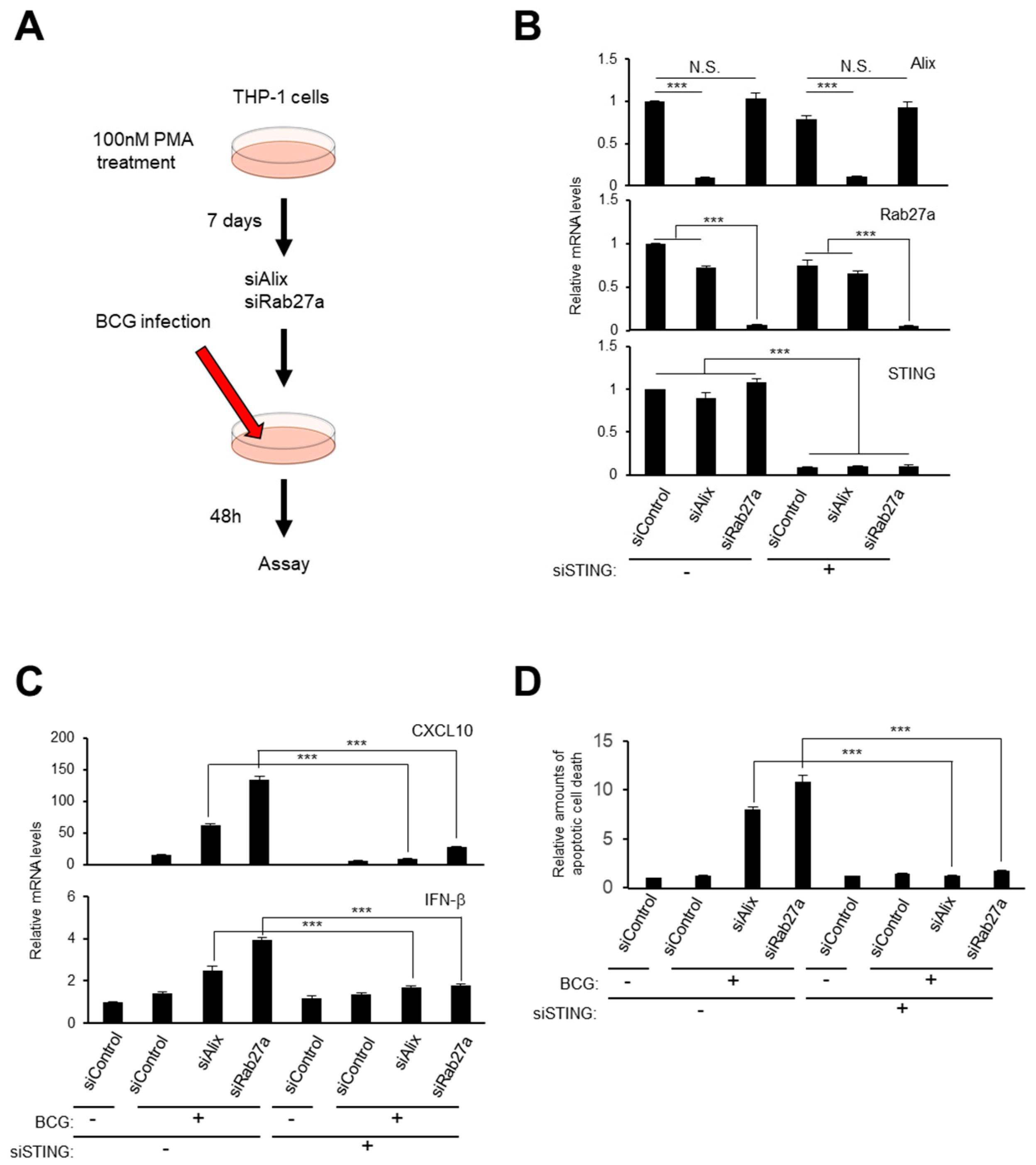
2.5. Small EV Release and Autophagy Cooperatively Prevent Cell Death Caused by Bacterial Infection
3. Discussion
4. Materials and Methods
4.1. Cell Culture
4.2. Cell Proliferation Assay
4.3. Apoptosis Assay
4.4. Cytoplasmic Nuclear DNA Analysis
4.5. Small Extracellular Vesicle Isolation from Cells
4.6. Fluorescence Microscopic Analysis
4.7. Electron Microscopy
4.8. RNAi
4.9. Plasmids
4.10. Quantitative Real-Time PCR
4.11. Western Blotting
4.12. Animal Experiments
4.13. Statistical Analysis
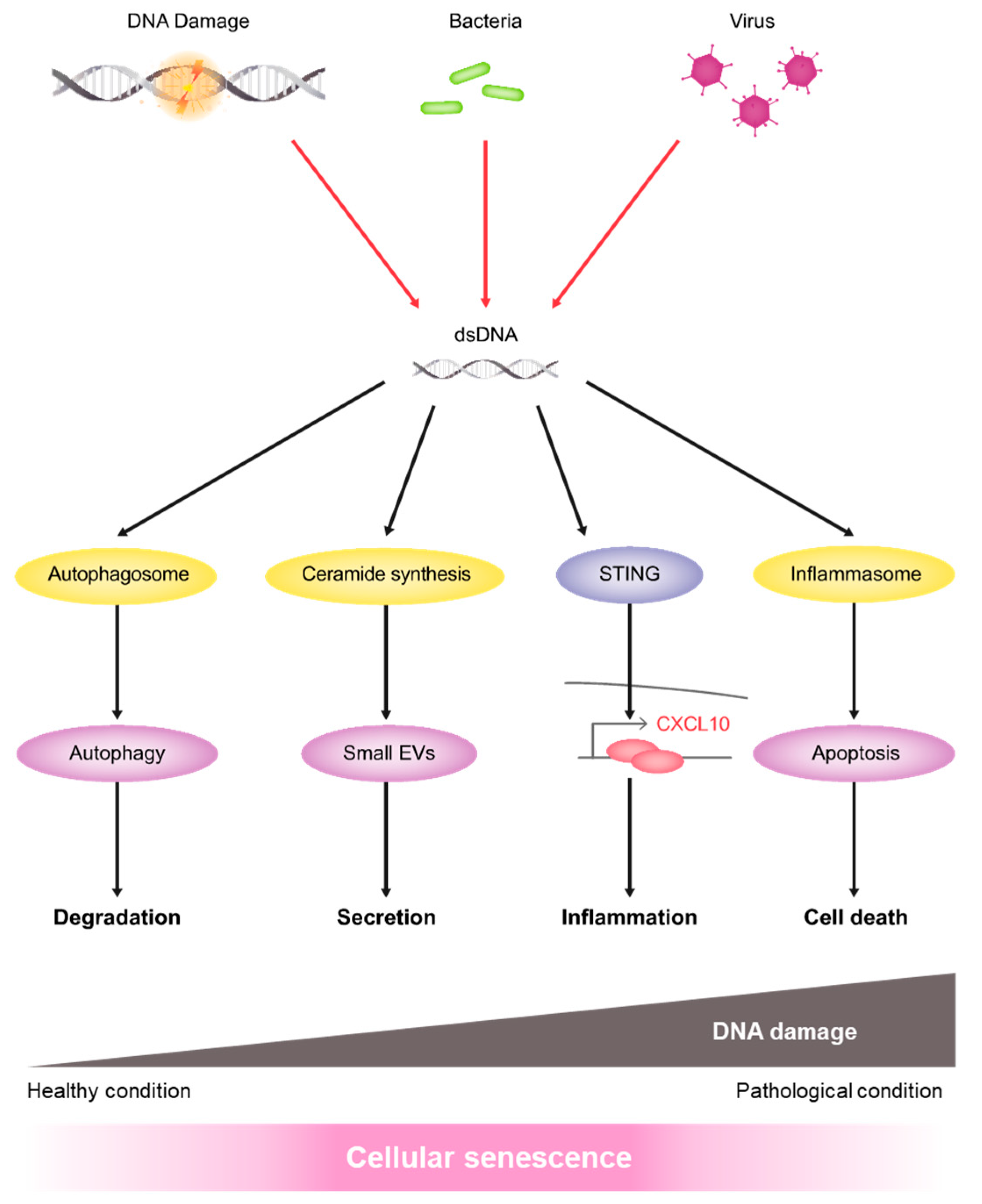
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| DDR | DNA damage response |
| EV | Extracellular vesicle |
| NTA | Nanoparticle tracking analysis |
| SASP | Senescence-associated secretory phenotype |
| cGAS | The cyclic GMP-AMP synthase |
| STING | Stimulator of interferon genes |
| WT | Wild-type |
| dsDNA | Double strand DNA |
References
- Campisi, J.; d’Adda di Fagagna, F. Cellular senescence: When bad things happen to good cells. Nat. Rev. Mol. Cell Biol. 2007, 8, 729–740. [Google Scholar] [CrossRef] [PubMed]
- White, R.R.; Vijg, J. Do DNA Double-Strand Breaks Drive Aging? Mol. Cell 2016, 63, 729–738. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Takeuchi, S.; Takahashi, A.; Motoi, N.; Yoshimoto, S.; Tajima, T.; Yamakoshi, K.; Hirao, A.; Yanagi, S.; Fukami, K.; Ishikawa, Y.; et al. Intrinsic cooperation between p16INK4a and p21Waf1/Cip1 in the onset of cellular senescence and tumor suppression in vivo. Cancer Res. 2010, 70, 9381–9390. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- He, S.; Sharpless, N.E. Senescence in Health and Disease. Cell 2017, 169, 1000–1011. [Google Scholar] [CrossRef]
- Gorgoulis, V.; Adams, P.D.; Alimonti, A.; Bennett, D.C.; Bischof, O.; Bishop, C.; Campisi, J.; Collado, M.; Evangelou, K.; Ferbeyre, G.; et al. Cellular Senescence: Defining a Path Forward. Cell 2019, 179, 813–827. [Google Scholar] [CrossRef]
- Maehara, K.; Yamakoshi, K.; Ohtani, N.; Kubo, Y.; Takahashi, A.; Arase, S.; Jones, N.; Hara, E. Reduction of total E2F/DP activity induces senescence-like cell cycle arrest in cancer cells lacking functional pRB and p53. J. Cell Biol. 2005, 168, 553–560. [Google Scholar] [CrossRef] [Green Version]
- Takahashi, A.; Ohtani, N.; Yamakoshi, K.; Iida, S.; Tahara, H.; Nakayama, K.; Nakayama, K.I.; Ide, T.; Saya, H.; Hara, E. Mitogenic signalling and the p16INK4a-Rb pathway cooperate to enforce irreversible cellular senescence. Nat. Cell Biol. 2006, 8, 1291–1297. [Google Scholar] [CrossRef]
