CRISPR/Cas9-Mediated Gene Correction to Understand ALS

Abstract
:1. Introduction
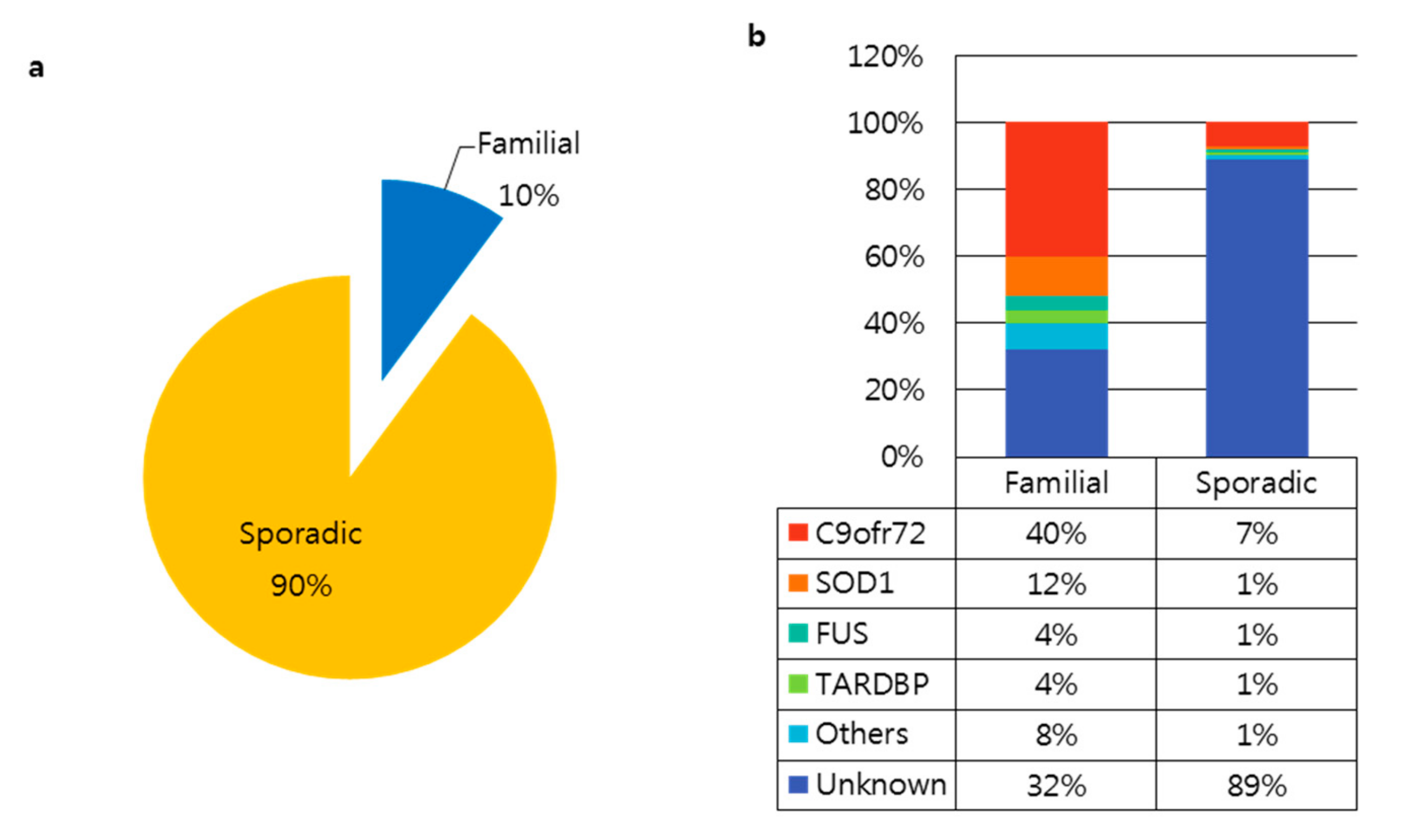
2. Amyotrophic Lateral Sclerosis (ALS)
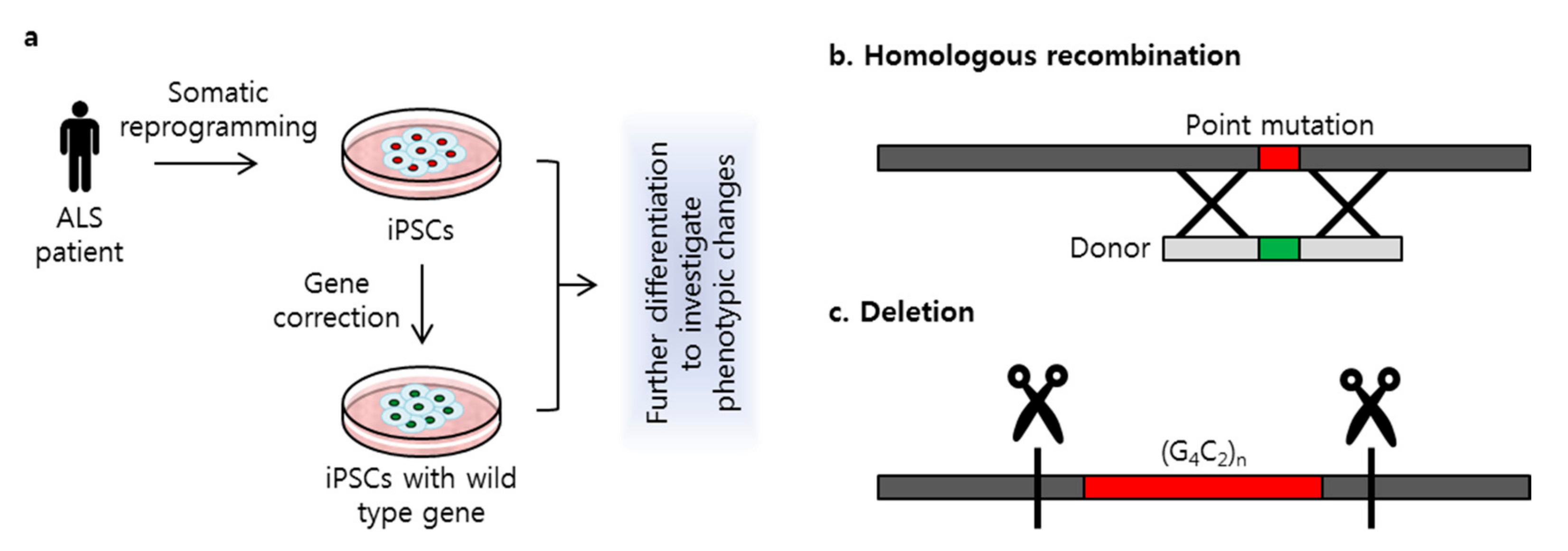
3. Targeted Genome Editing Using CRISPR/Cas9
3.1. Superoxide Dismutase 1 (SOD1)
3.2. C9orf72
3.3. Fused in Sarcoma (FUS)
3.4. TAR-Binding Protein Gene (TARDBP)
4. CRISPR/Cas9-Mediated In Vivo Therapeutic Approach

{kind=link}
{kind=link}
| Study | Gene | Variants | Model | Gene Editing Efficiency |
|---|---|---|---|---|
| Homologous recombination | ||||
| Wang et al., 2017 [46] | SOD1 | A272C | Human ALS patient-derived iPSCs | 20% |
| Bhinge et al., 2017 [47] | SOD1 | E100G | Human ALS patient-derived iPSCs | 0.5% (one of ~ 180 colonies) |
| Imamura et al., 2017 [48] | SOD1 | L144FVX | Human ALS patient-derived iPSCs | Selected with puromycin |
| Guo et al., 2017 [66] | FUS | R521H | Human ALS patient-derived iPSCs | Selected with puromycin |
| Wang et al., 2017 [46] | FUS | G1566A | Human ALS patient-derived iPSCs | 1% |
| Bhinge et al., 2017 [47] | FUS | H517Q | Human ALS patient-derived iPSCs | 0.5% (one of ~ 180 colonies) |
| Wang et al., 2018 [70] | FUS | R521H, P525L | Human ALS patient-derived iPSCs | Selected with puromycin |
| Vandoorne et al., 2019 [5] | FUS | R521H, P525L | Human ALS patient-derived iPSCs | Selected with puromycin |
| Tann et al., 2019 [77] | TARDBP | M337V | Human ALS patient-derived iPSCs | FACS sorting |
| Deletion | ||||
| Pribadi et al., 2016 [56] | C9orf72 | GGGGCC | Human FTD/ALS patient-derived iPSCs | 11.1% (66 of 593 colonies) |
| Selvaraj et al., 2018 [58] | C9orf72 | GGGGCC | Human ALS patient-derived iPSCs | 0.6% ~ 4.5% |
| Lopez-Gonzalez et al., 2019 [61] | C9orf72 | GGGGCC | C9orf72 patient-derived iPSCs | N/R |
| Duan et al., 2019 [79] | SOD1 | G93A | hSOD1 G93A transgenic mouse | N/R |
| Indel formation | ||||
| Gaj et al., 2017 [78] | SOD1 | G93A | hSOD1 G93A transgenic mouse | N/R |
5. Conclusions and Future
Author Contributions
Funding
Conflicts of Interest
References
- Hulisz, D. Amyotrophic lateral sclerosis: Disease state overview. Am. J. Manag. Care 2018, 24, S320–S326. [Google Scholar]
- Oskarsson, B.; Gendron, T.F.; Staff, N.P. Amyotrophic lateral sclerosis: An update for 2018. Mayo Clin. Proc. 2018, 93, 1617–1628. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rosen, D.R.; Siddique, T.; Patterson, D.; Figlewicz, D.A.; Sapp, P.; Hentati, A.; Donaldson, D.; Goto, J.; O’Regan, J.P.; Deng, H.X.; et al. Mutations in cu/zn superoxide dismutase gene are associated with familial amyotrophic lateral sclerosis. Nature 1993, 362, 59–62. [Google Scholar] [CrossRef] [PubMed]
- Wroe, R.; Wai-Ling Butler, A.; Andersen, P.M.; Powell, J.F.; Al-Chalabi, A. Alsod: The amyotrophic lateral sclerosis online database. Amyotroph. Lateral Scler. Off. Publ. World Fed. Neurol. Res. Group Mot. Neuron Dis. 2008, 9, 249–250. [Google Scholar] [CrossRef]
- Vandoorne, T.; Veys, K.; Guo, W.; Sicart, A.; Vints, K.; Swijsen, A.; Moisse, M.; Eelen, G.; Gounko, N.V.; Fumagalli, L.; et al. Differentiation but not als mutations in fus rewires motor neuron metabolism. Nat. Commun. 2019, 10, 4147. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rothstein, J.D. Edaravone: A new drug approved for als. Cell 2017, 171, 725. [Google Scholar] [CrossRef] [PubMed]
- Miller, R.G.; Appel, S.H. Introduction to supplement: The current status of treatment for als. Amyotroph. Lateral Scler. Front. Degener. 2017, 18, 1–4. [Google Scholar] [CrossRef]
- Bensimon, G.; Lacomblez, L.; Meininger, V. A controlled trial of riluzole in amyotrophic lateral sclerosis. Als/riluzole study group. N. Engl. J. Med. 1994, 330, 585–591. [Google Scholar] [CrossRef]
