Adherent-Invasive E. coli: Update on the Lifestyle of a Troublemaker in Crohn’s Disease



{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. The Challenging Identification of AIEC Bacteria in CD Patients
3. Access to the Mucosa
3.1. Mucus Crossing
3.2. Flagellum Regulation
3.3. Resistance to Antimicrobial Peptides
4. Interaction with the Intestinal Epithelium: Adhesion to IECs
4.1. Type 1 pili-CEACAM6 Interaction
4.2. ChiA-Chitinase 3-Like-1 Interaction
4.3. AIEC-M-cells Interaction
4.4. Invasion of IECs by AIEC Bacteria
5. Consequences of AIEC Interaction with Epithelial Layer
5.1. Effects of AIEC on Epithelial Barrier Function
5.2. Effects of AIEC on Host Glycosylation
5.3. Induction of Fibrosis by AIEC
5.4. Induction of Inflammation during AIEC Colonization
5.4.1. Through the TLR Pathway
5.4.2. Through Survival and Replication within Macrophages
5.4.3. Through the Release of Exosomes
5.5. Control of AIEC Intracellular Replication
5.5.1. Role of Autophagy in the Control of AIEC Replication
5.5.2. Role of micro-RNAs in the Control of AIEC Replication
5.5.3. Role of SUMOylation in the Control of AIEC Replication
6. AIEC in the Lumen
6.1. Intestinal Environment Favoring AIEC Virulence and Colonization
6.1.1. Regulation of Virulence Genes by Luminal Molecules
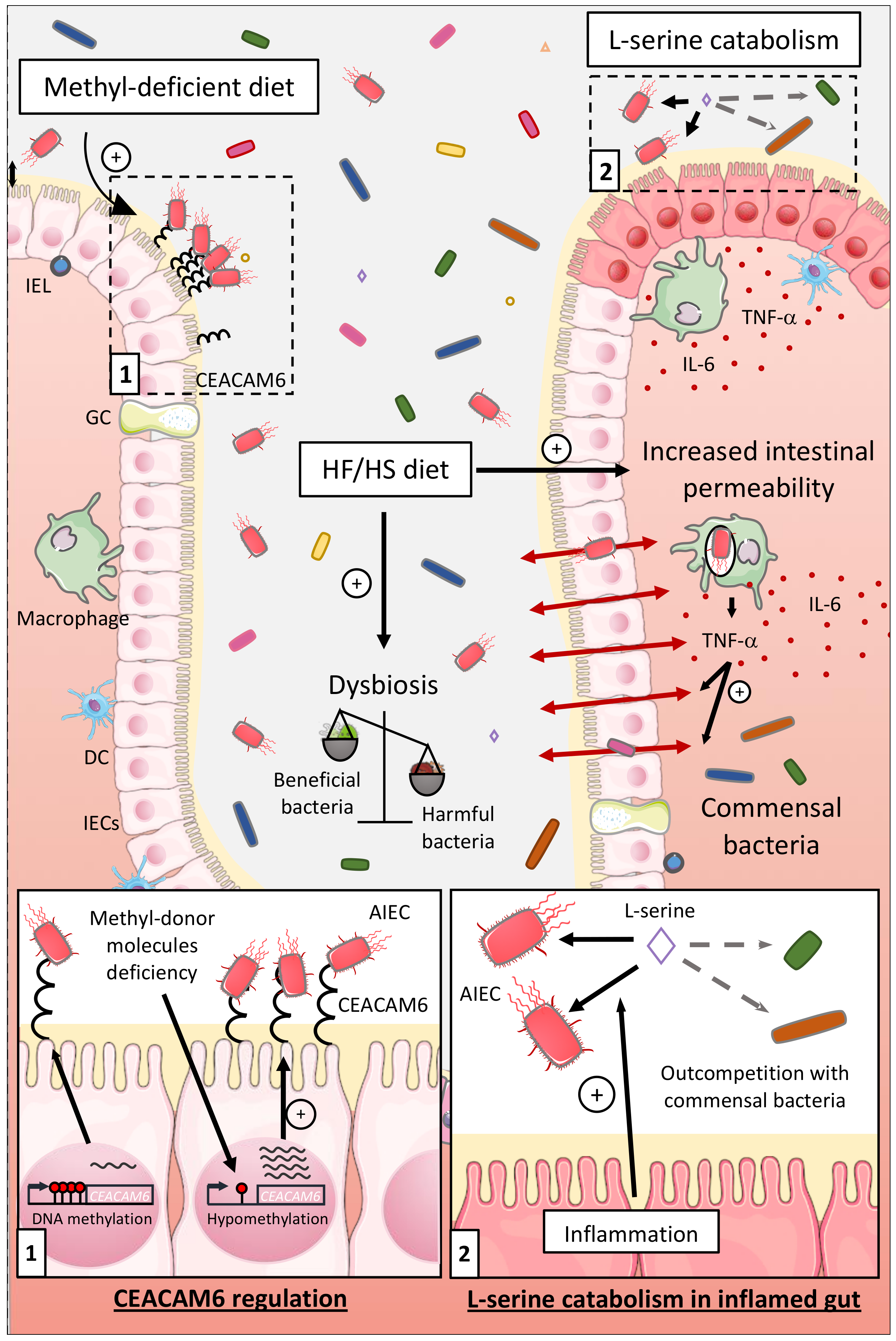
6.1.2. Influence of the Diet on the Ability of AIEC to Colonize in Intestinal Mucosa
6.1.3. Influence of the Nutrients and Carbon Sources on the Fitness of AIEC
6.2. Impact of AIEC Infection on Microbiota Composition
7. Targeting AIEC
7.1. Inhibition of AIEC–IECs Interaction
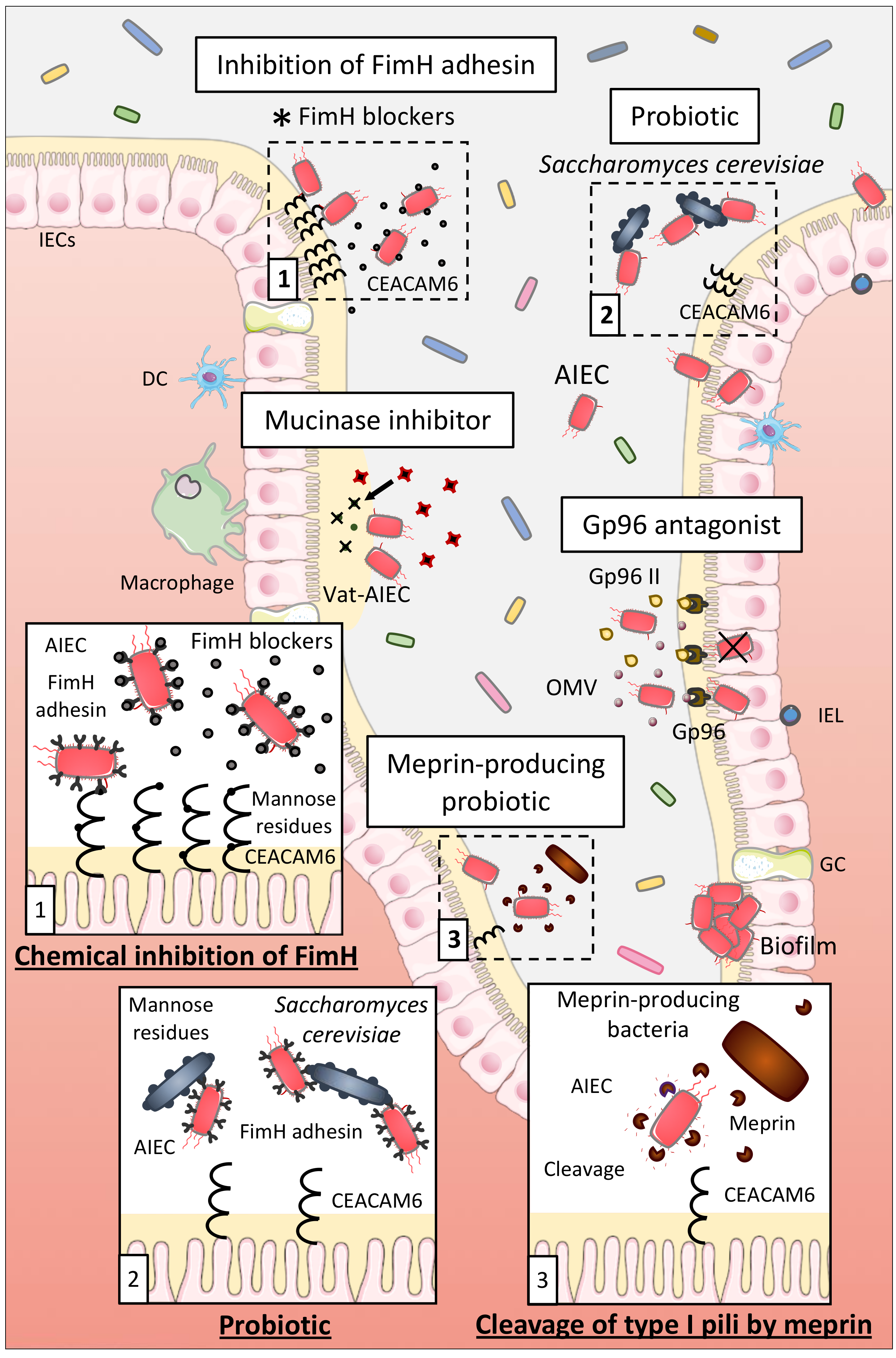
7.1.1. Probiotics
7.1.2. Inhibition of AIEC Adhesion Using Chemical Compounds
7.1.3. Other Strategies
7.2. Elimination of AIEC
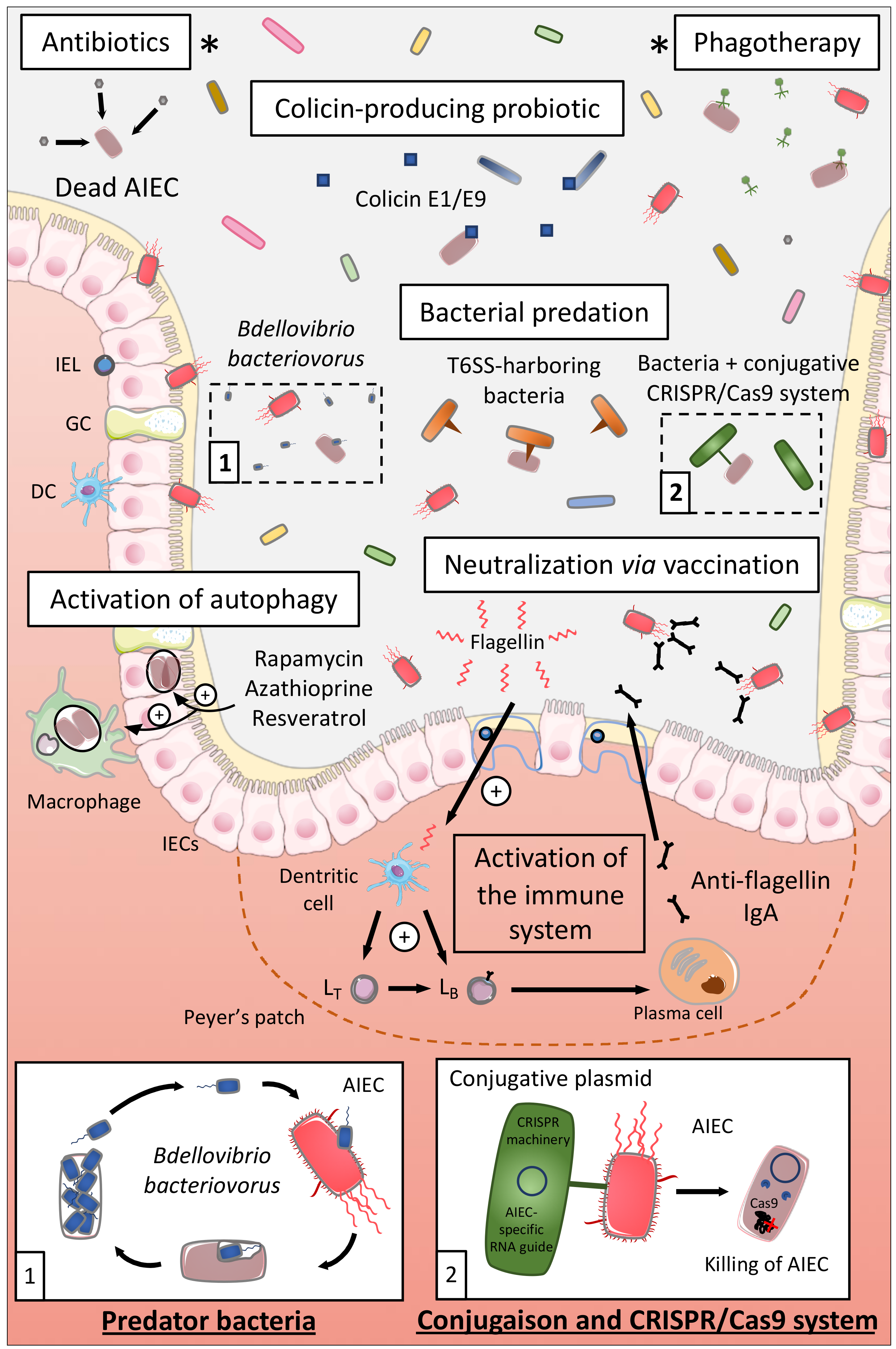
7.2.1. Antibiotics
7.2.2. Use of Colicin
7.2.3. Phagotherapy
7.2.4. Bacterial Predation/Competition
Bacterial Predation
Type VI Secretion System
Conjugative Bacteria and CRISPR
7.3. Activation of Autophagy
7.4. Nutritional Interventions
7.5. Flagellin Vaccination
8. Future Directions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| AIEC | Adherent-invasive Escherichia coli |
| AJC | Apical junctional complex |
| ASF | Altered Schaedler Flora |
| ATG16L1 | Autophagy Related 16 Like 1 |
| CBD | Chitin-binding domain |
| CD | Crohn’s disease |
| CEABAC10 | Carcinoembryonic antigen bacterial artificial chromosome 10 |
| CEACAM6 | Carcinoembryonic antigen-related cell adhesion molecule 6 |
| CHI3L1 | Chitinase 3-like-1 |
| CRISPR | Clustered regularly interspaced short palindromic repeat |
| DSS | Dextran sulfate sodium |
| EAEC | Enteroaggregative Escherichia coli |
| ETEC | Enterotoxigenic Escherichia coli |
| FAE | Follicle-associated epithelium |
| GI | Gastrointestinal tract |
| GP2 | Glycoprotein 2 |
| GPI | Glycosylphosphatidylinositol |
| HF/HS | High-fat/high sugar |
| HIF-1 | Hypoxia-inducible factor-1 |
| HM | n-Heptyl α-D-mannose |
| IBD | Inflammatory bowel disease |
| IECs | Intestinal epithelial cells |
| IL | Interleukin |
| IRGM | Immunity-related GTPase family M |
| LP | Lamina propria |
| LPF | Long polar fimbriae |
| LPS | Lipopolysaccharide |
| LRR | Leucin rich repeat |
| MAMP | Microbe-associated molecular pattern |
| MAPK | Mitogen-activated protein kinase |
| MDM | Monocyte-derived macrophages |
| MDP | Muramyl dipeptide |
| MEF | Mouse embryonic fibroblasts |
| miRNAs | MicroRNAs |
| MOBIDIC | MOlecular BIomarkers and Adherent and Invasive Escherichia coli (AIEC) Detection Study in Crohn’s Disease Patients |
| NF-κB | Nuclear factor-kappa B |
| NLR | NOD-like receptor |
| NLRC4 | NLR family CARD domain-containing protein 4 |
| NOD2 | Nucleotide-binding oligomerization domain 2 |
| O-GlcNAc | O-linked β-N-acetylglucosamine |
| OMV | Outer membrane vesicles |
| PA | Propionic acid |
| PBMC | Peripheral blood mononuclear cells |
| PP | Peyer’s patches |
| PRR | Pattern recognition receptors |
| SCFA | Short-chain fatty acids |
| SNP | Single nucleotide polymorphisms |
| T5KO | Toll-like receptor 5-deficient |
| T6SS | Type IV secretion system |
| TGF-β | Transforming growth factor beta |
| TJ | Tight junctions |
| TLR | Toll-like receptor |
| TNF-α | Tumor necrosis factor alpha |
| Tnseq | Transposon mutagenesis coupled with next-generation sequencing |
| UC | Ulcerative colitis |
| WT | Wild-type |
References
- Crohn, B.B. Regional ileitis: A pathologic and clinical entity. JAMA 1932, 99, 1323. [Google Scholar] [CrossRef]
- Torres, J.; Mehandru, S.; Colombel, J.-F.; Peyrin-Biroulet, L. Crohn’s disease. Lancet 2017, 389, 1741–1755. [Google Scholar] [CrossRef]
- Hugot, J.P.; Chamaillard, M.; Zouali, H.; Lesage, S.; Cézard, J.P.; Belaiche, J.; Almer, S.; Tysk, C.; O’Morain, C.A.; Gassull, M.; et al. Association of NOD2 leucine-rich repeat variants with susceptibility to Crohn’s disease. Nature 2001, 411, 599–603. [Google Scholar] [CrossRef] [PubMed]
- Ogura, Y.; Bonen, D.K.; Inohara, N.; Nicolae, D.L.; Chen, F.F.; Ramos, R.; Britton, H.; Moran, T.; Karaliuskas, R.; Duerr, R.H.; et al. A frameshift mutation in NOD2 associated with susceptibility to Crohn’s disease. Nature 2001, 411, 603–606. [Google Scholar] [CrossRef] [PubMed]
- Hampe, J.; Franke, A.; Rosenstiel, P.; Till, A.; Teuber, M.; Huse, K.; Albrecht, M.; Mayr, G.; De La Vega, F.M.; Briggs, J.; et al. A genome-wide association scan of nonsynonymous SNPs identifies a susceptibility variant for Crohn disease in ATG16L1. Nat. Genet. 2007, 39, 207–211. [Google Scholar] [CrossRef]
- Parkes, M.; Barrett, J.C.; Prescott, N.J.; Tremelling, M.; Anderson, C.A.; Fisher, S.A.; Roberts, R.G.; Nimmo, E.R.; Cummings, F.R.; Soars, D.; et al. Sequence variants in the autophagy gene IRGM and multiple other replicating loci contribute to Crohn’s disease susceptibility. Nat. Genet. 2007, 39, 830–832. [Google Scholar] [CrossRef]
- Henderson, P.; Stevens, C. The Role of Autophagy in Crohn’s Disease. Cells 2012, 1, 492–519. [Google Scholar] [CrossRef] [Green Version]
- Thompson, N.P.; Driscoll, R.; Pounder, R.E.; Wakefield, A.J. Genetics versus environment in inflammatory bowel disease: Results of a British twin study. BMJ 1996, 312, 95–96. [Google Scholar] [CrossRef] [Green Version]
- Orholm, M.; Binder, V.; Sørensen, T.I.; Rasmussen, L.P.; Kyvik, K.O. Concordance of inflammatory bowel disease among Danish twins. Results of a nationwide study. Scand. J. Gastroenterol. 2000, 35, 1075–1081. [Google Scholar] [CrossRef]
- Halfvarson, J.; Jess, T.; Bodin, L.; Järnerot, G.; Munkholm, P.; Binder, V.; Tysk, C. Longitudinal concordance for clinical characteristics in a Swedish-Danish twin population with inflammatory bowel disease. Inflamm. Bowel Dis. 2007, 13, 1536–1544. [Google Scholar] [CrossRef]
- Mahid, S.S.; Minor, K.S.; Soto, R.E.; Hornung, C.A.; Galandiuk, S. Smoking and inflammatory bowel disease: A meta-analysis. Mayo Clin. Proc. 2006, 81, 1462–1471. [Google Scholar] [CrossRef]
- Chapman-Kiddell, C.A.; Davies, P.S.W.; Gillen, L.; Radford-Smith, G.L. Role of diet in the development of inflammatory bowel disease. Inflamm. Bowel Dis. 2010, 16, 137–151. [Google Scholar] [CrossRef]
- Chiba, M.; Nakane, K.; Komatsu, M. Westernized Diet is the Most Ubiquitous Environmental Factor in Inflammatory Bowel Disease. Perm. J. 2019, 23. [Google Scholar] [CrossRef] [PubMed]
