A Tale of Two Proteolytic Machines: Matrix Metalloproteinases and the Ubiquitin–Proteasome System in Pulmonary Fibrosis


Abstract
:1. Introduction
2. Matrix Metalloproteinases
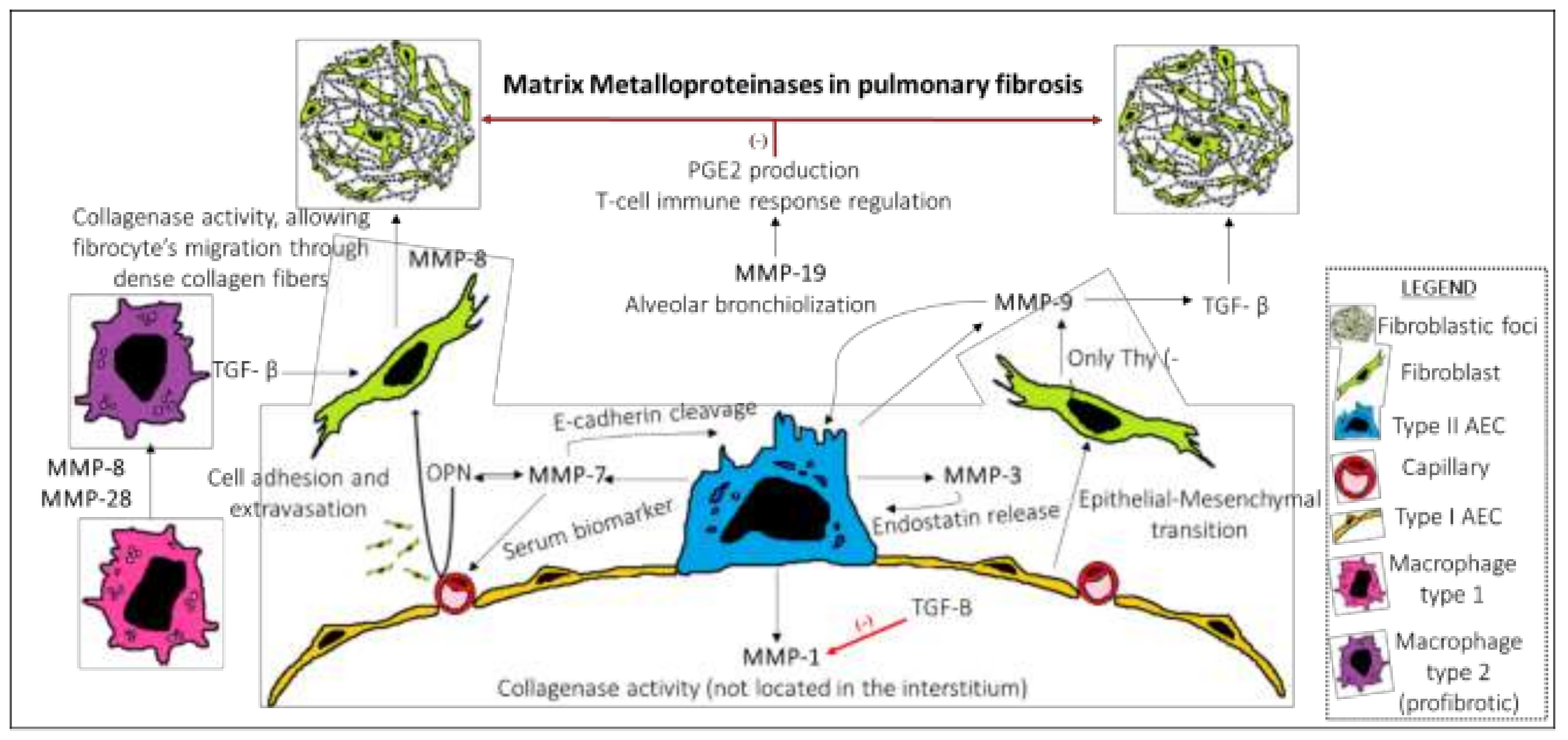
3. MMPs in Pulmonary Fibrosis
4. The Ubiquitin-Proteasome System
5. UPS in Pulmonary Fibrosis
6. The Interplay between the UPS and MMPs: Therapeutic Perspective
7. Concluding Remarks
Funding
Conflicts of Interest
References
- Raghu, G.; Remy-Jardin, M.; Myers, J.; Richeldi, L.; Ryerson, C.; Lederer, D. European respiratory society, Japanese respiratory society, and latin American thoracic society. Diagnosis of idiopathic pulmonary fibrosis. An official ATS. Am. J. Respir. Crit. Care Med. 2018, 198, e44–e68. [Google Scholar] [PubMed]
- Raghu, G.; Rochwerg, B.; Zhang, Y.; Garcia, C.A.C.; Azuma, A.; Behr, J.; Brozek, J.L.; Collard, H.R.; Cunningham, W.; Homma, S.; et al. An Official ATS/ERS/JRS/ALAT Clinical Practice Guideline: Treatment of Idiopathic Pulmonary Fibrosis. An Update of the 2011 Clinical Practice Guideline. Am. J. Respir. Crit. Care Med. 2015, 192, e3–e19. [Google Scholar] [CrossRef]
- Maher, T.; Wells, A.; Laurent, G. Idiopathic pulmonary fibrosis: Multiple causes and multiple mechanisms? Eur. Respir. J. 2007, 30, 835–839. [Google Scholar] [PubMed] [Green Version]
- Vicary, G.W.; Vergne, Y.; Santiago-Cornier, A.; Young, L.R.; Roman, J. Pulmonary Fibrosis in Hermansky-Pudlak Syndrome. Ann. Am. Thorac. Soc. 2016, 13, 1839–1846. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pardo, A.; Cabrera, S.; Maldonado, M.; Selman, M. Role of matrix metalloproteinases in the pathogenesis of idiopathic pulmonary fibrosis. Respir. Res. 2016, 17, 23. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mao, X.; Pan, X.; Cheng, T.; Zhang, X. Therapeutic potential of the proteasome inhibitor Bortezomib on titanium particle-induced inflammation in a murine model. Inflammation 2012, 35, 905–912. [Google Scholar]
- Goffin, L.; Seguin-Estévez, Q.; Alvarez, M.; Reith, W.; Chizzolini, C. Transcriptional regulation of matrix metalloproteinase-1 and collagen 1A2 explains the anti-fibrotic effect exerted by proteasome inhibition in human dermal fibroblasts. Arthritis Res. & Ther. 2010, 12, R73. [Google Scholar]
- Corbel, M.; Caulet-Maugendre, S.; Germain, N.; Molet, S.; Lagente, V.; Boichot, E. Inhibition of bleomycin-induced pulmonary fibrosis in mice by the matrix metalloproteinase inhibitor batimastat. J. Pathol. 2001, 193, 538–545. [Google Scholar] [CrossRef]