- Jaiswal, M.K. Riluzole and edaravone: A tale of two amyotrophic lateral sclerosis drugs. Med. Res. Rev. 2019, 39, 733–748. [Google Scholar] [CrossRef] [PubMed]
- You, L.; Tong, R.; Li, M.; Liu, Y.; Xue, J.; Lu, Y. Advancements and obstacles of crispr-cas9 technology in translational research. Mol. Ther. Methods Clin. Dev. 2019, 13, 359–370. [Google Scholar] [CrossRef] [Green Version]
- Maeder, M.L.; Gersbach, C.A. Genome-editing technologies for gene and cell therapy. Mol. Ther. J. Am. Soc. Gene Ther. 2016, 24, 430–446. [Google Scholar] [CrossRef] [Green Version]
- Gaj, T.; Gersbach, C.A.; Barbas, C.F., 3rd. Zfn, talen, and crispr/cas-based methods for genome engineering. Trends Biotechnol. 2013, 31, 397–405. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ragagnin, A.M.G.; Shadfar, S.; Vidal, M.; Jamali, M.S.; Atkin, J.D. Motor neuron susceptibility in als/ftd. Front. Neurosci. 2019, 13, 532. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Statland, J.M.; Barohn, R.J.; McVey, A.L.; Katz, J.S.; Dimachkie, M.M. Patterns of weakness, classification of motor neuron disease, and clinical diagnosis of sporadic amyotrophic lateral sclerosis. Neurol. Clin. 2015, 33, 735–748. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grad, L.I.; Rouleau, G.A.; Ravits, J.; Cashman, N.R. Clinical spectrum of amyotrophic lateral sclerosis (als). Cold Spring Harb. Perspect. Med. 2017, 7, a024117. [Google Scholar] [CrossRef] [Green Version]
- Marin, B.; Boumédiene, F.; Logroscino, G.; Couratier, P.; Babron, M.C.; Leutenegger, A.L.; Copetti, M.; Preux, P.M.; Beghi, E. Variation in worldwide incidence of amyotrophic lateral sclerosis: A meta-analysis. Int. J. Epidemiol. 2017, 46, 57–74. [Google Scholar] [CrossRef] [Green Version]
- Mehta, P.; Kaye, W.; Raymond, J.; Wu, R.; Larson, T.; Punjani, R.; Heller, D.; Cohen, J.; Peters, T.; Muravov, O.; et al. Prevalence of amyotrophic lateral sclerosis—United states, 2014. Mmwr. Morb. Mortal. Wkly. Rep. 2018, 67, 216–218. [Google Scholar] [CrossRef] [Green Version]
- Arthur, K.C.; Calvo, A.; Price, T.R.; Geiger, J.T.; Chiò, A.; Traynor, B.J. Projected increase in amyotrophic lateral sclerosis from 2015 to 2040. Nat. Commun. 2016, 7, 12408. [Google Scholar] [CrossRef] [Green Version]
- Brown, R.H.; Al-Chalabi, A. Amyotrophic lateral sclerosis. N. Engl. J. Med. 2017, 377, 162–172. [Google Scholar] [CrossRef] [Green Version]
- Mehta, P.; Horton, D.K.; Kasarskis, E.J.; Tessaro, E.; Eisenberg, M.S.; Laird, S.; Iskander, J. Cdc grand rounds: National amyotrophic lateral sclerosis (als) registry impact, challenges, and future directions. Mmwr. Morb. Mortal. Wkly. Rep. 2017, 66, 1379–1382. [Google Scholar] [CrossRef] [Green Version]
- Abel, O.; Shatunov, A.; Jones, A.R.; Andersen, P.M.; Powell, J.F.; Al-Chalabi, A. Development of a smartphone app for a genetics website: The amyotrophic lateral sclerosis online genetics database (alsod). Jmir Mhealth Uhealth 2013, 1, e18. [Google Scholar] [CrossRef] [PubMed]
- Abel, O.; Powell, J.F.; Andersen, P.M.; Al-Chalabi, A. Alsod: A user-friendly online bioinformatics tool for amyotrophic lateral sclerosis genetics. Hum. Mutat. 2012, 33, 1345–1351. [Google Scholar] [CrossRef]
- Richards, S.; Aziz, N.; Bale, S.; Bick, D.; Das, S.; Gastier-Foster, J.; Grody, W.W.; Hegde, M.; Lyon, E.; Spector, E.; et al. Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the american college of medical genetics and genomics and the association for molecular pathology. Genet. Med. Off. J. Am. Coll. Med. Genet. 2015, 17, 405–424. [Google Scholar] [CrossRef] [PubMed]
- Mejzini, R.; Flynn, L.L.; Pitout, I.L.; Fletcher, S.; Wilton, S.D.; Akkari, P.A. Als genetics, mechanisms, and therapeutics: Where are we now? Front. Neurosci. 2019, 13, 1310. [Google Scholar] [CrossRef] [Green Version]