- Levine, A.; Rhodes, J.M.; Lindsay, J.O.; Abreu, M.T.; Kamm, M.A.; Gibson, P.R.; Gasche, C.; Silverberg, M.S.; Mahadevan, U.; Sigall, B.R.; et al. Dietary Guidance for Patients With Inflammatory Bowel Disease from the International Organization for the Study of Inflammatory Bowel Disease. Clin. Gastroenterol. Hepatol. 2020. [Google Scholar] [CrossRef] [PubMed]
- Sokol, H.; Seksik, P.; Rigottier-Gois, L.; Lay, C.; Lepage, P.; Podglajen, I.; Marteau, P.; Doré, J. Specificities of the fecal microbiota in inflammatory bowel disease. Inflamm. Bowel Dis. 2006, 12, 106–111. [Google Scholar] [CrossRef] [PubMed]
- Frank, D.N.; Amand, A.L.S.; Feldman, R.A.; Boedeker, E.C.; Harpaz, N.; Pace, N.R. Molecular-phylogenetic characterization of microbial community imbalances in human inflammatory bowel diseases. Proc. Natl. Acad. Sci. USA 2007, 104, 13780–13785. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Walker, A.W.; Sanderson, J.D.; Churcher, C.; Parkes, G.C.; Hudspith, B.N.; Rayment, N.; Brostoff, J.; Parkhill, J.; Dougan, G.; Petrovska, L. High-throughput clone library analysis of the mucosa-associated microbiota reveals dysbiosis and differences between inflamed and non-inflamed regions of the intestine in inflammatory bowel disease. BMC Microbiol. 2011, 11, 7. [Google Scholar] [CrossRef] [Green Version]
- Fujimoto, T.; Imaeda, H.; Takahashi, K.; Kasumi, E.; Bamba, S.; Fujiyama, Y.; Andoh, A. Decreased abundance of Faecalibacterium prausnitzii in the gut microbiota of Crohn’s disease. J. Gastroenterol. Hepatol. 2013, 28, 613–619. [Google Scholar] [CrossRef]
- Alhagamhmad, M.H.; Day, A.S.; Lemberg, D.A.; Leach, S.T. An overview of the bacterial contribution to Crohn disease pathogenesis. J. Med. Microbiol. 2016, 65, 1049–1059. [Google Scholar] [CrossRef]
- Sokol, H.; Brot, L.; Stefanescu, C.; Auzolle, C.; Barnich, N.; Buisson, A.; Fumery, M.; Pariente, B.; Le Bourhis, L.; Treton, X.; et al. Prominence of ileal mucosa-associated microbiota to predict postoperative endoscopic recurrence in Crohn’s disease. Gut 2020, 69, 462–472. [Google Scholar] [CrossRef]
- Darfeuille-Michaud, A.; Neut, C.; Barnich, N.; Lederman, E.; Di Martino, P.; Desreumaux, P.; Gambiez, L.; Joly, B.; Cortot, A.; Colombel, J.F. Presence of adherent Escherichia coli strains in ileal mucosa of patients with Crohn’s disease. Gastroenterology 1998, 115, 1405–1413. [Google Scholar] [CrossRef]
- Darfeuille-Michaud, A.; Boudeau, J.; Bulois, P.; Neut, C.; Glasser, A.-L.; Barnich, N.; Bringer, M.-A.; Swidsinski, A.; Beaugerie, L.; Colombel, J.-F. High prevalence of adherent-invasive Escherichia coli associated with ileal mucosa in Crohn’s disease. Gastroenterology 2004, 127, 412–421. [Google Scholar] [CrossRef]
- Baumgart, M.; Dogan, B.; Rishniw, M.; Weitzman, G.; Bosworth, B.; Yantiss, R.; Orsi, R.H.; Wiedmann, M.; McDonough, P.; Kim, S.G.; et al. Culture independent analysis of ileal mucosa reveals a selective increase in invasive Escherichia coli of novel phylogeny relative to depletion of Clostridiales in Crohn’s disease involving the ileum. ISME J. 2007, 1, 403–418. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martinez-Medina, M.; Aldeguer, X.; Lopez-Siles, M.; González-Huix, F.; López-Oliu, C.; Dahbi, G.; Blanco, J.E.; Blanco, J.; Garcia-Gil, L.J.; Darfeuille-Michaud, A. Molecular diversity of Escherichia coli in the human gut: New ecological evidence supporting the role of adherent-invasive E. coli (AIEC) in Crohn’s disease. Inflamm. Bowel Dis. 2009, 15, 872–882. [Google Scholar] [CrossRef] [PubMed]
- Sepehri, S.; Khafipour, E.; Bernstein, C.N.; Coombes, B.K.; Pilar, A.V.; Karmali, M.; Ziebell, K.; Krause, D.O. Characterization of Escherichia coli isolated from gut biopsies of newly diagnosed patients with inflammatory bowel disease. Inflamm. Bowel Dis. 2011, 17, 1451–1463. [Google Scholar] [CrossRef] [PubMed]
- Raso, T.; Crivellaro, S.; Chirillo, M.G.; Pais, P.; Gaia, E.; Savoia, D. Analysis of Escherichia coli isolated from patients affected by Crohn’s disease. Curr. Microbiol. 2011, 63, 131–137. [Google Scholar] [CrossRef]
- Dogan, B.; Scherl, E.; Bosworth, B.; Yantiss, R.; Altier, C.; McDonough, P.L.; Jiang, Z.-D.; Dupont, H.L.; Garneau, P.; Harel, J.; et al. Multidrug resistance is common in Escherichia coli associated with ileal Crohn’s disease. Inflamm. Bowel Dis. 2013, 19, 141–150. [Google Scholar] [CrossRef]
- Boudeau, J.; Glasser, A.L.; Masseret, E.; Joly, B.; Darfeuille-Michaud, A. Invasive ability of an Escherichia coli strain isolated from the ileal mucosa of a patient with Crohn’s disease. Infect. Immun. 1999, 67, 4499–4509. [Google Scholar] [CrossRef] [Green Version]
- Carvalho, F.A.; Barnich, N.; Sivignon, A.; Darcha, C.; Chan, C.H.F.; Stanners, C.P.; Darfeuille-Michaud, A. Crohn’s disease adherent-invasive Escherichia coli colonize and induce strong gut inflammation in transgenic mice expressing human CEACAM. J. Exp. Med. 2009, 206, 2179–2189. [Google Scholar] [CrossRef] [Green Version]
- Chassaing, B.; Koren, O.; Carvalho, F.A.; Ley, R.E.; Gewirtz, A.T. AIEC pathobiont instigates chronic colitis in susceptible hosts by altering microbiota composition. Gut 2014, 63, 1069–1080. [Google Scholar] [CrossRef]
- Schmitz, J.M.; Tonkonogy, S.L.; Dogan, B.; Leblond, A.; Whitehead, K.J.; Kim, S.C.; Simpson, K.W.; Sartor, R.B. Murine Adherent and Invasive E. coli Induces Chronic Inflammation and Immune Responses in the Small and Large Intestines of Monoassociated IL-10-/- Mice Independent of Long Polar Fimbriae Adhesin, A. Inflamm. Bowel Dis. 2019, 25, 875–885. [Google Scholar] [CrossRef] [PubMed]
- Palmela, C.; Chevarin, C.; Xu, Z.; Torres, J.; Sevrin, G.; Hirten, R.; Barnich, N.; Ng, S.C.; Colombel, J.-F. Adherent-invasive Escherichia coli in inflammatory bowel disease. Gut 2018, 67, 574–587. [Google Scholar] [CrossRef] [PubMed]
- O’Brien, C.L.; Bringer, M.-A.; Holt, K.E.; Gordon, D.M.; Dubois, A.L.; Barnich, N.; Darfeuille-Michaud, A.; Pavli, P. Comparative genomics of Crohn’s disease-associated adherent-invasive Escherichia coli. Gut 2017, 66, 1382–1389. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Camprubí-Font, C.; Lopez-Siles, M.; Ferrer-Guixeras, M.; Niubó-Carulla, L.; Abellà-Ametller, C.; Garcia-Gil, L.J.; Martinez-Medina, M. Comparative genomics reveals new single-nucleotide polymorphisms that can assist in identification of adherent-invasive Escherichia coli. Sci. Rep. 2018, 8. [Google Scholar] [CrossRef]
- Camprubí-Font, C.; Bustamante, P.; Vidal, R.M.; O’Brien, C.L.; Barnich, N.; Martinez-Medina, M. Study of a classification algorithm for AIEC identification in geographically distinct E. coli strains. Sci. Rep. 2020, 10, 8094. [Google Scholar] [CrossRef]
- Forstner, G. Signal transduction, packaging and secretion of mucins. Annu. Rev. Physiol. 1995, 57, 585–605. [Google Scholar] [CrossRef]
- Mantle, M.; Rombough, C. Growth in and breakdown of purified rabbit small intestinal mucin by Yersinia enterocolitica. Infect. Immun. 1993, 61, 4131–4138. [Google Scholar] [CrossRef] [Green Version]
- Arike, L.; Hansson, G.C. The Densely O-glycosylated MUC2 Mucin Protects the Intestine and Provides Food for the Commensal Bacteria. J. Mol. Biol. 2016, 428, 3221–3229. [Google Scholar] [CrossRef] [Green Version]
- Derrien, M.; van Passel, M.W.; van de Bovenkamp, J.H.; Schipper, R.G.; de Vos, W.M.; Dekker, J. Mucin-bacterial interactions in the human oral cavity and digestive tract. Gut Microbes 2010, 1, 254–268. [Google Scholar] [CrossRef] [Green Version]