- Sela-Passwell, N.; Kikkeri, R.; Dym, O.; Rozenberg, H.; Margalit, R.; Arad-Yellin, R.; Eisenstein, M.; Brenner, O.; Shoham, T.; Danon, T. Antibodies targeting the catalytic zinc complex of activated matrix metalloproteinases show therapeutic potential. Nat. Med. 2012, 18, 143. [Google Scholar] [CrossRef]
- Marshall, D.C.; Lyman, S.K.; McCauley, S.; Kovalenko, M.; Spangler, R.; Liu, C.; Lee, M.; O’Sullivan, C.; Barry-Hamilton, V.; Ghermazien, H. Selective allosteric inhibition of MMP9 is efficacious in preclinical models of ulcerative colitis and colorectal cancer. PLoS ONE 2015, 10, e0127063. [Google Scholar] [CrossRef] [Green Version]
- Baker, T.A.; Bach Iv, H.; Gamelli, R.L.; Love, R.B.; Majetschak, M. Proteasomes in lungs from organ donors and patients with end-stage pulmonary diseases. Physiol. Res. 2014, 63, 311. [Google Scholar] [PubMed]
- Roque, W.; Summer, R.; Romero, F. Fine-tuning the ubiquitin-proteasome system to treat pulmonary fibrosis. Connect. Tissue Res. 2019, 60, 50–61. [Google Scholar] [CrossRef] [PubMed]
- Nandi, D.; Tahiliani, P.; Kumar, A.; Chandu, D. The ubiquitin-proteasome system. J. Biosci. 2006, 31, 137–155. [Google Scholar] [CrossRef] [PubMed]
- Page-McCaw, A.; Ewald, A.J.; Werb, Z. Matrix metalloproteinases and the regulation of tissue remodelling. Nat. Rev. Mol. Cell Biol. 2007, 8, 221–233. [Google Scholar] [CrossRef] [PubMed]
- Grünwald, B.; Schlage, P.; Krüger, A.; Auf Dem Keller, U. MMPs: From Genomics to Degradomics; Sagi, I., Gaffney, J.P., Eds.; Wiley Blackwell: Hoboken, NJ, USA, 2015; p. 181. [Google Scholar]
- Elkins, P.A.; Ho, Y.S.; Smith, W.W.; Janson, C.A.; D’Alessio, K.J.; McQueney, M.S.; Cummings, M.D.; Romanic, A.M. Structure of the C-terminally truncated human ProMMP9, a gelatin-binding matrix metalloproteinase. Acta Crystallogr. Sect. D Biol. Crystallogr. 2002, 58 (Pt 7), 1182–1192. [Google Scholar] [CrossRef] [Green Version]
- Morgunova, E.; Tuuttila, A.; Bergmann, U.; Isupov, M.; Lindqvist, Y.; Schneider, G.; Tryggvason, K. Structure of human pro-matrix metalloproteinase-2: Activation mechanism revealed. Sci. N.Y. 1999, 284, 1667–1670. [Google Scholar] [CrossRef]
- Shiomi, T.; Inoki, I.; Kataoka, F.; Ohtsuka, T.; Hashimoto, G.; Nemori, R.; Okada, Y. Pericellular activation of proMMP-7 (promatrilysin-1) through interaction with CD151. Lab. Investig. J. Tech. Methods Pathol. 2005, 85, 1489–1506. [Google Scholar] [CrossRef] [Green Version]
- Nagase, H.; Visse, R.; Murphy, G. Structure and function of matrix metalloproteinases and TIMPs. Cardiovasc. Res. 2006, 69, 562–573. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Visse, R.; Nagase, H. Matrix metalloproteinases and tissue inhibitors of metalloproteinases: Structure, function, and biochemistry. Circ. Res. 2003, 92, 827–839. [Google Scholar] [CrossRef] [Green Version]