- Scarrott, J.M.; Herranz-Martín, S.; Alrafiah, A.R.; Shaw, P.J.; Azzouz, M. Current developments in gene therapy for amyotrophic lateral sclerosis. Expert Opin. Biol. Ther. 2015, 15, 935–947. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van der Spek, R.A.A.; Van Rheenen, W.; Pulit, S.L.; Kenna, K.P.; Van den Berg, L.H.; Veldink, J.H. The project mine databrowser: Bringing large-scale whole-genome sequencing in als to researchers and the public. Amyotroph. Lateral Scler. Front. Degener. 2019, 20, 432–440. [Google Scholar] [CrossRef] [Green Version]
- Rheenen, W.V.; Pulit, S.L.; Dekker, A.M.; Khleifat, A.A.; Brands, W.J.; Iacoangeli, A.; Kenna, K.P.; Kooyman, M.; McLaughlin, R.L.; Middelkoop, B.; et al. Project mine: Study design and pilot analyses of a large-scale whole genome sequencing study in amyotrophic lateral sclerosis. J. bioRxiv 2017, 152553. [Google Scholar] [CrossRef] [Green Version]
- Chen, S.; Sayana, P.; Zhang, X.; Le, W. Genetics of amyotrophic lateral sclerosis: An update. Mol. Neurodegener. 2013, 8, 28. [Google Scholar] [CrossRef] [Green Version]
- Charpentier, E.; Marraffini, L.A. Harnessing crispr-cas9 immunity for genetic engineering. Curr. Opin. Microbiol. 2014, 19, 114–119. [Google Scholar] [CrossRef] [Green Version]
- Hryhorowicz, M.; Lipiński, D.; Zeyland, J.; Słomski, R. Crispr/cas9 immune system as a tool for genome engineering. Arch. Immunol. Et Ther. Exp. 2017, 65, 233–240. [Google Scholar] [CrossRef] [Green Version]
- Ran, F.A.; Hsu, P.D.; Wright, J.; Agarwala, V.; Scott, D.A.; Zhang, F. Genome engineering using the crispr-cas9 system. Nat. Protoc. 2013, 8, 2281–2308. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, J.; Chung, J.H.; Kim, H.M.; Kim, D.W.; Kim, H. Designed nucleases for targeted genome editing. Plant. Biotechnol. J. 2016, 14, 448–462. [Google Scholar] [CrossRef] [PubMed]
- Nakade, S.; Yamamoto, T.; Sakuma, T. Cas9, cpf1 and c2c1/2/3-what’s next? Bioengineered 2017, 8, 265–273. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, K.; Cai, D.; Wang, Z.; He, Z.; Chen, S. Development of an efficient genome editing tool in bacillus licheniformis using crispr-cas9 nickase. Appl. Environ. Microbiol. 2018, 84, e02608-17. [Google Scholar] [CrossRef] [Green Version]
- Dominguez, A.A.; Lim, W.A.; Qi, L.S. Beyond editing: Repurposing crispr-cas9 for precision genome regulation and interrogation. Nat. Rev. Mol. Cell Biol. 2016, 17, 5–15. [Google Scholar] [CrossRef] [Green Version]
- Pearson, M.M.; Himpsl, S.D.; Mobley, H.L.T. Insertional mutagenesis protocol for constructing single or sequential mutations. Methods Mol. Biol. (CliftonN.J.) 2019, 2021, 61–76. [Google Scholar]
- Takahashi, K.; Yamanaka, S. Induction of pluripotent stem cells from mouse embryonic and adult fibroblast cultures by defined factors. Cell 2006, 126, 663–676. [Google Scholar] [CrossRef] [Green Version]
- Chen, H.; Qian, K.; Du, Z.; Cao, J.; Petersen, A.; Liu, H.; Blackbourn, L.W.T.; Huang, C.L.; Errigo, A.; Yin, Y.; et al. Modeling als with ipscs reveals that mutant sod1 misregulates neurofilament balance in motor neurons. Cell Stem Cell 2014, 14, 796–809. [Google Scholar] [CrossRef] [Green Version]
- Kiskinis, E.; Sandoe, J.; Williams, L.A.; Boulting, G.L.; Moccia, R.; Wainger, B.J.; Han, S.; Peng, T.; Thams, S.; Mikkilineni, S.; et al. Pathways disrupted in human als motor neurons identified through genetic correction of mutant sod1. Cell Stem Cell 2014, 14, 781–795. [Google Scholar] [CrossRef] [Green Version]
- Liu, X.; Chen, J.; Liu, W.; Li, X.; Chen, Q.; Liu, T.; Gao, S.; Deng, M. The fused in sarcoma protein forms cytoplasmic aggregates in motor neurons derived from integration-free induced pluripotent stem cells generated from a patient with familial amyotrophic lateral sclerosis carrying the fus-p525l mutation. Neurogenetics 2015, 16, 223–231. [Google Scholar] [CrossRef]