- Strugala, V.; Dettmar, P.W.; Pearson, J.P. Thickness and continuity of the adherent colonic mucus barrier in active and quiescent ulcerative colitis and Crohn’s disease. Int. J. Clin. Pract. 2008, 62, 762–769. [Google Scholar] [CrossRef]
- Buisine, M.; Desreumaux, P.; Leteurtre, E.; Copin, M.; Colombel, J.; Porchet, N.; Aubert, J. Mucin gene expression in intestinal epithelial cells in Crohn’s disease. Gut 2001, 49, 544–551. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moehle, C.; Ackermann, N.; Langmann, T.; Aslanidis, C.; Kel, A.; Kel-Margoulis, O.; Schmitz-Madry, A.; Zahn, A.; Stremmel, W.; Schmitz, G. Aberrant intestinal expression and allelic variants of mucin genes associated with inflammatory bowel disease. J. Mol. Med. 2006, 84, 1055–1066. [Google Scholar] [CrossRef] [PubMed]
- Dorofeyev, A.E.; Vasilenko, I.V.; Rassokhina, O.A.; Kondratiuk, R.B. Mucosal barrier in ulcerative colitis and Crohn’s disease. Gastroenterol. Res. Pract. 2013, 2013, 431231. [Google Scholar] [CrossRef] [PubMed]
- Sicard, J.-F.; Le Bihan, G.; Vogeleer, P.; Jacques, M.; Harel, J. Interactions of Intestinal Bacteria with Components of the Intestinal Mucus. Front. Cell. Infect. Microbiol. 2017, 7. [Google Scholar] [CrossRef] [PubMed]
- Elatrech, I.; Marzaioli, V.; Boukemara, H.; Bournier, O.; Neut, C.; Darfeuille-Michaud, A.; Luis, J.; Dubuquoy, L.; El-Benna, J.; My-Chan Dang, P.; et al. Escherichia coli LF82 differentially regulates ROS production and mucin expression in intestinal epithelial T84 cells: Implication of NOX1. Inflamm. Bowel Dis. 2015, 21, 1018–1026. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gibold, L.; Garenaux, E.; Dalmasso, G.; Gallucci, C.; Cia, D.; Mottet-Auselo, B.; Faïs, T.; Darfeuille-Michaud, A.; Nguyen, H.T.T.; Barnich, N.; et al. The Vat-AIEC protease promotes crossing of the intestinal mucus layer by Crohn’s disease-associated Escherichia coli. Cell. Microbiol. 2016, 18, 617–631. [Google Scholar] [CrossRef]
- Barnich, N.; Boudeau, J.; Claret, L.; Darfeuille-Michaud, A. Regulatory and functional co-operation of flagella and type 1 pili in adhesive and invasive abilities of AIEC strain LF82 isolated from a patient with Crohn’s disease. Mol. Microbiol. 2003, 48, 781–794. [Google Scholar] [CrossRef]
- Nakamura, S.; Minamino, T. Flagella-Driven Motility of Bacteria. Biomolecules 2019, 9, 279. [Google Scholar] [CrossRef] [Green Version]
- Elhenawy, W.; Tsai, C.N.; Coombes, B.K. Host-Specific Adaptive Diversification of Crohn’s Disease-Associated Adherent-Invasive Escherichia coli. Cell Host Microbe 2019, 25, 301–312. [Google Scholar] [CrossRef] [Green Version]
- Sevrin, G.; Massier, S.; Chassaing, B.; Agus, A.; Delmas, J.; Denizot, J.; Billard, E.; Barnich, N. Adaptation of adherent-invasive E. coli to gut environment: Impact on flagellum expression and bacterial colonization ability. Gut Microbes 2018, 1–17. [Google Scholar] [CrossRef]
- Bevins, C.L.; Salzman, N.H. Paneth cells, antimicrobial peptides and maintenance of intestinal homeostasis. Nat. Rev. Microbiol. 2011, 9, 356–368. [Google Scholar] [CrossRef] [PubMed]
- Zasloff, M. Antimicrobial peptides of multicellular organisms. Nature 2002, 415, 389–395. [Google Scholar] [CrossRef] [PubMed]
- McPhee, J.B.; Small, C.L.; Reid-Yu, S.A.; Brannon, J.R.; Le Moual, H.; Coombes, B.K. Host Defense Peptide Resistance Contributes to Colonization and Maximal Intestinal Pathology by Crohn’s Disease-Associated Adherent-Invasive Escherichia coli. Infect. Immun. 2014, 82, 3383–3393. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boudeau, J.; Barnich, N.; Darfeuille-Michaud, A. Type 1 pili-mediated adherence of Escherichia coli strain LF82 isolated from Crohn’s disease is involved in bacterial invasion of intestinal epithelial cells. Mol. Microbiol. 2001, 39, 1272–1284. [Google Scholar] [CrossRef] [PubMed]
- Dreux, N.; Denizot, J.; Martinez-Medina, M.; Mellmann, A.; Billig, M.; Kisiela, D.; Chattopadhyay, S.; Sokurenko, E.; Neut, C.; Gower-Rousseau, C.; et al. Point mutations in FimH adhesin of Crohn’s disease-associated adherent-invasive Escherichia coli enhance intestinal inflammatory response. PLoS Pathog. 2013, 9, e1003141. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barnich, N.; Carvalho, F.A.; Glasser, A.-L.; Darcha, C.; Jantscheff, P.; Allez, M.; Peeters, H.; Bommelaer, G.; Desreumaux, P.; Colombel, J.-F.; et al. CEACAM6 acts as a receptor for adherent-invasive E. coli, supporting ileal mucosa colonization in Crohn disease. J. Clin. Investig. 2007, 117, 1566–1574. [Google Scholar] [CrossRef] [Green Version]
- Dumych, T.; Yamakawa, N.; Sivignon, A.; Garenaux, E.; Robakiewicz, S.; Coddeville, B.; Bongiovanni, A.; Bray, F.; Barnich, N.; Szunerits, S.; et al. Oligomannose-Rich Membranes of Dying Intestinal Epithelial Cells Promote Host Colonization by Adherent-Invasive E. coli. Front. Microbiol. 2018, 9, 742. [Google Scholar] [CrossRef] [Green Version]
- Barnich, N.; Darfeuille-Michaud, A. Abnormal CEACAM6 expression in Crohn disease patients favors gut colonization and inflammation by adherent-invasive E. coli. Virulence 2010, 1, 281–282. [Google Scholar] [CrossRef] [Green Version]
- Mimouna, S.; Gonçalvès, D.; Barnich, N.; Darfeuille-Michaud, A.; Hofman, P.; Vouret-Craviari, V. Crohn disease-associated Escherichia coli promote gastrointestinal inflammatory disorders by activation of HIF-dependent responses. Gut Microbes 2011, 2, 335–346. [Google Scholar] [CrossRef]
- Denizot, J.; Desrichard, A.; Agus, A.; Uhrhammer, N.; Dreux, N.; Vouret-Craviari, V.; Hofman, P.; Darfeuille-Michaud, A.; Barnich, N. Diet-induced hypoxia responsive element demethylation increases CEACAM6 expression, favouring Crohn’s disease-associated Escherichia coli colonisation. Gut 2015, 64, 428–437. [Google Scholar] [CrossRef]
- Mizoguchi, E. Chitinase 3-like-1 exacerbates intestinal inflammation by enhancing bacterial adhesion and invasion in colonic epithelial cells. Gastroenterology 2006, 130, 398–411. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.-C.; Pekow, J.; Llado, V.; Kanneganti, M.; Lau, C.W.; Mizoguchi, A.; Mino-Kenudson, M.; Bissonnette, M.; Mizoguchi, E. Chitinase 3-like-1 expression in colonic epithelial cells as a potentially novel marker for colitis-associated neoplasia. Am. J. Pathol. 2011, 179, 1494–1503. [Google Scholar] [CrossRef] [PubMed]
- Low, D.; Tran, H.T.; Lee, I.-A.; Dreux, N.; Kamba, A.; Reinecker, H.-C.; Darfeuille-Michaud, A.; Barnich, N.; Mizoguchi, E. Chitin-binding domains of Escherichia coli ChiA mediate interactions with intestinal epithelial cells in mice with colitis. Gastroenterology 2013, 145, 602–612.e9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chassaing, B.; Rolhion, N.; de Vallée, A.; Salim, S.Y.; Prorok-Hamon, M.; Neut, C.; Campbell, B.J.; Söderholm, J.D.; Hugot, J.-P.; Colombel, J.-F.; et al. Crohn disease--associated adherent-invasive E. coli bacteria target mouse and human Peyer’s patches via long polar fimbriae. J. Clin. Investig. 2011, 121, 966–975. [Google Scholar] [CrossRef]
- Keita, Å.V.; Alkaissi, L.Y.; Holm, E.B.; Heil, S.D.S.; Chassaing, B.; Darfeuille-Michaud, A.; McKay, D.M.; Söderholm, J.D. Enhanced E. coli LF82 Translocation through the Follicle-associated Epithelium in Crohn’s Disease is Dependent on Long Polar Fimbriae and CEACAM6 expression, and Increases Paracellular Permeability. J. Crohns Colitis 2020, 14, 216–229. [Google Scholar] [CrossRef]