- Imai, Y.; Takahashi, A.; Hanyu, A.; Hori, S.; Sato, S.; Naka, K.; Hirao, A.; Ohtani, N.; Hara, E. Crosstalk between the Rb pathway and AKT signaling forms a quiescence-senescence switch. Cell Rep. 2014, 7, 194–207. [Google Scholar] [CrossRef] [Green Version]
- Ohtani, N.; Imamura, Y.; Yamakoshi, K.; Hirota, F.; Nakayama, R.; Kubo, Y.; Ishimaru, N.; Takahashi, A.; Hirao, A.; Shimizu, T.; et al. Visualizing the dynamics of p21(Waf1/Cip1) cyclin-dependent kinase inhibitor expression in living animals. Proc. Natl. Acad. Sci. USA 2007, 104, 15034–15039. [Google Scholar] [CrossRef] [Green Version]
- Yamakoshi, K.; Takahashi, A.; Hirota, F.; Nakayama, R.; Ishimaru, N.; Kubo, Y.; Mann, D.J.; Ohmura, M.; Hirao, A.; Saya, H.; et al. Real-time in vivo imaging of p16Ink4a reveals cross talk with p53. J. Cell Biol. 2009, 186, 393–407. [Google Scholar] [CrossRef] [Green Version]
- Burd, C.E.; Sorrentino, J.A.; Clark, K.S.; Darr, D.B.; Krishnamurthy, J.; Deal, A.M.; Bardeesy, N.; Castrillon, D.H.; Beach, D.H.; Sharpless, N.E. Monitoring tumorigenesis and senescence in vivo with a p16(INK4a)-luciferase model. Cell 2013, 152, 340–351. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kuilman, T.; Michaloglou, C.; Mooi, W.J.; Peeper, D.S. The essence of senescence. Genes Dev. 2010, 24, 2463–2479. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Munoz-Espin, D.; Serrano, M. Cellular senescence: From physiology to pathology. Nat. Rev. Mol. Cell Biol. 2014, 15, 482–496. [Google Scholar] [CrossRef]
- Childs, B.G.; Gluscevic, M.; Baker, D.J.; Laberge, R.M.; Marquess, D.; Dananberg, J.; van Deursen, J.M. Senescent cells: An emerging target for diseases of ageing. Nat. Rev. Drug Discov. 2017, 16, 718–735. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Faget, D.V.; Ren, Q.; Stewart, S.A. Unmasking senescence: Context-dependent effects of SASP in cancer. Nat. Rev. Cancer 2019, 19, 439–453. [Google Scholar] [CrossRef]
- Loo, T.M.; Miyata, K.; Tanaka, Y.; Takahashi, A. Cellular senescence and senescence-associated secretory phenotype via the cGAS-STING signaling pathway in cancer. Cancer Sci. 2020, 111, 304–311. [Google Scholar] [CrossRef] [Green Version]
- Rodier, F.; Campisi, J. Four faces of cellular senescence. J. Cell Biol. 2011, 192, 547–556. [Google Scholar] [CrossRef]
- Takahashi, A.; Imai, Y.; Yamakoshi, K.; Kuninaka, S.; Ohtani, N.; Yoshimoto, S.; Hori, S.; Tachibana, M.; Anderton, E.; Takeuchi, T.; et al. DNA damage signaling triggers degradation of histone methyltransferases through APC/C(Cdh1) in senescent cells. Mol. Cell 2012, 45, 123–131. [Google Scholar] [CrossRef] [Green Version]