- Fujimori, K.; Ishikawa, M.; Otomo, A.; Atsuta, N.; Nakamura, R.; Akiyama, T.; Hadano, S.; Aoki, M.; Saya, H.; Sobue, G.; et al. Modeling sporadic als in ipsc-derived motor neurons identifies a potential therapeutic agent. Nat. Med. 2018, 24, 1579–1589. [Google Scholar] [CrossRef]
- Yamashita, S.; Ando, Y. Genotype-phenotype relationship in hereditary amyotrophic lateral sclerosis. Transl. Neurodegener. 2015, 4, 13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zou, Z.Y.; Zhou, Z.R.; Che, C.H.; Liu, C.Y.; He, R.L.; Huang, H.P. Genetic epidemiology of amyotrophic lateral sclerosis: A systematic review and meta-analysis. J. Neurol. Neurosurg. Psychiatry 2017, 88, 540–549. [Google Scholar] [CrossRef]
- Kaur, S.J.; McKeown, S.R.; Rashid, S. Mutant sod1 mediated pathogenesis of amyotrophic lateral sclerosis. Gene 2016, 577, 109–118. [Google Scholar] [CrossRef]
- Kato, S.; Kato, M.; Abe, Y.; Matsumura, T.; Nishino, T.; Aoki, M.; Itoyama, Y.; Asayama, K.; Awaya, A.; Hirano, A.; et al. Redox system expression in the motor neurons in amyotrophic lateral sclerosis (als): Immunohistochemical studies on sporadic als, superoxide dismutase 1 (sod1)-mutated familial als, and sod1-mutated als animal models. Acta Neuropathol. 2005, 110, 101–112. [Google Scholar] [CrossRef]
- Wang, L.; Yi, F.; Fu, L.; Yang, J.; Wang, S.; Wang, Z.; Suzuki, K.; Sun, L.; Xu, X.; Yu, Y.; et al. Crispr/cas9-mediated targeted gene correction in amyotrophic lateral sclerosis patient ipscs. Protein Cell 2017, 8, 365–378. [Google Scholar] [CrossRef]
- Bhinge, A.; Namboori, S.C.; Zhang, X.; VanDongen, A.M.J.; Stanton, L.W. Genetic correction of sod1 mutant ipscs reveals erk and jnk activated ap1 as a driver of neurodegeneration in amyotrophic lateral sclerosis. Stem Cell Rep. 2017, 8, 856–869. [Google Scholar] [CrossRef] [Green Version]
- Imamura, K.; Izumi, Y.; Watanabe, A.; Tsukita, K.; Woltjen, K.; Yamamoto, T.; Hotta, A.; Kondo, T.; Kitaoka, S.; Ohta, A.; et al. The src/c-abl pathway is a potential therapeutic target in amyotrophic lateral sclerosis. Sci. Transl. Med. 2017, 9, eaaf3962. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Majounie, E.; Renton, A.E.; Mok, K.; Dopper, E.G.; Waite, A.; Rollinson, S.; Chiò, A.; Restagno, G.; Nicolaou, N.; Simon-Sanchez, J.; et al. Frequency of the c9orf72 hexanucleotide repeat expansion in patients with amyotrophic lateral sclerosis and frontotemporal dementia: A cross-sectional study. Lancet Neurol. 2012, 11, 323–330. [Google Scholar] [CrossRef]
- Gami, P.; Murray, C.; Schottlaender, L.; Bettencourt, C.; De Pablo Fernandez, E.; Mudanohwo, E.; Mizielinska, S.; Polke, J.M.; Holton, J.L.; Isaacs, A.M.; et al. A 30-unit hexanucleotide repeat expansion in c9orf72 induces pathological lesions with dipeptide-repeat proteins and rna foci, but not tdp-43 inclusions and clinical disease. Acta Neuropathol. 2015, 130, 599–601. [Google Scholar] [CrossRef] [PubMed]
- Zhang, K.; Donnelly, C.J.; Haeusler, A.R.; Grima, J.C.; Machamer, J.B.; Steinwald, P.; Daley, E.L.; Miller, S.J.; Cunningham, K.M.; Vidensky, S.; et al. The c9orf72 repeat expansion disrupts nucleocytoplasmic transport. Nature 2015, 525, 56–61. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lagier-Tourenne, C.; Baughn, M.; Rigo, F.; Sun, S.; Liu, P.; Li, H.R.; Jiang, J.; Watt, A.T.; Chun, S.; Katz, M.; et al. Targeted degradation of sense and antisense c9orf72 rna foci as therapy for als and frontotemporal degeneration. Proc. Natl. Acad. Sci. USA 2013, 110, E4530–E4539. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Westergard, T.; McAvoy, K.; Russell, K.; Wen, X.; Pang, Y.; Morris, B.; Pasinelli, P.; Trotti, D.; Haeusler, A. Repeat-associated non-aug translation in c9orf72-als/ftd is driven by neuronal excitation and stress. Embo Mol. Med. 2019, 11, e9423. [Google Scholar] [CrossRef] [PubMed]