- Chassaing, B.; Etienne-Mesmin, L.; Bonnet, R.; Darfeuille-Michaud, A. Bile salts induce long polar fimbriae expression favouring Crohn’s disease-associated adherent-invasive Escherichia coli interaction with Peyer’s patches. Environ. Microbiol. 2013, 15, 355–371. [Google Scholar] [CrossRef]
- Hase, K.; Kawano, K.; Nochi, T.; Pontes, G.S.; Fukuda, S.; Ebisawa, M.; Kadokura, K.; Tobe, T.; Fujimura, Y.; Kawano, S.; et al. Uptake through glycoprotein 2 of FimH(+) bacteria by M cells initiates mucosal immune response. Nature 2009, 462, 226–230. [Google Scholar] [CrossRef]
- Baranov, V.; Hammarström, S. Carcinoembryonic antigen (CEA) and CEA-related cell adhesion molecule 1 (CEACAM1), apically expressed on human colonic M cells, are potential receptors for microbial adhesion. Histochem. Cell Biol. 2004, 121, 83–89. [Google Scholar] [CrossRef]
- Rolhion, N.; Barnich, N.; Claret, L.; Darfeuille-Michaud, A. Strong decrease in invasive ability and outer membrane vesicle release in Crohn’s disease-associated adherent-invasive Escherichia coli strain LF82 with the yfgL gene deleted. J. Bacteriol. 2005, 187, 2286–2296. [Google Scholar] [CrossRef] [Green Version]
- Rolhion, N.; Barnich, N.; Bringer, M.-A.; Glasser, A.-L.; Ranc, J.; Hébuterne, X.; Hofman, P.; Darfeuille-Michaud, A. Abnormally expressed ER stress response chaperone Gp96 in CD favours adherent-invasive Escherichia coli invasion. Gut 2010, 59, 1355–1362. [Google Scholar] [CrossRef]
- Rolhion, N.; Hofman, P.; Darfeuille-Michaud, A. The endoplasmic reticulum stress response chaperone: Gp96, a host receptor for Crohn disease-associated adherent-invasive Escherichia coli. Gut Microbes 2011, 2, 115–119. [Google Scholar] [CrossRef] [PubMed]
- Cieza, R.J.; Hu, J.; Ross, B.N.; Sbrana, E.; Torres, A.G. The IbeA invasin of adherent-invasive Escherichia coli mediates interaction with intestinal epithelia and macrophages. Infect. Immun. 2015, 83, 1904–1918. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van Opijnen, T.; Bodi, K.L.; Camilli, A. Tn-seq: High-throughput parallel sequencing for fitness and genetic interaction studies in microorganisms. Nat. Methods 2009, 6, 767–772. [Google Scholar] [CrossRef] [PubMed]
- Turner, J.R. Intestinal mucosal barrier function in health and disease. Nat. Rev. Immunol. 2009, 9, 799–809. [Google Scholar] [CrossRef] [PubMed]
- Peterson, L.W.; Artis, D. Intestinal epithelial cells: Regulators of barrier function and immune homeostasis. Nat. Rev. Immunol. 2014, 14, 141–153. [Google Scholar] [CrossRef]
- Michielan, A.; D’Incà, R. Intestinal Permeability in Inflammatory Bowel Disease: Pathogenesis, Clinical Evaluation, and Therapy of Leaky Gut. Mediators Inflamm. 2015, 2015, 628157. [Google Scholar] [CrossRef] [Green Version]
- Sasaki, M.; Sitaraman, S.V.; Babbin, B.A.; Gerner-Smidt, P.; Ribot, E.M.; Garrett, N.; Alpern, J.A.; Akyildiz, A.; Theiss, A.L.; Nusrat, A.; et al. Invasive Escherichia coli are a feature of Crohn’s disease. Lab. Investig. 2007, 87, 1042–1054. [Google Scholar] [CrossRef]
- Wine, E.; Ossa, J.C.; Gray-Owen, S.D.; Sherman, P.M. Adherent-invasive Escherichia coli, strain LF82 disrupts apical junctional complexes in polarized epithelia. BMC Microbiol. 2009, 9, 180. [Google Scholar] [CrossRef] [Green Version]
- Denizot, J.; Sivignon, A.; Barreau, F.; Darcha, C.; Chan, H.F.C.; Stanners, C.P.; Hofman, P.; Darfeuille-Michaud, A.; Barnich, N. Adherent-invasive Escherichia coli induce claudin-2 expression and barrier defect in CEABAC10 mice and Crohn’s disease patients. Inflamm. Bowel Dis. 2012, 18, 294–304. [Google Scholar] [CrossRef]
- Assa, A.; Vong, L.; Pinnell, L.J.; Rautava, J.; Avitzur, N.; Johnson-Henry, K.C.; Sherman, P.M. Vitamin D deficiency predisposes to adherent-invasive Escherichia coli-induced barrier dysfunction and experimental colonic injury. Inflamm. Bowel Dis. 2015, 21, 297–306. [Google Scholar] [CrossRef]
- Martinez-Medina, M.; Denizot, J.; Dreux, N.; Robin, F.; Billard, E.; Bonnet, R.; Darfeuille-Michaud, A.; Barnich, N. Western diet induces dysbiosis with increased E coli in CEABAC10 mice, alters host barrier function favouring AIEC colonisation. Gut 2014, 63, 116–124. [Google Scholar] [CrossRef] [PubMed]
- Hanić, M.; Trbojević-Akmačić, I.; Lauc, G. Inflammatory bowel disease—Glycomics perspective. Biochim. Biophys. Acta Gen. Subj. 2019, 1863, 1595–1601. [Google Scholar] [CrossRef] [PubMed]
- Sun, Q.-H.; Wang, Y.-S.; Liu, G.; Zhou, H.-L.; Jian, Y.-P.; Liu, M.-D.; Zhang, D.; Ding, Q.; Zhao, R.-X.; Chen, J.-F.; et al. Enhanced O-linked Glcnacylation in Crohn’s disease promotes intestinal inflammation. EBioMedicine 2020, 53, 102693. [Google Scholar] [CrossRef]
- Rieder, F.; Fiocchi, C.; Rogler, G. Mechanisms, Management, and Treatment of Fibrosis in Patients With Inflammatory Bowel Diseases. Gastroenterology 2017, 152, 340–350. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Small, C.-L.N.; Reid-Yu, S.A.; McPhee, J.B.; Coombes, B.K. Persistent infection with Crohn’s disease-associated adherent-invasive Escherichia coli leads to chronic inflammation and intestinal fibrosis. Nat. Commun. 2013, 4, 1957. [Google Scholar] [CrossRef]
- Ellermann, M.; Gharaibeh, R.Z.; Fulbright, L.; Dogan, B.; Moore, L.N.; Broberg, C.A.; Lopez, L.R.; Rothemich, A.M.; Herzog, J.W.; Rogala, A.; et al. Yersiniabactin-Producing Adherent/Invasive Escherichia coli Promotes Inflammation-Associated Fibrosis in Gnotobiotic Il10−/− Mice. Infect. Immun. 2019, 87. [Google Scholar] [CrossRef] [Green Version]
- Imai, J.; Kitamoto, S.; Sugihara, K.; Nagao-Kitamoto, H.; Hayashi, A.; Morhardt, T.L.; Kuffa, P.; Higgins, P.D.R.; Barnich, N.; Kamada, N. Flagellin-mediated activation of IL-33-ST2 signaling by a pathobiont promotes intestinal fibrosis. Mucosal Immunol. 2019, 12, 632–643. [Google Scholar] [CrossRef]
- Schmitz, J.; Owyang, A.; Oldham, E.; Song, Y.; Murphy, E.; McClanahan, T.K.; Zurawski, G.; Moshrefi, M.; Qin, J.; Li, X.; et al. IL-33, an interleukin-1-like cytokine that signals via the IL-1 receptor-related protein ST2 and induces T helper type 2-associated cytokines. Immunity 2005, 23, 479–490. [Google Scholar] [CrossRef] [Green Version]
- Fichtner-Feigl, S.; Fuss, I.J.; Young, C.A.; Watanabe, T.; Geissler, E.K.; Schlitt, H.-J.; Kitani, A.; Strober, W. Induction of IL-13 triggers TGF-beta1-dependent tissue fibrosis in chronic 2,4,6-trinitrobenzene sulfonic acid colitis. J. Immunol. 2007, 178, 5859–5870. [Google Scholar] [CrossRef] [Green Version]
- Gewirtz, A.T.; Navas, T.A.; Lyons, S.; Godowski, P.J.; Madara, J.L. Cutting edge: Bacterial flagellin activates basolaterally expressed TLR5 to induce epithelial proinflammatory gene expression. J. Immunol. 2001, 167, 1882–1885. [Google Scholar] [CrossRef] [Green Version]
- Zhao, Y.; Shao, F. The NAIP-NLRC4 inflammasome in innate immune detection of bacterial flagellin and type III secretion apparatus. Immunol. Rev. 2015, 265, 85–102. [Google Scholar] [CrossRef] [PubMed]
- Carvalho, F.A.; Barnich, N.; Sauvanet, P.; Darcha, C.; Gelot, A.; Darfeuille-Michaud, A. Crohn’s disease-associated Escherichia coli LF82 aggravates colitis in injured mouse colon via signaling by flagellin. Inflamm. Bowel Dis. 2008, 14, 1051–1060. [Google Scholar] [CrossRef] [PubMed]