- Dou, Z.; Ghosh, K.; Vizioli, M.G.; Zhu, J.; Sen, P.; Wangensteen, K.J.; Simithy, J.; Lan, Y.; Lin, Y.; Zhou, Z.; et al. Cytoplasmic chromatin triggers inflammation in senescence and cancer. Nature 2017, 550, 402–406. [Google Scholar] [CrossRef] [Green Version]
- Gluck, S.; Guey, B.; Gulen, M.F.; Wolter, K.; Kang, T.W.; Schmacke, N.A.; Bridgeman, A.; Rehwinkel, J.; Zender, L.; Ablasser, A. Innate immune sensing of cytosolic chromatin fragments through cGAS promotes senescence. Nat. Cell Biol. 2017, 19, 1061–1070. [Google Scholar] [CrossRef]
- Yang, H.; Wang, H.; Ren, J.; Chen, Q.; Chen, Z.J. cGAS is essential for cellular senescence. Proc. Natl. Acad. Sci. USA 2017, 114, 4612–4620. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Takahashi, A.; Loo, T.M.; Okada, R.; Kamachi, F.; Watanabe, Y.; Wakita, M.; Watanabe, S.; Kawamoto, S.; Miyata, K.; Barber, G.N.; et al. Downregulation of cytoplasmic DNases is implicated in cytoplasmic DNA accumulation and SASP in senescent cells. Nat. Commun. 2018, 9, 1249. [Google Scholar] [CrossRef] [PubMed]
- De Cecco, M.; Ito, T.; Petrashen, A.P.; Elias, A.E.; Skvir, N.J.; Criscione, S.W.; Caligiana, A.; Brocculi, G.; Adney, E.M.; Boeke, J.D.; et al. L1 drives IFN in senescent cells and promotes age-associated inflammation. Nature 2019, 566, 73–78. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, A.; Okada, R.; Nagao, K.; Kawamata, Y.; Hanyu, A.; Yoshimoto, S.; Takasugi, M.; Watanabe, S.; Kanemaki, M.T.; Obuse, C.; et al. Exosomes maintain cellular homeostasis by excreting harmful DNA from cells. Nat. Commun. 2017, 8, 15287. [Google Scholar] [CrossRef] [Green Version]
- Takasugi, M.; Okada, R.; Takahashi, A.; Virya Chen, D.; Watanabe, S.; Hara, E. Small extracellular vesicles secreted from senescent cells promote cancer cell proliferation through EphA2. Nat. Commun. 2017, 8, 15729. [Google Scholar] [CrossRef]
- Riquelme, J.A.; Takov, K.; Santiago-Fernandez, C.; Rossello, X.; Lavandero, S.; Yellon, D.M.; Davidson, S.M. Increased production of functional small extracellular vesicles in senescent endothelial cells. J. Cell. Mol. Med. 2020, 24, 4871–4876. [Google Scholar] [CrossRef] [Green Version]
- Valadi, H.; Ekstrom, K.; Bossios, A.; Sjostrand, M.; Lee, J.J.; Lotvall, J.O. Exosome-mediated transfer of mRNAs and microRNAs is a novel mechanism of genetic exchange between cells. Nat. Cell Biol. 2007, 9, 654–659. [Google Scholar] [CrossRef] [Green Version]
- Raposo, G.; Stoorvogel, W. Extracellular vesicles: Exosomes, microvesicles, and friends. J. Cell Biol. 2013, 200, 373–383. [Google Scholar] [CrossRef] [Green Version]
- Tkach, M.; Thery, C. Communication by Extracellular Vesicles: Where We Are and Where We Need to Go. Cell 2016, 164, 1226–1232. [Google Scholar] [CrossRef] [Green Version]