- Saberi, S.; Stauffer, J.E.; Jiang, J.; Garcia, S.D.; Taylor, A.E.; Schulte, D.; Ohkubo, T.; Schloffman, C.L.; Maldonado, M.; Baughn, M.; et al. Sense-encoded poly-gr dipeptide repeat proteins correlate to neurodegeneration and uniquely co-localize with tdp-43 in dendrites of repeat-expanded c9orf72 amyotrophic lateral sclerosis. Acta Neuropathol. 2018, 135, 459–474. [Google Scholar] [CrossRef]
- Sakae, N.; Bieniek, K.F.; Zhang, Y.J.; Ross, K.; Gendron, T.F.; Murray, M.E.; Rademakers, R.; Petrucelli, L.; Dickson, D.W. Poly-gr dipeptide repeat polymers correlate with neurodegeneration and clinicopathological subtypes in c9orf72-related brain disease. Acta Neuropathol. Commun. 2018, 6, 63. [Google Scholar] [CrossRef]
- Pribadi, M.; Yang, Z.; Kim, T.S.; Swartz, E.W.; Huang, A.Y.; Chen, J.A.; Dokuru, D.; Baek, J.; Gao, F.; Fua, A.T.; et al. Crispr-cas9 targeted deletion of the c9orf72 repeat expansion mutation corrects cellular phenotypes in patient-derived ips cells. J. bioRxiv 2016, 051193. [Google Scholar] [CrossRef] [Green Version]
- Joshi, D.C.; Tewari, B.P.; Singh, M.; Joshi, P.G.; Joshi, N.B. Ampa receptor activation causes preferential mitochondrial ca2+ load and oxidative stress in motor neurons. Brain Res. 2015, 1616, 1–9. [Google Scholar] [CrossRef]
- Selvaraj, B.T.; Livesey, M.R.; Zhao, C.; Gregory, J.M.; James, O.T.; Cleary, E.M.; Chouhan, A.K.; Gane, A.B.; Perkins, E.M.; Dando, O.; et al. C9orf72 repeat expansion causes vulnerability of motor neurons to ca(2+)-permeable ampa receptor-mediated excitotoxicity. Nat. Commun. 2018, 9, 347. [Google Scholar] [CrossRef] [Green Version]
- Koppers, M.; Blokhuis, A.M.; Westeneng, H.J.; Terpstra, M.L.; Zundel, C.A.; Vieira de Sá, R.; Schellevis, R.D.; Waite, A.J.; Blake, D.J.; Veldink, J.H.; et al. C9orf72 ablation in mice does not cause motor neuron degeneration or motor deficits. Ann. Neurol. 2015, 78, 426–438. [Google Scholar] [CrossRef] [Green Version]
- Freibaum, B.D.; Lu, Y.; Lopez-Gonzalez, R.; Kim, N.C.; Almeida, S.; Lee, K.H.; Badders, N.; Valentine, M.; Miller, B.L.; Wong, P.C.; et al. Ggggcc repeat expansion in c9orf72 compromises nucleocytoplasmic transport. Nature 2015, 525, 129–133. [Google Scholar] [CrossRef]
- Lopez-Gonzalez, R.; Yang, D.; Pribadi, M.; Kim, T.S.; Krishnan, G.; Choi, S.Y.; Lee, S.; Coppola, G.; Gao, F.B. Partial inhibition of the overactivated ku80-dependent DNA repair pathway rescues neurodegeneration in c9orf72-als/ftd. Proc. Natl. Acad. Sci. USA 2019, 116, 9628–9633. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shang, Y.; Huang, E.J. Mechanisms of fus mutations in familial amyotrophic lateral sclerosis. Brain Res. 2016, 1647, 65–78. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- An, H.; Skelt, L.; Notaro, A.; Highley, J.R.; Fox, A.H.; La Bella, V.; Buchman, V.L.; Shelkovnikova, T.A. Als-linked fus mutations confer loss and gain of function in the nucleus by promoting excessive formation of dysfunctional paraspeckles. Acta Neuropathol. Commun. 2019, 7, 7. [Google Scholar] [CrossRef] [Green Version]
- Sharma, A.; Lyashchenko, A.K.; Lu, L.; Nasrabady, S.E.; Elmaleh, M.; Mendelsohn, M.; Nemes, A.; Tapia, J.C.; Mentis, G.Z.; Shneider, N.A. Als-associated mutant fus induces selective motor neuron degeneration through toxic gain of function. Nat. Commun. 2016, 7, 10465. [Google Scholar] [CrossRef] [PubMed]