- Abreu, M.T.; Vora, P.; Faure, E.; Thomas, L.S.; Arnold, E.T.; Arditi, M. Decreased expression of Toll-like receptor-4 and MD-2 correlates with intestinal epithelial cell protection against dysregulated proinflammatory gene expression in response to bacterial lipopolysaccharide. J. Immunol. 2001, 167, 1609–1616. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Raisch, J.; Darfeuille-Michaud, A.; Nguyen, H.T.T. Role of microRNAs in the immune system, inflammation and cancer. World J. Gastroenterol. 2013, 19, 2985–2996. [Google Scholar] [CrossRef] [PubMed]
- Mohammadi, A.; Kelly, O.B.; Smith, M.I.; Kabakchiev, B.; Silverberg, M.S. Differential miRNA Expression in Ileal and Colonic Tissues Reveals an Altered Immunoregulatory Molecular Profile in Individuals With Crohn’s Disease versus Healthy Subjects. J. Crohns Colitis 2019, 13, 1459–1469. [Google Scholar] [CrossRef]
- Guo, Z.; Cai, X.; Guo, X.; Xu, Y.; Gong, J.; Li, Y.; Zhu, W. Let-7b ameliorates Crohn’s disease-associated adherent-invasive E coli induced intestinal inflammation via modulating Toll-Like Receptor 4 expression in intestinal epithelial cells. Biochem. Pharmacol. 2018, 156, 196–203. [Google Scholar] [CrossRef]
- Steinbach, E.C.; Plevy, S.E. The role of macrophages and dendritic cells in the initiation of inflammation in IBD. Inflamm. Bowel Dis. 2014, 20, 166–175. [Google Scholar] [CrossRef] [Green Version]
- Naser, S.A.; Romero, C.; Urbina, P.; Naser, N.; Valentine, J. Cellular infiltration and cytokine expression correlate with fistulizing state in Crohn’s disease. Clin. Vaccine Immunol. 2011, 18, 1416–1419. [Google Scholar] [CrossRef]
- Glasser, A.L.; Boudeau, J.; Barnich, N.; Perruchot, M.H.; Colombel, J.F.; Darfeuille-Michaud, A. Adherent invasive Escherichia coli strains from patients with Crohn’s disease survive and replicate within macrophages without inducing host cell death. Infect. Immun. 2001, 69, 5529–5537. [Google Scholar] [CrossRef] [Green Version]
- Bringer, M.-A.; Barnich, N.; Glasser, A.-L.; Bardot, O.; Darfeuille-Michaud, A. HtrA stress protein is involved in intramacrophagic replication of adherent and invasive Escherichia coli strain LF82 isolated from a patient with Crohn’s disease. Infect. Immun. 2005, 73, 712–721. [Google Scholar] [CrossRef] [Green Version]
- Bringer, M.-A.; Rolhion, N.; Glasser, A.-L.; Darfeuille-Michaud, A. The oxidoreductase DsbA plays a key role in the ability of the Crohn’s disease-associated adherent-invasive Escherichia coli strain LF82 to resist macrophage killing. J. Bacteriol. 2007, 189, 4860–4871. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vazeille, E.; Chassaing, B.; Buisson, A.; Dubois, A.; de Vallée, A.; Billard, E.; Neut, C.; Bommelaer, G.; Colombel, J.-F.; Barnich, N.; et al. GipA Factor Supports Colonization of Peyer’s Patches by Crohn’s Disease-associated Escherichia Coli. Inflamm. Bowel Dis. 2016, 22, 68–81. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Demarre, G.; Prudent, V.; Schenk, H.; Rousseau, E.; Bringer, M.-A.; Barnich, N.; Tran Van Nhieu, G.; Rimsky, S.; De Monte, S.; Espéli, O. The Crohn’s disease-associated Escherichia coli strain LF82 relies on SOS and stringent responses to survive, multiply and tolerate antibiotics within macrophages. PLoS Pathog. 2019, 15. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bringer, M.-A.; Billard, E.; Glasser, A.-L.; Colombel, J.-F.; Darfeuille-Michaud, A. Replication of Crohn’s disease-associated AIEC within macrophages is dependent on TNF-α secretion. Lab. Investig. 2012, 92, 411–419. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van der Grein, S.G.; Nolte-’t Hoen, E.N.M. “Small Talk” in the Innate Immune System via RNA-Containing Extracellular Vesicles. Front. Immunol. 2014, 5, 542. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Greening, D.W.; Gopal, S.K.; Xu, R.; Simpson, R.J.; Chen, W. Exosomes and their roles in immune regulation and cancer. Semin. Cell Dev. Biol. 2015, 40, 72–81. [Google Scholar] [CrossRef]
- Larabi, A.; Barnich, N.; Nguyen, H.T.T. Emerging Role of Exosomes in Diagnosis and Treatment of Infectious and Inflammatory Bowel Diseases. Cells 2020, 9, 1111. [Google Scholar] [CrossRef]
- Bhatnagar, S.; Shinagawa, K.; Castellino, F.J.; Schorey, J.S. Exosomes released from macrophages infected with intracellular pathogens stimulate a proinflammatory response in vitro and in vivo. Blood 2007, 110, 3234–3244. [Google Scholar] [CrossRef] [Green Version]
- Carrière, J.; Bretin, A.; Darfeuille-Michaud, A.; Barnich, N.; Nguyen, H.T.T. Exosomes Released from Cells Infected with Crohnʼs Disease–associated Adherent-Invasive Escherichia coli Activate Host Innate Immune Responses and Enhance Bacterial Intracellular Replication. Inflamm. Bowel Dis. 2016, 22, 516–528. [Google Scholar] [CrossRef] [Green Version]
- Glick, D.; Barth, S.; Macleod, K.F. Autophagy: Cellular and molecular mechanisms. J. Pathol. 2010, 221, 3–12. [Google Scholar] [CrossRef] [Green Version]
- Lapaquette, P.; Glasser, A.-L.; Huett, A.; Xavier, R.J.; Darfeuille-Michaud, A. Crohn’s disease-associated adherent-invasive E. coli are selectively favoured by impaired autophagy to replicate intracellularly. Cell. Microbiol. 2010, 12, 99–113. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bretin, A.; Carrière, J.; Dalmasso, G.; Bergougnoux, A.; B’chir, W.; Maurin, A.-C.; Müller, S.; Seibold, F.; Barnich, N.; Bruhat, A.; et al. Activation of the EIF2AK4-EIF2A/eIF2α-ATF4 pathway triggers autophagy response to Crohn disease-associated adherent-invasive Escherichia coli infection. Autophagy 2016, 12, 770–783. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Homer, C.R.; Richmond, A.L.; Rebert, N.A.; Achkar, J.-P.; McDonald, C. ATG16L1 and NOD2 interact in an autophagy-dependent antibacterial pathway implicated in Crohn’s disease pathogenesis. Gastroenterology 2010, 139, 1630–1641. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Travassos, L.H.; Carneiro, L.A.M.; Ramjeet, M.; Hussey, S.; Kim, Y.-G.; Magalhães, J.G.; Yuan, L.; Soares, F.; Chea, E.; Le Bourhis, L.; et al. Nod1 and Nod2 direct autophagy by recruiting ATG16L1 to the plasma membrane at the site of bacterial entry. Nat. Immunol. 2010, 11, 55–62. [Google Scholar] [CrossRef]
- Negroni, A.; Colantoni, E.; Vitali, R.; Palone, F.; Pierdomenico, M.; Costanzo, M.; Cesi, V.; Cucchiara, S.; Stronati, L. NOD2 induces autophagy to control AIEC bacteria infectiveness in intestinal epithelial cells. Inflamm. Res. 2016, 65, 803–813. [Google Scholar] [CrossRef]
- Henderson, P.; Wilson, D.C.; Satsangi, J.; Stevens, C. A role for vimentin in Crohn disease. Autophagy 2012, 8, 1695–1696. [Google Scholar] [CrossRef] [Green Version]
- Stevens, C.; Henderson, P.; Nimmo, E.R.; Soares, D.C.; Dogan, B.; Simpson, K.W.; Barrett, J.C.; International Inflammatory Bowel Disease Genetics Consortium; Wilson, D.C.; Satsangi, J. The intermediate filament protein, vimentin, is a regulator of NOD2 activity. Gut 2013, 62, 695–707. [Google Scholar] [CrossRef]
- Vazeille, E.; Buisson, A.; Bringer, M.-A.; Goutte, M.; Ouchchane, L.; Hugot, J.-P.; de Vallée, A.; Barnich, N.; Bommelaer, G.; Darfeuille-Michaud, A. Monocyte-derived macrophages from Crohn’s disease patients are impaired in the ability to control intracellular adherent-invasive Escherichia coli and exhibit disordered cytokine secretion profile. J. Crohns Colitis 2015, 9, 410–420. [Google Scholar] [CrossRef] [Green Version]