- Jakhar, R.; Crasta, K. Exosomes as Emerging Pro-Tumorigenic Mediators of the Senescence-Associated Secretory Phenotype. Int. J. Mol. Sci. 2019, 20, 2547. [Google Scholar] [CrossRef] [Green Version]
- Miyazoe, Y.; Miuma, S.; Miyaaki, H.; Kanda, Y.; Nakashiki, S.; Sasaki, R.; Haraguchi, M.; Shibata, H.; Honda, T.; Taura, N.; et al. Extracellular vesicles from senescent hepatic stellate cells promote cell viability of hepatoma cells through increasing EGF secretion from differentiated THP-1 cells. Biomed. Rep. 2020, 12, 163–170. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lehmann, B.D.; Paine, M.S.; Brooks, A.M.; McCubrey, J.A.; Renegar, R.H.; Wang, R.; Terrian, D.M. Senescence-associated exosome release from human prostate cancer cells. Cancer Res. 2008, 68, 7864–7871. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moertl, S.; Buschmann, D.; Azimzadeh, O.; Schneider, M.; Kell, R.; Winkler, K.; Tapio, S.; Hornhardt, S.; Merl-Pham, J.; Pfaffl, M.W.; et al. Radiation Exposure of Peripheral Mononuclear Blood Cells Alters the Composition and Function of Secreted Extracellular Vesicles. Int. J. Mol. Sci. 2020, 21, 2336. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Trajkovic, K.; Hsu, C.; Chiantia, S.; Rajendran, L.; Wenzel, D.; Wieland, F.; Schwille, P.; Brugger, B.; Simons, M. Ceramide triggers budding of exosome vesicles into multivesicular endosomes. Science 2008, 319, 1244–1247. [Google Scholar] [CrossRef] [PubMed]
- Ostrowski, M.; Carmo, N.B.; Krumeich, S.; Fanget, I.; Raposo, G.; Savina, A.; Moita, C.F.; Schauer, K.; Hume, A.N.; Freitas, R.P.; et al. Rab27a and Rab27b control different steps of the exosome secretion pathway. Nat. Cell Biol. 2010, 12, 19–30. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baietti, M.F.; Zhang, Z.; Mortier, E.; Melchior, A.; Degeest, G.; Geeraerts, A.; Ivarsson, Y.; Depoortere, F.; Coomans, C.; Vermeiren, E.; et al. Syndecan–syntenin–ALIX regulates the biogenesis of exosomes. Nat. Cell Biol. 2012, 14, 677–685. [Google Scholar] [CrossRef]
- Colombo, M.; Moita, C.; van Niel, G.; Kowal, J.; Vigneron, J.; Benaroch, P.; Manel, N.; Moita, L.F.; Thery, C.; Raposo, G. Analysis of ESCRT functions in exosome biogenesis, composition and secretion highlights the heterogeneity of extracellular vesicles. J. Cell Sci. 2013, 126, 5553–5565. [Google Scholar] [CrossRef] [Green Version]
- Kosaka, N.; Iguchi, H.; Yoshioka, Y.; Takeshita, F.; Matsuki, Y.; Ochiya, T. Secretory mechanisms and intercellular transfer of microRNAs in living cells. J. Biol. Chem. 2010, 285, 17442–17452. [Google Scholar] [CrossRef] [Green Version]