- Selman, M.; Ruiz, V.; Cabrera, S.; Segura, L.; Ramírez, R.; Barrios, R.; Pardo, A. TIMP-1, -2, -3, and -4 in idiopathic pulmonary fibrosis. A prevailing nondegradative lung microenvironment? Am. J. Physiol. -Lung Cell. Mol. Physiol. 2000, 279, L562–L574. [Google Scholar] [CrossRef]
- Madtes, D.K.; Elston, A.L.; Kaback, L.A.; Clark, J.G. Selective Induction of Tissue Inhibitor of Metalloproteinase-1 in Bleomycin-Induced Pulmonary Fibrosis. Am. J. Respir. Cell Mol. Biol. 2001, 24, 599–607. [Google Scholar] [CrossRef] [PubMed]
- Dancer, R.C.A.; Wood, A.M.; Thickett, D.R. Metalloproteinases in idiopathic pulmonary fibrosis. Eur. Respir. J. 2011, 38, 1461–1467. [Google Scholar] [PubMed] [Green Version]
- Pirici, D.; Pirici, I.; Mogoanta, L.; Margaritescu, O.; Tudorica, V.; Margaritescu, C.; Ion, D.A.; Simionescu, C.; Coconu, M. Matrix metalloproteinase-9 expression in the nuclear compartment of neurons and glial cells in aging and stroke. Neuropathology 2012, 32, 492–504. [Google Scholar] [PubMed]
- Giannandrea, M.; Parks, W.C. Diverse functions of matrix metalloproteinases during fibrosis. Dis. Models & Mech. 2014, 7, 193–203. [Google Scholar]
- Egeblad, M.; Werb, Z. New functions for the matrix metalloproteinases in cancer progression. Nat. Rev. Cancer 2002, 2, 161–174. [Google Scholar] [CrossRef] [PubMed]
- Selman, M.; Pardo, A. Revealing the Pathogenic and Aging-related Mechanisms of the Enigmatic Idiopathic Pulmonary Fibrosis. An Integral Model. Am. J. Respir. Crit. Care Med. 2014, 189, 1161–1172. [Google Scholar] [CrossRef]
- Selman, M.; Pardo, A. Role of Epithelial Cells in Idiopathic Pulmonary Fibrosis. Proc. Am. Thorac. Soc. 2006, 3, 364–372. [Google Scholar]
- Checa, M.; Ruiz, V.; Montaño, M.; Velázquez-Cruz, R.; Selman, M.; Pardo, A. MMP-1 polymorphisms and the risk of idiopathic pulmonary fibrosis. Hum. Genet. 2008, 124, 465–472. [Google Scholar] [CrossRef]
- Herrera, I.; Cisneros, J.; Maldonado, M.; Ramírez, R.; Ortiz-Quintero, B.; Anso, E.; Chandel, N.S.; Selman, M.; Pardo, A. Matrix metalloproteinase (MMP)-1 induces lung alveolar epithelial cell migration and proliferation, protects from apoptosis, and represses mitochondrial oxygen consumption. J. Biol. Chem. 2013, 288, 25964–25975. [Google Scholar] [CrossRef] [Green Version]
- Trask, B.C.; Malone, M.J.; Lum, E.H.; Welgus, H.G.; Crouch, E.C.; Shapiro, S.D. Induction of macrophage matrix metalloproteinase biosynthesis by surfactant protein D. J. Biol. Chem. 2001, 276, 37846–37852. [Google Scholar]