- Elliott, T.R.; Hudspith, B.N.; Rayment, N.B.; Prescott, N.J.; Petrovska, L.; Hermon-Taylor, J.; Brostoff, J.; Boussioutas, A.; Mathew, C.G.; Sanderson, J.D. Defective macrophage handling of Escherichia coli in Crohn’s disease. J. Gastroenterol. Hepatol. 2015, 30, 1265–1274. [Google Scholar] [CrossRef]
- Lapaquette, P.; Bringer, M.-A.; Darfeuille-Michaud, A. Defects in autophagy favour adherent-invasive Escherichia coli persistence within macrophages leading to increased pro-inflammatory response: Autophagy controls AIEC replication within macrophages. Cell. Microbiol. 2012, 14, 791–807. [Google Scholar] [CrossRef] [Green Version]
- Buisson, A.; Douadi, C.; Ouchchane, L.; Goutte, M.; Hugot, J.-P.; Dubois, A.; Minet-Quinard, R.; Bouvier, D.; Bommelaer, G.; Vazeille, E.; et al. Macrophages Inability to Mediate Adherent-Invasive E. coli Replication is Linked to Autophagy in Crohn’s Disease Patients. Cells 2019, 8, 1394. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fasseu, M.; Tréton, X.; Guichard, C.; Pedruzzi, E.; Cazals-Hatem, D.; Richard, C.; Aparicio, T.; Daniel, F.; Soulé, J.-C.; Moreau, R.; et al. Identification of restricted subsets of mature microRNA abnormally expressed in inactive colonic mucosa of patients with inflammatory bowel disease. PLoS ONE 2010, 5. [Google Scholar] [CrossRef] [PubMed]
- Brest, P.; Lapaquette, P.; Souidi, M.; Lebrigand, K.; Cesaro, A.; Vouret-Craviari, V.; Mari, B.; Barbry, P.; Mosnier, J.-F.; Hébuterne, X.; et al. A synonymous variant in IRGM alters a binding site for miR-196 and causes deregulation of IRGM-dependent xenophagy in Crohn’s disease. Nat. Genet. 2011, 43, 242–245. [Google Scholar] [CrossRef] [PubMed]
- Lu, C.; Chen, J.; Xu, H.-G.; Zhou, X.; He, Q.; Li, Y.-L.; Jiang, G.; Shan, Y.; Xue, B.; Zhao, R.-X.; et al. MIR106B and MIR93 prevent removal of bacteria from epithelial cells by disrupting ATG16L1-mediated autophagy. Gastroenterology 2014, 146, 188–199. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nguyen, H.T.T.; Dalmasso, G.; Müller, S.; Carrière, J.; Seibold, F.; Darfeuille–Michaud, A. Crohn’s Disease–Associated Adherent Invasive Escherichia coli Modulate Levels of microRNAs in Intestinal Epithelial Cells to Reduce Autophagy. Gastroenterology 2014, 146, 508–519. [Google Scholar] [CrossRef]
- Larabi, A.; Dalmasso, G.; Delmas, J.; Barnich, N.; Nguyen, H.T.T. Exosomes transfer miRNAs from cell-to-cell to inhibit autophagy during infection with Crohn’s disease- associated adherent-invasive E. coli. Gut Microbes 2020. accepted. [Google Scholar]
- Flotho, A.; Melchior, F. Sumoylation: A regulatory protein modification in health and disease. Annu. Rev. Biochem. 2013, 82, 357–385. [Google Scholar] [CrossRef]
- Fritah, S.; Lhocine, N.; Golebiowski, F.; Mounier, J.; Andrieux, A.; Jouvion, G.; Hay, R.T.; Sansonetti, P.; Dejean, A. Sumoylation controls host anti-bacterial response to the gut invasive pathogen Shigella flexneri. EMBO Rep. 2014, 15, 965–972. [Google Scholar] [CrossRef] [Green Version]
- Sidik, S.M.; Salsman, J.; Dellaire, G.; Rohde, J.R. Shigella Infection Interferes with SUMOylation and Increases PML-NB Number. PLoS ONE 2015, 10. [Google Scholar] [CrossRef] [Green Version]
- Ribet, D.; Hamon, M.; Gouin, E.; Nahori, M.-A.; Impens, F.; Neyret-Kahn, H.; Gevaert, K.; Vandekerckhove, J.; Dejean, A.; Cossart, P. Listeria monocytogenes impairs SUMOylation for efficient infection. Nature 2010, 464, 1192–1195. [Google Scholar] [CrossRef] [Green Version]
- Dalmasso, G.; Nguyen, H.T.T.; Faïs, T.; Massier, S.; Barnich, N.; Delmas, J.; Bonnet, R. Crohn’s Disease-Associated Adherent-Invasive Escherichia coli Manipulate Host Autophagy by Impairing SUMOylation. Cells 2019, 8, 35. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Delmas, J.; Gibold, L.; Faïs, T.; Batista, S.; Leremboure, M.; Sinel, C.; Vazeille, E.; Cattoir, V.; Buisson, A.; Barnich, N.; et al. Metabolic adaptation of adherent-invasive Escherichia coli to exposure to bile salts. Sci. Rep. 2019, 9. [Google Scholar] [CrossRef] [PubMed]
- Ormsby, M.J.; Logan, M.; Johnson, S.A.; McIntosh, A.; Fallata, G.; Papadopoulou, R.; Papachristou, E.; Hold, G.L.; Hansen, R.; Ijaz, U.Z.; et al. Inflammation associated ethanolamine facilitates infection by Crohn’s disease-linked adherent-invasive Escherichia coli. EBioMedicine 2019, 43, 325–332. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Duboc, H.; Rajca, S.; Rainteau, D.; Benarous, D.; Maubert, M.-A.; Quervain, E.; Thomas, G.; Barbu, V.; Humbert, L.; Despras, G.; et al. Connecting dysbiosis, bile-acid dysmetabolism and gut inflammation in inflammatory bowel diseases. Gut 2013, 62, 531–539. [Google Scholar] [CrossRef]
- Tedelind, S.; Westberg, F.; Kjerrulf, M.; Vidal, A. Anti-inflammatory properties of the short-chain fatty acids acetate and propionate: A study with relevance to inflammatory bowel disease. World J. Gastroenterol. 2007, 13, 2826–2832. [Google Scholar] [CrossRef]
- Tan, J.; McKenzie, C.; Potamitis, M.; Thorburn, A.N.; Mackay, C.R.; Macia, L. The role of short-chain fatty acids in health and disease. Adv. Immunol. 2014, 121, 91–119. [Google Scholar] [CrossRef]
- Hung, C.-C.; Garner, C.D.; Slauch, J.M.; Dwyer, Z.W.; Lawhon, S.D.; Frye, J.G.; McClelland, M.; Ahmer, B.M.M.; Altier, C. The Intestinal Fatty Acid Propionate Inhibits Salmonella Invasion through the Post-translational Control of HilD. Mol. Microbiol. 2013, 87, 1045–1060. [Google Scholar] [CrossRef] [Green Version]
- González-Fandos, E.; Maya, N.; Pérez-Arnedo, I. Effect of propionic acid on Campylobacter jejuni attached to chicken skin during refrigerated storage. Int. Microbiol. 2015, 18, 171–175. [Google Scholar] [CrossRef]
- Ormsby, M.J.; Johnson, S.A.; Carpena, N.; Meikle, L.M.; Goldstone, R.J.; McIntosh, A.; Wessel, H.M.; Hulme, H.E.; McConnachie, C.C.; Connolly, J.P.R.; et al. Propionic Acid Promotes the Virulent Phenotype of Crohn’s Disease-Associated Adherent-Invasive Escherichia coli. Cell Rep. 2020, 30, 2297–2305.e5. [Google Scholar] [CrossRef] [Green Version]
- Agus, A.; Denizot, J.; Thévenot, J.; Martinez-Medina, M.; Massier, S.; Sauvanet, P.; Bernalier-Donadille, A.; Denis, S.; Hofman, P.; Bonnet, R.; et al. Western diet induces a shift in microbiota composition enhancing susceptibility to Adherent-Invasive E. coli infection and intestinal inflammation. Sci. Rep. 2016, 6, 19032. [Google Scholar] [CrossRef] [Green Version]
- Kitamoto, S.; Alteri, C.J.; Rodrigues, M.; Nagao-Kitamoto, H.; Sugihara, K.; Himpsl, S.D.; Bazzi, M.; Miyoshi, M.; Nishioka, T.; Hayashi, A.; et al. Dietary l-serine confers a competitive fitness advantage to Enterobacteriaceae in the inflamed gut. Nat. Microbiol. 2019. [Google Scholar] [CrossRef] [PubMed]
- Chassaing, B.; Koren, O.; Goodrich, J.K.; Poole, A.C.; Srinivasan, S.; Ley, R.E.; Gewirtz, A.T. Dietary emulsifiers impact the mouse gut microbiota promoting colitis and metabolic syndrome. Nature 2015, 519, 92–96. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chassaing, B.; Van de Wiele, T.; De Bodt, J.; Marzorati, M.; Gewirtz, A.T. Dietary emulsifiers directly alter human microbiota composition and gene expression ex vivo potentiating intestinal inflammation. Gut 2017, 66, 1414–1427. [Google Scholar] [CrossRef] [PubMed]