- Mittelbrunn, M.; Sánchez-Madrid, F. Intercellular communication: Diverse structures for exchange of genetic information. Nat. Rev. Mol. Cell Biol. 2012, 13, 328–335. [Google Scholar] [CrossRef]
- Yuyama, K.; Sun, H.; Mitsutake, S.; Igarashi, Y. Sphingolipid-modulated exosome secretion promotes clearance of amyloid-beta by microglia. J. Biol. Chem. 2012, 287, 10977–10989. [Google Scholar] [CrossRef] [Green Version]
- Li, J.; Liu, K.; Liu, Y.; Xu, Y.; Zhang, F.; Yang, H.; Liu, J.; Pan, T.; Chen, J.; Wu, M.; et al. Exosomes mediate the cell-to-cell transmission of IFN-alpha-induced antiviral activity. Nat. Immunol. 2013, 14, 793–803. [Google Scholar] [CrossRef] [PubMed]
- Pols, M.S.; Klumperman, J. Trafficking and function of the tetraspanin CD63. Exp. Cell Res. 2009, 315, 1584–1592. [Google Scholar] [CrossRef] [PubMed]
- Mitsutake, S.; Zama, K.; Yokota, H.; Yoshida, T.; Tanaka, M.; Mitsui, M.; Ikawa, M.; Okabe, M.; Tanaka, Y.; Yamashita, T.; et al. Dynamic modification of sphingomyelin in lipid microdomains controls development of obesity, fatty liver, and type 2 diabetes. J. Biol. Chem. 2011, 286, 28544–28555. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lan, Y.Y.; Londono, D.; Bouley, R.; Rooney, M.S.; Hacohen, N. Dnase2a deficiency uncovers lysosomal clearance of damaged nuclear DNA via autophagy. Cell Rep. 2014, 9, 180–192. [Google Scholar] [CrossRef] [Green Version]
- Asada, K.; Ito, K.; Yui, D.; Tagaya, H.; Yokota, T. Cytosolic Genomic DNA functions as a Natural Antisense. Sci. Rep. 2018, 8, 8551. [Google Scholar] [CrossRef]
- Orsini, M.; Chateauvieux, S.; Rhim, J.; Gaigneaux, A.; Cheillan, D.; Christov, C.; Dicato, M.; Morceau, F.; Diederich, M. Sphingolipid-mediated inflammatory signaling leading to autophagy inhibition converts erythropoiesis to myelopoiesis in human hematopoietic stem/progenitor cells. Cell Death Differ. 2019, 26, 1796–1812. [Google Scholar] [CrossRef] [Green Version]
- Redmann, M.; Benavides, G.A.; Berryhill, T.F.; Wani, W.Y.; Ouyang, X.; Johnson, M.S.; Ravi, S.; Barnes, S.; Darley-Usmar, V.M.; Zhang, J. Inhibition of autophagy with bafilomycin and chloroquine decreases mitochondrial quality and bioenergetic function in primary neurons. Redox Biol. 2017, 11, 73–81. [Google Scholar] [CrossRef] [Green Version]
- Kuma, A.; Hatano, M.; Matsui, M.; Yamamoto, A.; Nakaya, H.; Yoshimori, T.; Ohsumi, Y.; Tokuhisa, T.; Mizushima, N. The role of autophagy during the early neonatal starvation period. Nature 2004, 432, 1032–1036. [Google Scholar] [CrossRef]
- Kang, C.; Xu, Q.; Martin, T.D.; Li, M.Z.; Demaria, M.; Aron, L.; Lu, T.; Yankner, B.A.; Campisi, J.; Elledge, S.J. The DNA damage response induces inflammation and senescence by inhibiting autophagy of GATA4. Science 2015, 349, aaa5612. [Google Scholar] [CrossRef] [Green Version]