- Morishita, A.; Gerber, A.; Gow, C.H.; Zelonina, T.; Chada, K.; D’Armiento, J. Cell Specific Matrix Metalloproteinase-1 Regulates Lung Metastasis Synergistically with Smoke Exposure. J. Cancer Res. Forecast. 2018, 1, 1014. [Google Scholar] [PubMed]
- Yamashita, C.M.; Dolgonos, L.; Zemans, R.L.; Young, S.K.; Robertson, J.; Briones, N.; Suzuki, T.; Campbell, M.N.; Gauldie, J.; Radisky, D.C. Matrix metalloproteinase 3 is a mediator of pulmonary fibrosis. Am. J. Pathol. 2011, 179, 1733–1745. [Google Scholar] [CrossRef] [PubMed]
- Quy, P.N.; Kuma, A.; Pierre, P.; Mizushima, N. Proteasome-dependent activation of mammalian target of rapamycin complex 1 (mTORC1) is essential for autophagy suppression and muscle remodeling following denervation. J. Biol. Chem. 2013, 288, 1125–1134. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Agnihotri, R.; Crawford, H.C.; Haro, H.; Matrisian, L.M.; Havrda, M.C.; Liaw, L. Osteopontin, a novel substrate for matrix metalloproteinase-3 (stromelysin-1) and matrix metalloproteinase-7 (matrilysin). J. Biol. Chem. 2001, 276, 28261–28267. [Google Scholar] [PubMed] [Green Version]
- Manicone, A.M.; Huizar, I.; McGuire, J.K. Matrilysin (Matrix Metalloproteinase-7) regulates anti-inflammatory and antifibrotic pulmonary dendritic cells that express CD103 (alpha(E)beta(7)-integrin). Am. J. Pathol. 2009, 175, 2319–2331. [Google Scholar] [PubMed] [Green Version]
- Pardo, A.; Gibson, K.; Cisneros, J.; Richards, T.J.; Yang, Y.; Becerril, C.; Yousem, S.; Herrera, I.; Ruiz, V.; Selman, M. Up-regulation and profibrotic role of osteopontin in human idiopathic pulmonary fibrosis. PLoS Med. 2005, 2, e251. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McGuire, J.K.; Li, Q.; Parks, W.C. Matrilysin (matrix metalloproteinase-7) mediates E-cadherin ectodomain shedding in injured lung epithelium. Am. J. Pathol. 2003, 162, 1831–1843. [Google Scholar] [CrossRef] [Green Version]
- Chen, P.; Abacherli, L.E.; Nadler, S.T.; Wang, Y.; Li, Q.; Parks, W.C. MMP7 shedding of syndecan-1 facilitates re-epithelialization by affecting alpha(2)beta(1) integrin activation. PLoS ONE 2009, 4, e6565. [Google Scholar]
- Betsuyaku, T.; Fukuda, Y.; Parks, W.C.; Shipley, J.M.; Senior, R.M. Gelatinase B is required for alveolar bronchiolization after intratracheal bleomycin. Am. J. Pathol. 2000, 157, 525–535. [Google Scholar]