- Small, C.L.; Xing, L.; McPhee, J.B.; Law, H.T.; Coombes, B.K. Acute Infectious Gastroenteritis Potentiates a Crohn’s Disease Pathobiont to Fuel Ongoing Inflammation in the Post-Infectious Period. PLoS Pathog. 2016, 12. [Google Scholar] [CrossRef] [PubMed]
- Vijay-Kumar, M.; Sanders, C.J.; Taylor, R.T.; Kumar, A.; Aitken, J.D.; Sitaraman, S.V.; Neish, A.S.; Uematsu, S.; Akira, S.; Williams, I.R.; et al. Deletion of TLR5 results in spontaneous colitis in mice. J. Clin. Investig. 2007, 117, 3909–3921. [Google Scholar] [CrossRef] [Green Version]
- Carvalho, F.A.; Koren, O.; Goodrich, J.K.; Johansson, M.E.V.; Nalbantoglu, I.; Aitken, J.D.; Su, Y.; Chassaing, B.; Walters, W.A.; González, A.; et al. Transient inability to manage proteobacteria promotes chronic gut inflammation in TLR5-deficient mice. Cell Host Microbe 2012, 12, 139–152. [Google Scholar] [CrossRef] [Green Version]
- Wymore Brand, M.; Wannemuehler, M.J.; Phillips, G.J.; Proctor, A.; Overstreet, A.-M.; Jergens, A.E.; Orcutt, R.P.; Fox, J.G. The Altered Schaedler Flora: Continued Applications of a Defined Murine Microbial Community. ILAR J. 2015, 56, 169–178. [Google Scholar] [CrossRef] [Green Version]
- Chassaing, B.; Gewirtz, A.T. Mice harboring pathobiont-free microbiota do not develop intestinal inflammation that normally results from an innate immune deficiency. PLoS ONE 2018, 13. [Google Scholar] [CrossRef]
- Bretin, A.; Lucas, C.; Larabi, A.; Dalmasso, G.; Billard, E.; Barnich, N.; Bonnet, R.; Nguyen, H.T.T. AIEC infection triggers modification of gut microbiota composition in genetically predisposed mice, contributing to intestinal inflammation. Sci. Rep. 2018, 8. [Google Scholar] [CrossRef]
- Van den Abbeele, P.; Marzorati, M.; Derde, M.; De Weirdt, R.; Joan, V.; Possemiers, S.; Van de Wiele, T. Arabinoxylans, inulin and Lactobacillus reuteri 1063 repress the adherent-invasive Escherichia coli from mucus in a mucosa-comprising gut model. NPJ Biofilms Microbiomes 2016, 2, 16016. [Google Scholar] [CrossRef]
- Boudeau, J.; Glasser, A.-L.; Julien, S.; Colombel, J.-F.; Darfeuille-Michaud, A. Inhibitory effect of probiotic Escherichia coli strain Nissle 1917 on adhesion to and invasion of intestinal epithelial cells by adherent-invasive E. coli strains isolated from patients with Crohn’s disease. Aliment. Pharmacol. Ther. 2003, 18, 45–56. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sivignon, A.; de Vallée, A.; Barnich, N.; Denizot, J.; Darcha, C.; Pignède, G.; Vandekerckove, P.; Darfeuille-Michaud, A. Saccharomyces cerevisiae CNCM I-3856 Prevents Colitis Induced by AIEC Bacteria in the Transgenic Mouse Model Mimicking Crohnʼs Disease. Inflamm. Bowel Dis. 2015, 21, 276–286. [Google Scholar] [CrossRef] [PubMed]
- Brument, S.; Sivignon, A.; Dumych, T.I.; Moreau, N.; Roos, G.; Guérardel, Y.; Chalopin, T.; Deniaud, D.; Bilyy, R.O.; Darfeuille-Michaud, A.; et al. Thiazolylaminomannosides As Potent Antiadhesives of Type 1 Piliated Escherichia coli Isolated from Crohn’s Disease Patients. J. Med. Chem. 2013, 56, 5395–5406. [Google Scholar] [CrossRef]
- Yan, X.; Sivignon, A.; Yamakawa, N.; Crepet, A.; Travelet, C.; Borsali, R.; Dumych, T.; Li, Z.; Bilyy, R.; Deniaud, D.; et al. Glycopolymers as Antiadhesives of E. coli Strains Inducing Inflammatory Bowel Diseases. Biomacromolecules 2015, 16, 1827–1836. [Google Scholar] [CrossRef] [PubMed]
- Chalopin, T.; Alvarez Dorta, D.; Sivignon, A.; Caudan, M.; Dumych, T.I.; Bilyy, R.O.; Deniaud, D.; Barnich, N.; Bouckaert, J.; Gouin, S.G. Second generation of thiazolylmannosides, FimH antagonists for E. coli-induced Crohn’s disease. Org. Biomol. Chem. 2016, 14, 3913–3925. [Google Scholar] [CrossRef]
- Sivignon, A.; Yan, X.; Alvarez Dorta, D.; Bonnet, R.; Bouckaert, J.; Fleury, E.; Bernard, J.; Gouin, S.G.; Darfeuille-Michaud, A.; Barnich, N. Development of Heptylmannoside-Based Glycoconjugate Antiadhesive Compounds against Adherent-Invasive Escherichia coli Bacteria Associated with Crohn’s Disease. mBio 2015, 6. [Google Scholar] [CrossRef] [Green Version]
- Alvarez Dorta, D.; Sivignon, A.; Chalopin, T.; Dumych, T.I.; Roos, G.; Bilyy, R.O.; Deniaud, D.; Krammer, E.-M.; de Ruyck, J.; Lensink, M.F.; et al. The Antiadhesive Strategy in Crohn’s Disease: Orally Active Mannosides to Decolonize Pathogenic Escherichia coli from the Gut. ChemBioChem 2016, 17, 936–952. [Google Scholar] [CrossRef]
- Cauwel, M.; Sivignon, A.; Bridot, C.; Nongbe, M.C.; Deniaud, D.; Roubinet, B.; Landemarre, L.; Felpin, F.-X.; Bouckaert, J.; Barnich, N.; et al. Heptylmannose-functionalized cellulose for the binding and specific detection of pathogenic E. coli. Chem. Commun. 2019, 55, 10158–10161. [Google Scholar] [CrossRef]
- Vazeille, E.; Bringer, M.-A.; Gardarin, A.; Chambon, C.; Becker-Pauly, C.; Pender, S.L.F.; Jakob, C.; Müller, S.; Lottaz, D.; Darfeuille-Michaud, A. Role of meprins to protect ileal mucosa of Crohn’s disease patients from colonization by adherent-invasive E. coli. PLoS ONE 2011, 6, e21199. [Google Scholar] [CrossRef]
- Nold-Petry, C.A.; Nold, M.F.; Levy, O.; Kliger, Y.; Oren, A.; Borukhov, I.; Becker, C.; Wirtz, S.; Sandhu, M.K.; Neurath, M.; et al. Gp96 Peptide Antagonist gp96-II Confers Therapeutic Effects in Murine Intestinal Inflammation. Front. Immunol. 2017, 8. [Google Scholar] [CrossRef] [Green Version]
- Png, C.W.; Lindén, S.K.; Gilshenan, K.S.; Zoetendal, E.G.; McSweeney, C.S.; Sly, L.I.; McGuckin, M.A.; Florin, T.H.J. Mucolytic bacteria with increased prevalence in IBD mucosa augment in vitro utilization of mucin by other bacteria. Am. J. Gastroenterol. 2010, 105, 2420–2428. [Google Scholar] [CrossRef] [PubMed]
- Kerman, D.H.; Deshpande, A.R. Gut microbiota and inflammatory bowel disease: The role of antibiotics in disease management. Postgrad. Med. 2014, 126, 7–19. [Google Scholar] [CrossRef] [PubMed]
- Nitzan, O.; Elias, M.; Peretz, A.; Saliba, W. Role of antibiotics for treatment of inflammatory bowel disease. World J. Gastroenterol. 2016, 22, 1078–1087. [Google Scholar] [CrossRef] [PubMed]
- Dogan, B.; Fu, J.; Zhang, S.; Scherl, E.J.; Simpson, K.W. Rifaximin decreases virulence of Crohn’s disease-associated Escherichia coli and epithelial inflammatory responses. J. Antibiot. 2018, 71, 485–494. [Google Scholar] [CrossRef]
- Kleanthous, C.; Hemmings, A.M.; Moore, G.R.; James, R. Immunity proteins and their specificity for endonuclease colicins: Telling right from wrong in protein-protein recognition. Mol. Microbiol. 1998, 28, 227–233. [Google Scholar] [CrossRef] [Green Version]
- Riley, M.A.; Wertz, J.E. Bacteriocin diversity: Ecological and evolutionary perspectives. Biochimie 2002, 84, 357–364. [Google Scholar] [CrossRef]
- Cascales, E.; Buchanan, S.K.; Duché, D.; Kleanthous, C.; Lloubès, R.; Postle, K.; Riley, M.; Slatin, S.; Cavard, D. Colicin biology. Microbiol. Mol. Biol. Rev. 2007, 71, 158–229. [Google Scholar] [CrossRef] [Green Version]
- Brown, C.L.; Smith, K.; Wall, D.M.; Walker, D. Activity of Species-specific Antibiotics Against Crohnʼs Disease–Associated Adherent-invasive Escherichia coli. Inflamm. Bowel Dis. 2015, 21, 2372–2382. [Google Scholar] [CrossRef] [Green Version]
- Kotłowski, R. Use of Escherichia coli Nissle 1917 producing recombinant colicins for treatment of IBD patients. Med Hypotheses 2016, 93, 8–10. [Google Scholar] [CrossRef]
- Reyes, A.; Haynes, M.; Hanson, N.; Angly, F.E.; Heath, A.C.; Rohwer, F.; Gordon, J.I. Viruses in the fecal microbiota of monozygotic twins and their mothers. Nature 2010, 466, 334–338. [Google Scholar] [CrossRef]