- Leidal, A.M.; Levine, B.; Debnath, J. Autophagy and the cell biology of age-related disease. Nat. Cell Biol. 2018, 20, 1338–1348. [Google Scholar] [CrossRef]
- Jung, C.H.; Ro, S.H.; Cao, J.; Otto, N.M.; Kim, D.H. mTOR regulation of autophagy. FEBS Lett. 2010, 584, 1287–1295. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Herranz, N.; Gallage, S.; Mellone, M.; Wuestefeld, T.; Klotz, S.; Hanley, C.J.; Raguz, S.; Acosta, J.C.; Innes, A.J.; Banito, A.; et al. mTOR regulates MAPKAPK2 translation to control the senescence-associated secretory phenotype. Nat. Cell Biol. 2015, 17, 1205–1217. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Laberge, R.M.; Sun, Y.; Orjalo, A.V.; Patil, C.K.; Freund, A.; Zhou, L.; Curran, S.C.; Davalos, A.R.; Wilson-Edell, K.A.; Liu, S.; et al. MTOR regulates the pro-tumorigenic senescence-associated secretory phenotype by promoting IL1A translation. Nat. Cell Biol. 2015, 17, 1049–1061. [Google Scholar] [CrossRef] [PubMed]
- Collins, A.C.; Cai, H.; Li, T.; Franco, L.H.; Li, X.D.; Nair, V.R.; Scharn, C.R.; Stamm, C.E.; Levine, B.; Chen, Z.J.; et al. Cyclic GMP-AMP Synthase Is an Innate Immune DNA Sensor for Mycobacterium tuberculosis. Cell Host Microbe 2015, 17, 820–828. [Google Scholar] [CrossRef] [Green Version]
- Wassermann, R.; Gulen, M.F.; Sala, C.; Perin, S.G.; Lou, Y.; Rybniker, J.; Schmid-Burgk, J.L.; Schmidt, T.; Hornung, V.; Cole, S.T.; et al. Mycobacterium tuberculosis Differentially Activates cGAS- and Inflammasome-Dependent Intracellular Immune Responses through ESX-1. Cell Host Microbe 2015, 17, 799–810. [Google Scholar] [CrossRef] [Green Version]
- Watson, R.O.; Bell, S.L.; MacDuff, D.A.; Kimmey, J.M.; Diner, E.J.; Olivas, J.; Vance, R.E.; Stallings, C.L.; Virgin, H.W.; Cox, J.S. The Cytosolic Sensor cGAS Detects Mycobacterium tuberculosis DNA to Induce Type I Interferons and Activate Autophagy. Cell Host Microbe 2015, 17, 811–819. [Google Scholar] [CrossRef] [Green Version]
- Barber, G.N. STING-dependent cytosolic DNA sensing pathways. Trends Immunol. 2014, 35, 88–93. [Google Scholar] [CrossRef]
- Sergeeva, O.A.; van der Goot, F.G. Kicking Out Pathogens in Exosomes. Cell 2015, 161, 1241–1242. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, Y.T.; Tan, H.L.; Shui, G.; Bauvy, C.; Huang, Q.; Wenk, M.R.; Ong, C.N.; Codogno, P.; Shen, H.M. Dual role of 3-methyladenine in modulation of autophagy via different temporal patterns of inhibition on class I and III phosphoinositide 3-kinase. J. Biol. Chem. 2010, 285, 10850–10861. [Google Scholar] [CrossRef] [Green Version]