- Nakaya, T.; Maragkakis, M. Amyotrophic lateral sclerosis associated fus mutation shortens mitochondria and induces neurotoxicity. Sci. Rep. 2018, 8, 15575. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guo, W.; Naujock, M.; Fumagalli, L.; Vandoorne, T.; Baatsen, P.; Boon, R.; Ordovás, L.; Patel, A.; Welters, M.; Vanwelden, T.; et al. Hdac6 inhibition reverses axonal transport defects in motor neurons derived from fus-als patients. Nat. Commun. 2017, 8, 861. [Google Scholar] [CrossRef] [PubMed]
- Barber, S.C.; Shaw, P.J. Oxidative stress in als: Key role in motor neuron injury and therapeutic target. Free Radic. Biol. Med. 2010, 48, 629–641. [Google Scholar] [CrossRef]
- Formella, I.; Svahn, A.J.; Radford, R.A.W.; Don, E.K.; Cole, N.J.; Hogan, A.; Lee, A.; Chung, R.S.; Morsch, M. Real-time visualization of oxidative stress-mediated neurodegeneration of individual spinal motor neurons in vivo. Redox Biol. 2018, 19, 226–234. [Google Scholar] [CrossRef]
- Wang, H.; Dharmalingam, P.; Vasquez, V.; Mitra, J.; Boldogh, I.; Rao, K.S.; Kent, T.A.; Mitra, S.; Hegde, M.L. Chronic oxidative damage together with genome repair deficiency in the neurons is a double whammy for neurodegeneration: Is damage response signaling a potential therapeutic target? Mech. Ageing Dev. 2017, 161, 163–176. [Google Scholar] [CrossRef] [Green Version]
- Wang, H.; Guo, W.; Mitra, J.; Hegde, P.M.; Vandoorne, T.; Eckelmann, B.J.; Mitra, S.; Tomkinson, A.E.; Van Den Bosch, L.; Hegde, M.L. Mutant fus causes DNA ligation defects to inhibit oxidative damage repair in amyotrophic lateral sclerosis. Nat. Commun. 2018, 9, 3683. [Google Scholar] [CrossRef] [Green Version]
- Naumann, M.; Pal, A.; Goswami, A.; Lojewski, X.; Japtok, J.; Vehlow, A.; Naujock, M.; Günther, R.; Jin, M.; Stanslowsky, N.; et al. Impaired DNA damage response signaling by fus-nls mutations leads to neurodegeneration and fus aggregate formation. Nat. Commun. 2018, 9, 335. [Google Scholar] [CrossRef] [PubMed]
- Naujock, M.; Stanslowsky, N.; Bufler, S.; Naumann, M.; Reinhardt, P.; Sterneckert, J.; Kefalakes, E.; Kassebaum, C.; Bursch, F.; Lojewski, X.; et al. 4-aminopyridine induced activity rescues hypoexcitable motor neurons from amyotrophic lateral sclerosis patient-derived induced pluripotent stem cells. Stem Cells (Dayt. Ohio) 2016, 34, 1563–1575. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, T.; Jiang, X.; Chen, G.; Xu, J. Interaction of amyotrophic lateral sclerosis/frontotemporal lobar degeneration-associated fused-in-sarcoma with proteins involved in metabolic and protein degradation pathways. Neurobiol. Aging 2015, 36, 527–535. [Google Scholar] [CrossRef] [PubMed]
- Sreedharan, J.; Blair, I.P.; Tripathi, V.B.; Hu, X.; Vance, C.; Rogelj, B.; Ackerley, S.; Durnall, J.C.; Williams, K.L.; Buratti, E.; et al. Tdp-43 mutations in familial and sporadic amyotrophic lateral sclerosis. Science 2008, 319, 1668–1672. [Google Scholar] [CrossRef]
- Prasad, A.; Bharathi, V.; Sivalingam, V.; Girdhar, A.; Patel, B.K. Molecular mechanisms of tdp-43 misfolding and pathology in amyotrophic lateral sclerosis. Front. Mol. Neurosci. 2019, 12, 25. [Google Scholar] [CrossRef]
- Johnson, B.S.; Snead, D.; Lee, J.J.; McCaffery, J.M.; Shorter, J.; Gitler, A.D. Tdp-43 is intrinsically aggregation-prone, and amyotrophic lateral sclerosis-linked mutations accelerate aggregation and increase toxicity. J. Biol. Chem. 2009, 284, 20329–20339. [Google Scholar] [CrossRef] [Green Version]
- Tann, J.Y.; Wong, L.W.; Sajikumar, S.; Ibáñez, C.F. Abnormal tdp-43 function impairs activity-dependent bdnf secretion, synaptic plasticity, and cognitive behavior through altered sortilin splicing. Embo J. 2019, 38. [Google Scholar] [CrossRef]
- Gaj, T.; Ojala, D.S.; Ekman, F.K.; Byrne, L.C.; Limsirichai, P.; Schaffer, D.V. In vivo genome editing improves motor function and extends survival in a mouse model of als. Sci. Adv. 2017, 3, eaar3952. [Google Scholar] [CrossRef] [Green Version]