- Hoyles, L.; McCartney, A.L.; Neve, H.; Gibson, G.R.; Sanderson, J.D.; Heller, K.J.; van Sinderen, D. Characterization of virus-like particles associated with the human faecal and caecal microbiota. Res. Microbiol. 2014, 165, 803–812. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sarker, S.A.; Sultana, S.; Reuteler, G.; Moine, D.; Descombes, P.; Charton, F.; Bourdin, G.; McCallin, S.; Ngom-Bru, C.; Neville, T.; et al. Oral Phage Therapy of Acute Bacterial Diarrhea With Two Coliphage Preparations: A Randomized Trial in Children From Bangladesh. EBioMedicine 2016, 4, 124–137. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Galtier, M.; De Sordi, L.; Maura, D.; Arachchi, H.; Volant, S.; Dillies, M.-A.; Debarbieux, L. Bacteriophages to reduce gut carriage of antibiotic resistant uropathogens with low impact on microbiota composition. Environ. Microbiol. 2016, 18, 2237–2245. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Galtier, M.; De Sordi, L.; Sivignon, A.; de Vallée, A.; Maura, D.; Neut, C.; Rahmouni, O.; Wannerberger, K.; Darfeuille-Michaud, A.; Desreumaux, P.; et al. Bacteriophages targeting adherent invasive Escherichia coli strains as a promising new treatment for Crohn’s disease. J. Crohns Colitis 2017, 11, 840–847. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stolp, H.; Starr, M.P. Bdellovibrio bacteriovorus gen. et sp. n., a predatory, ectoparasitic, and bacteriolytic microorganism. Antonie Leeuwenhoek 1963, 29, 217–248. [Google Scholar] [CrossRef]
- Iebba, V.; Santangelo, F.; Totino, V.; Nicoletti, M.; Gagliardi, A.; De Biase, R.V.; Cucchiara, S.; Nencioni, L.; Conte, M.P.; Schippa, S. Higher prevalence and abundance of Bdellovibrio bacteriovorus in the human gut of healthy subjects. PLoS ONE 2013, 8, e61608. [Google Scholar] [CrossRef]
- Rendulic, S.; Jagtap, P.; Rosinus, A.; Eppinger, M.; Baar, C.; Lanz, C.; Keller, H.; Lambert, C.; Evans, K.J.; Goesmann, A.; et al. A predator unmasked: Life cycle of Bdellovibrio bacteriovorus from a genomic perspective. Science 2004, 303, 689–692. [Google Scholar] [CrossRef]
- Lambert, C.; Morehouse, K.A.; Chang, C.-Y.; Sockett, R.E. Bdellovibrio: Growth and development during the predatory cycle. Curr. Opin. Microbiol. 2006, 9, 639–644. [Google Scholar] [CrossRef]
- Bonfiglio, G.; Neroni, B.; Radocchia, G.; Pompilio, A.; Mura, F.; Trancassini, M.; Di Bonaventura, G.; Pantanella, F.; Schippa, S. Growth Control of Adherent-Invasive Escherichia coli (AIEC) by the Predator Bacteria Bdellovibrio bacteriovorus: A New Therapeutic Approach for Crohn’s Disease Patients. Microorganisms 2019, 8, 17. [Google Scholar] [CrossRef] [Green Version]
- Cianfanelli, F.R.; Monlezun, L.; Coulthurst, S.J. Aim, Load, Fire: The Type VI Secretion System, a Bacterial Nanoweapon. Trends Microbiol. 2016, 24, 51–62. [Google Scholar] [CrossRef]
- Chassaing, B.; Cascales, E. Antibacterial Weapons: Targeted Destruction in the Microbiota. Trends Microbiol. 2018, 26, 329–338. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sana, T.G.; Flaugnatti, N.; Lugo, K.A.; Lam, L.H.; Jacobson, A.; Baylot, V.; Durand, E.; Journet, L.; Cascales, E.; Monack, D.M. Salmonella Typhimurium utilizes a T6SS-mediated antibacterial weapon to establish in the host gut. Proc. Natl. Acad. Sci. USA 2016, 113, E5044–E5051. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chatzidaki-Livanis, M.; Geva-Zatorsky, N.; Comstock, L.E. Bacteroides fragilis type VI secretion systems use novel effector and immunity proteins to antagonize human gut Bacteroidales species. Proc. Natl. Acad. Sci. USA 2016, 113, 3627–3632. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hecht, A.L.; Casterline, B.W.; Earley, Z.M.; Goo, Y.A.; Goodlett, D.R.; Bubeck Wardenburg, J. Strain competition restricts colonization of an enteric pathogen and prevents colitis. EMBO Rep. 2016, 17, 1281–1291. [Google Scholar] [CrossRef] [Green Version]
- Anderson, M.C.; Vonaesch, P.; Saffarian, A.; Marteyn, B.S.; Sansonetti, P.J. Shigella sonnei Encodes a Functional T6SS Used for Interbacterial Competition and Niche Occupancy. Cell Host Microbe 2017, 21, 769–776. [Google Scholar] [CrossRef] [Green Version]
- Wettstadt, S.; Filloux, A. Manipulating the type VI secretion system spike to shuttle passenger proteins. PLoS ONE 2020, 15. [Google Scholar] [CrossRef] [Green Version]
- Hamilton, T.A.; Pellegrino, G.M.; Therrien, J.A.; Ham, D.T.; Bartlett, P.C.; Karas, B.J.; Gloor, G.B.; Edgell, D.R. Efficient inter-species conjugative transfer of a CRISPR nuclease for targeted bacterial killing. Nat. Commun. 2019, 10, 4544. [Google Scholar] [CrossRef]
- Rodrigues, M.; McBride, S.W.; Hullahalli, K.; Palmer, K.L.; Duerkop, B.A. Conjugative Delivery of CRISPR-Cas9 for the Selective Depletion of Antibiotic-Resistant Enterococci. Antimicrob. Agents Chemother. 2019, 63. [Google Scholar] [CrossRef]
- Hooper, K.M.; Casanova, V.; Kemp, S.; Staines, K.A.; Satsangi, J.; Barlow, P.G.; Henderson, P.; Stevens, C. The Inflammatory Bowel Disease Drug Azathioprine Induces Autophagy via mTORC1 and the Unfolded Protein Response Sensor PERK. Inflamm. Bowel Dis. 2019, 25, 1481–1496. [Google Scholar] [CrossRef] [Green Version]
- Al Azzaz, J.; Rieu, A.; Aires, V.; Delmas, D.; Chluba, J.; Winckler, P.; Bringer, M.-A.; Lamarche, J.; Vervandier-Fasseur, D.; Dalle, F.; et al. Resveratrol-Induced Xenophagy Promotes Intracellular Bacteria Clearance in Intestinal Epithelial Cells and Macrophages. Front. Immunol. 2019, 9. [Google Scholar] [CrossRef]
- Nickerson, K.P.; McDonald, C. Crohn’s Disease-Associated Adherent-Invasive Escherichia coli Adhesion Is Enhanced by Exposure to the Ubiquitous Dietary Polysaccharide Maltodextrin. PLoS ONE 2012, 7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tran, H.Q.; Ley, R.E.; Gewirtz, A.T.; Chassaing, B. Flagellin-elicited adaptive immunity suppresses flagellated microbiota and vaccinates against chronic inflammatory diseases. Nat. Commun. 2019, 10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Viladomiu, M.; Kivolowitz, C.; Abdulhamid, A.; Dogan, B.; Victorio, D.; Castellanos, J.G.; Woo, V.; Teng, F.; Tran, N.L.; Sczesnak, A.; et al. IgA-coated E. coli enriched in Crohn’s disease spondyloarthritis promote TH 17-dependent inflammation. Sci. Transl. Med. 2017, 9, eaaf9655. [Google Scholar] [CrossRef] [PubMed]




© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chervy, M.; Barnich, N.; Denizot, J. Adherent-Invasive E. coli: Update on the Lifestyle of a Troublemaker in Crohn’s Disease. Int. J. Mol. Sci. 2020, 21, 3734. https://doi.org/10.3390/ijms21103734
Chervy M, Barnich N, Denizot J. Adherent-Invasive E. coli: Update on the Lifestyle of a Troublemaker in Crohn’s Disease. International Journal of Molecular Sciences. 2020; 21(10):3734. https://doi.org/10.3390/ijms21103734
Chicago/Turabian StyleChervy, Mélissa, Nicolas Barnich, and Jérémy Denizot. 2020. "Adherent-Invasive E. coli: Update on the Lifestyle of a Troublemaker in Crohn’s Disease" International Journal of Molecular Sciences 21, no. 10: 3734. https://doi.org/10.3390/ijms21103734
APA StyleChervy, M., Barnich, N., & Denizot, J. (2020). Adherent-Invasive E. coli: Update on the Lifestyle of a Troublemaker in Crohn’s Disease. International Journal of Molecular Sciences, 21(10), 3734. https://doi.org/10.3390/ijms21103734