- Buratta, S.; Urbanelli, L.; Sagini, K.; Giovagnoli, S.; Caponi, S.; Fioretto, D.; Mitro, N.; Caruso, D.; Emiliani, C. Extracellular vesicles released by fibroblasts undergoing H-Ras induced senescence show changes in lipid profile. PLoS ONE 2017, 12, e0188840. [Google Scholar] [CrossRef]
- Jeppesen, D.K.; Fenix, A.M.; Franklin, J.L.; Higginbotham, J.N.; Zhang, Q.; Zimmerman, L.J.; Liebler, D.C.; Ping, J.; Liu, Q.; Evans, R.; et al. Reassessment of Exosome Composition. Cell 2019, 177, 428–445.e18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alibhai, F.J.; Lim, F.; Yeganeh, A.; DiStefano, P.V.; Binesh-Marvasti, T.; Belfiore, A.; Wlodarek, L.; Gustafson, D.; Millar, S.; Li, S.H.; et al. Cellular senescence contributes to age-dependent changes in circulating extracellular vesicle cargo and function. Aging Cell 2020, 19, e13103. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Basisty, N.; Kale, A.; Jeon, O.H.; Kuehnemann, C.; Payne, T.; Rao, C.; Holtz, A.; Shah, S.; Sharma, V.; Ferrucci, L.; et al. A proteomic atlas of senescence-associated secretomes for aging biomarker development. PLoS Biol. 2020, 18, e3000599. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yoshida, M.; Satoh, A.; Lin, J.B.; Mills, K.F.; Sasaki, Y.; Rensing, N.; Wong, M.; Apte, R.S.; Imai, S.I. Extracellular Vesicle-Contained eNAMPT Delays Aging and Extends Lifespan in Mice. Cell Metab. 2019, 30, 329–342.e5. [Google Scholar] [CrossRef]
- Yu, X.; Riley, T.; Levine, A.J. The regulation of the endosomal compartment by p53 the tumor suppressor gene. FEBS J. 2009, 276, 2201–2212. [Google Scholar] [CrossRef] [PubMed]
- Shamseddine, A.A.; Clarke, C.J.; Carroll, B.; Airola, M.V.; Mohammed, S.; Rella, A.; Obeid, L.M.; Hannun, Y.A. P53-dependent upregulation of neutral sphingomyelinase-2: Role in doxorubicin-induced growth arrest. Cell Death Dis. 2015, 6, e1947. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wakita, M.; Takahashi, A.; Sano, O.; Loo, T.M.; Imai, Y.; Narukawa, M.; Iwata, H.; Matsudaira, T.; Kawamoto, S.; Ohtani, N.; et al. A BET family protein degrader provokes senolysis by targeting NHEJ and autophagy in senescent cells. Nat. Commun. 2020, 11, 1935. [Google Scholar] [CrossRef] [Green Version]
- Kawashima, A.; Tanigawa, K.; Akama, T.; Wu, H.; Sue, M.; Yoshihara, A.; Ishido, Y.; Kobiyama, K.; Takeshita, F.; Ishii, K.J.; et al. Fragments of genomic DNA released by injured cells activate innate immunity and suppress endocrine function in the thyroid. Endocrinology 2011, 152, 1702–1712. [Google Scholar] [CrossRef] [Green Version]