- Duan, W.; Guo, M.; Yi, L.; Liu, Y.; Li, Z.; Ma, Y.; Zhang, G.; Liu, Y.; Bu, H.; Song, X.; et al. The deletion of mutant sod1 via crispr/cas9/sgrna prolongs survival in an amyotrophic lateral sclerosis mouse model. Gene Ther. 2019. [Google Scholar] [CrossRef]
- Ran, F.A.; Cong, L.; Yan, W.X.; Scott, D.A.; Gootenberg, J.S.; Kriz, A.J.; Zetsche, B.; Shalem, O.; Wu, X.; Makarova, K.S.; et al. In vivo genome editing using staphylococcus aureus cas9. Nature 2015, 520, 186–191. [Google Scholar] [CrossRef]
- Chen, S.J. Minimizing off-target effects in crispr-cas9 genome editing. Cell Biol. Toxicol. 2019, 35, 399–401. [Google Scholar] [CrossRef] [Green Version]
- Yang, D.; Scavuzzo, M.A.; Chmielowiec, J.; Sharp, R.; Bajic, A.; Borowiak, M. Enrichment of g2/m cell cycle phase in human pluripotent stem cells enhances hdr-mediated gene repair with customizable endonucleases. Sci. Rep. 2016, 6, 21264. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lin, S.; Staahl, B.T.; Alla, R.K.; Doudna, J.A. Enhanced homology-directed human genome engineering by controlled timing of crispr/cas9 delivery. eLife 2014, 3, e04766. [Google Scholar] [CrossRef] [PubMed]
- Chu, V.T.; Weber, T.; Wefers, B.; Wurst, W.; Sander, S.; Rajewsky, K.; Kühn, R. Increasing the efficiency of homology-directed repair for crispr-cas9-induced precise gene editing in mammalian cells. Nat. Biotechnol. 2015, 33, 543–548. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Corcia, P.; Couratier, P.; Blasco, H.; Andres, C.R.; Beltran, S.; Meininger, V.; Vourc’h, P. Genetics of amyotrophic lateral sclerosis. Rev. Neurol. 2017, 173, 254–262. [Google Scholar] [CrossRef]
- Vildan, C.; Sule, D.; Turker, B.; Hilmi, U.; Sibel, K.B. Genetic alterations of c9orf72, sod1, tardbp, fus, and ubqln2 genes in patients with amyotrophic lateral sclerosis. Cogent Med. 2019, 6, 1. [Google Scholar] [CrossRef]


| Nucleotide | Protein | dbSNP | Mutation Type |
|---|---|---|---|
| SOD1 | |||
| NM_000454.4:c.14C >T | NP_000445.1:p.Ala5Val | rs121912442 | Missense |
| NM_000454.4:c.20G >T | NP_000445.1:p.Cys7Phe | rs121912448 | Missense |
| NM_000454.4:c.13G >A | NP_000445.1:p.Ala5Thr | rs121912444 | Missense |
| NM_000454.4:c.37G >C | NP_000445.1:p.Gly13Arg | rs121912456 | Missense |
| NM_000454.4:c.49G >A | NP_000445.1:p.Gly17Ser | rs121912453 | Missense |
| NM_000454.4:c.64G >A | NP_000445.1:p.Glu22Lys | rs121912450 | Missense |
| NM_000454.4:c.115C >G | NP_000445.1:p.Leu39Val | rs121912432 | Missense |
| NM_000454.4:c.112G >C | NP_000445.1:p.Gly38Arg | rs121912431 | Missense |
| NM_000454.4:c.112G >A | NP_000445.1:p.Gly38Arg | rs121912431 | Missense |
| NM_000454.4:c.131A >G | NP_000445.1:p.His44Arg | rs121912435 | Missense |
| NM_000454.4:c.125G >A | NP_000445.1:p.Gly42Asp | rs121912434 | Missense |
| NM_000454.4:c.140A >G | NP_000445.1:p.His47Arg | rs121912443 | Missense |
| NM_000454.4:c.124G >A | NP_000445.1:p.Gly42Ser | rs121912433 | Missense |
| NM_000454.4:c.137T >G | NP_000445.1:p.Phe46Cys | rs121912457 | Missense |
| NM_000454.4:c.217G >A | NP_000445.1:p.Gly73Ser | rs121912455 | Missense |
| NM_000454.4:c.242A >G | NP_000445.1:p.His81Arg | rs121912458 | Missense |
| NM_000454.4:c.256G >C | NP_000445.1:p.Gly86Arg | rs121912436 | Missense |
| NM_000454.4:c.253T >G | NP_000445.1:p.Leu85Val | rs121912452 | Missense |
| NM_000454.4:c.281G >C | NP_000445.1:p.Gly94Ala | rs121912438 | Missense |
| NM_000454.4:c.280G >T | NP_000445.1:p.Gly94Cys | rs121912437 | Missense |
| NM_000454.4:c.280G >C | NP_000445.1:p.Gly94Arg | rs121912437 | Missense |
| NM_000454.4:c.289G >A | NP_000445.1:p.Asp97Asn | rs121912459 | Missense |
| NM_000454.4:c.302A >G | NP_000445.1:p.Glu101Gly | rs121912439 | Missense |
| NM_000454.4:c.319C >G | NP_000445.1:p.Leu107Val | rs121912440 | Missense |
| LRG_652t1:c.317C >T | LRG_652p1:p.Ser106Leu | Missense | |
| NM_000454.4:c.313A >T | NP_000445.1:p.Ile105Phe | rs121912445 | Missense |
| NM_000454.4:c.341T >C | NP_000445.1:p.Ile114Thr | rs121912441 | Missense |
| NM_000454.4:c.338T >C | NP_000445.1:p.Ile113Thr | rs74315452 | Missense |
| NM_000454.4:c.358-10T >G | Intron variant | ||
| NM_000454.4:c.358-11A >G | Intron variant | ||
| NM_000454.4:c.380T >A | NP_000445.1:p.Leu127Ter | rs121912454 | Stop gain |
| NM_000454.4:c.404G >A | NP_000445.1:p.Ser135Asn | rs121912451 | Missense |
| NM_000454.4:c.436G >A | NP_000445.1:p.Ala146Thr | rs121912447 | Missense |
| NM_000454.4:c.435G >C | NP_000445.1:p.Leu145Phe | rs1482760341 | Missense |
| NM_000454.4:c.434T >C | NP_000445.1:p.Leu145Ser | rs121912446 | Missense |
| NM_000454.4:c.455T >C | NP_000445.1:p.Ile152Thr | rs121912449 | Missense |
| C9orf72 | |||
| NM_001256054.2:c. − 45 + 163GGGGCC[>24] | rs143561967 | Intron variant | |
| FUS | |||
| NM_004960.3:c.616G >A | NP_004951.1:p.Gly206Ser | rs387906628 | Missense |
| NM_004960.3:c.646C >T | NP_004951.1:p.Arg216Cys | rs267606832 | Missense |
| NM_004960.3:c.1507_1508AG [3] | NP_004951.1:p.Gly504fs | Frameshift | |
| NM_004960.3:c.1483C >T | NP_004951.1:p.Arg495Ter | rs387906627 | Stop gain |
| NM_004960.3:c.1520G >A | NP_004951.1:p.Gly507Asp | rs267606831 | Missense |
| NM_004960.3:c.1570A >T | NP_004951.1:p.Arg524Trp | rs267606833 | Missense |
| NM_004960.3:c.1562G >A | NP_004951.1:p.Arg521His | rs121909671 | Missense |
| NM_004960.3:c.1561C >T | NP_004951.1:p.Arg521Cys | rs121909668 | Missense |
| NM_004960.3:c.1561C >G | NP_004951.1:p.Arg521Gly | rs121909668 | Missense |
| NM_004960.3:c.1553G >A | NP_004951.1:p.Arg518Lys | rs121909669 | Missense |
| NM_004960.3:c.1551C >G | NP_004951.1:p.His517Gln | rs121909667 | Missense |
| TARDBP | |||
| NM_007375.3:c.800A >G | NP_031401.1:p.Asn267Ser | rs80356718 | Missense |
| NM_007375.3:c.869G >C | NP_031401.1:p.Gly290Ala | rs121908395 | Missense |
| NM_007375.3:c.881G >T | NP_031401.1:p.Gly294Val | rs80356721 | Missense |
| NM_007375.3:c.881G >C | NP_031401.1:p.Gly294Ala | rs80356721 | Missense |
| NM_007375.3:c.883G >A | NP_031401.1:p.Gly295Ser | rs80356723 | Missense |
| NM_007375.3:c.892G >A | NP_031401.1:p.Gly298Ser | rs4884357 | Missense |
| NM_007375.3:c.943G >A | NP_031401.1:p.Ala315Thr | rs80356726 | Missense |
| NM_007375.3:c.991C >A | NP_031401.1:p.Gln331Lys | rs80356727 | Missense |
| NM_007375.3:c.1009A >G | NP_031401.1:p.Met337Val | rs80356730 | Missense |
| NM_007375.3:c.1028A >G | NP_031401.1:p.Gln343Arg | rs80356731 | Missense |
| NM_007375.3:c.1042G >T | NP_031401.1:p.Gly348Cys | rs80356733 | Missense |
| NM_007375.3:c.1055A >G | NP_031401.1:p.Asn352Ser | rs80356734 | Missense |
| NM_007375.3:c.1144G >A | NP_031401.1:p.Ala382Thr | rs367543041 | Missense |
| NM_007375.3:c.1153T >G | NP_031401.1:p.Trp385Gly | rs797044595 | Missense |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yun, Y.; Ha, Y. CRISPR/Cas9-Mediated Gene Correction to Understand ALS. Int. J. Mol. Sci. 2020, 21, 3801. https://doi.org/10.3390/ijms21113801
Yun Y, Ha Y. CRISPR/Cas9-Mediated Gene Correction to Understand ALS. International Journal of Molecular Sciences. 2020; 21(11):3801. https://doi.org/10.3390/ijms21113801
Chicago/Turabian StyleYun, Yeomin, and Yoon Ha. 2020. "CRISPR/Cas9-Mediated Gene Correction to Understand ALS" International Journal of Molecular Sciences 21, no. 11: 3801. https://doi.org/10.3390/ijms21113801
APA StyleYun, Y., & Ha, Y. (2020). CRISPR/Cas9-Mediated Gene Correction to Understand ALS. International Journal of Molecular Sciences, 21(11), 3801. https://doi.org/10.3390/ijms21113801