- Thery, C.; Amigorena, S.; Raposo, G.; Clayton, A. Isolation and characterization of exosomes from cell culture supernatants and biological fluids. Curr. Protoc. Cell Biol. 2006, 30, 3.22.1–3.22.29. [Google Scholar] [CrossRef]
- Yoshida, T.; Ishidome, T.; Hanayama, R. High Purity Isolation and Sensitive Quantification of Extracellular Vesicles Using Affinity to TIM4. Curr. Protoc. Cell Biol. 2017, 77, 3.45.1–3.45.18. [Google Scholar] [CrossRef]
- Hartlova, A.; Erttmann, S.F.; Raffi, F.A.; Schmalz, A.M.; Resch, U.; Anugula, S.; Lienenklaus, S.; Nilsson, L.M.; Kroger, A.; Nilsson, J.A.; et al. DNA damage primes the type I interferon system via the cytosolic DNA sensor STING to promote anti-microbial innate immunity. Immunity 2015, 42, 332–343. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marchesini, N.; Osta, W.; Bielawski, J.; Luberto, C.; Obeid, L.M.; Hannun, Y.A. Role for mammalian neutral sphingomyelinase 2 in confluence-induced growth arrest of MCF7 cells. J. Biol. Chem. 2004, 279, 25101–25111. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Papaparaskevas, J.; Houhoula, D.P.; Siatelis, A.; Tsakris, A. Molecular-beacon-based real-time PCR for detection and quantification of Mycobacterium tuberculosis DNA in clinical samples. J. Clin. Microbiol. 2008, 46, 3177–3178. [Google Scholar] [CrossRef] [PubMed] [Green Version]






© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hitomi, K.; Okada, R.; Loo, T.M.; Miyata, K.; Nakamura, A.J.; Takahashi, A. DNA Damage Regulates Senescence-Associated Extracellular Vesicle Release via the Ceramide Pathway to Prevent Excessive Inflammatory Responses. Int. J. Mol. Sci. 2020, 21, 3720. https://doi.org/10.3390/ijms21103720
Hitomi K, Okada R, Loo TM, Miyata K, Nakamura AJ, Takahashi A. DNA Damage Regulates Senescence-Associated Extracellular Vesicle Release via the Ceramide Pathway to Prevent Excessive Inflammatory Responses. International Journal of Molecular Sciences. 2020; 21(10):3720. https://doi.org/10.3390/ijms21103720
Chicago/Turabian StyleHitomi, Kazuhiro, Ryo Okada, Tze Mun Loo, Kenichi Miyata, Asako J. Nakamura, and Akiko Takahashi. 2020. "DNA Damage Regulates Senescence-Associated Extracellular Vesicle Release via the Ceramide Pathway to Prevent Excessive Inflammatory Responses" International Journal of Molecular Sciences 21, no. 10: 3720. https://doi.org/10.3390/ijms21103720
APA StyleHitomi, K., Okada, R., Loo, T. M., Miyata, K., Nakamura, A. J., & Takahashi, A. (2020). DNA Damage Regulates Senescence-Associated Extracellular Vesicle Release via the Ceramide Pathway to Prevent Excessive Inflammatory Responses. International Journal of Molecular Sciences, 21(10), 3720. https://doi.org/10.3390/ijms21103720