- Ramírez, G.; Hagood, J.S.; Sanders, Y.; Ramírez, R.; Becerril, C.; Segura, L.; Barrera, L.; Selman, M.; Pardo, A. Absence of Thy-1 results in TGF-β induced MMP-9 expression and confers a profibrotic phenotype to human lung fibroblasts. Lab. Investig. 2011, 91, 1206–1218. [Google Scholar] [CrossRef] [Green Version]
- Gharib, S.A.; Johnston, L.K.; Huizar, I.; Birkland, T.P.; Hanson, J.; Wang, Y.; Parks, W.C.; Manicone, A.M. MMP28 promotes macrophage polarization toward M2 cells and augments pulmonary fibrosis. J. Leukoc. Biol. 2014, 95, 9–18. [Google Scholar] [CrossRef] [Green Version]
- García-de-Alba, C.; Becerril, C.; Ruiz, V.; González, Y.; Reyes, S.; García-Alvarez, J.; Selman, M.; Pardo, A. Expression of matrix metalloproteases by fibrocytes: Possible role in migration and homing. Am. J. Respir. Crit. Care Med. 2010, 182, 1144–1152. [Google Scholar]
- Hodges, R.J.; Jenkins, R.G.; Wheeler-Jones, C.P.; Copeman, D.M.; Bottoms, S.E.; Bellingan, G.J.; Nanthakumar, C.B.; Laurent, G.J.; Hart, S.L.; Foster, M.L. Severity of lung injury in cyclooxygenase-2-deficient mice is dependent on reduced prostaglandin E2 production. Am. J. Pathol. 2004, 165, 1663–1676. [Google Scholar]
- Beck, I.M.; Rückert, R.; Brandt, K.; Mueller, M.S.; Sadowski, T.; Brauer, R.; Schirmacher, P.; Mentlein, R.; Sedlacek, R. MMP19 is essential for T cell development and T cell-mediated cutaneous immune responses. PLoS ONE 2008, 3, e2343. [Google Scholar] [CrossRef]
- Nkyimbeng, T.; Ruppert, C.; Shiomi, T.; Dahal, B.; Lang, G.; Seeger, W.; Okada, Y.; D’Armiento, J.; Günther, A. Pivotal role of matrix metalloproteinase 13 in extracellular matrix turnover in idiopathic pulmonary fibrosis. PLoS ONE 2013, 8, e73279. [Google Scholar] [CrossRef]
- Flechsig, P.; Hartenstein, B.; Teurich, S.; Dadrich, M.; Hauser, K.; Abdollahi, A.; Gröne, H.-J.; Angel, P.; Huber, P.E. Loss of matrix metalloproteinase-13 attenuates murine radiation-induced pulmonary fibrosis. Int. J. Radiat. Oncol. Biol. Phys. 2010, 77, 582–590. [Google Scholar]
- Fukuda, Y.; Ishizaki, M.; Kudoh, S.; Kitaichi, M.; Yamanaka, N. Localization of matrix metalloproteinases-1, -2, and -9 and tissue inhibitor of metalloproteinase-2 in interstitial lung diseases. Lab. Investig. J. Tech. Methods Pathol. 1998, 78, 687–698. [Google Scholar]
- Lemjabbar, H.; Gosset, P.; Lechapt-Zalcman, E.; Franco-Montoya, M.L.; Wallaert, B.; Harf, A.; Lafuma, C. Overexpression of alveolar macrophage gelatinase B (MMP-9) in patients with idiopathic pulmonary fibrosis: Effects of steroid and immunosuppressive treatment. Am. J. Respir. Cell Mol. Biol. 1999, 20, 903–913. [Google Scholar] [CrossRef] [PubMed]
- Pérez-Ramos, J.; de Lourdes Segura-Valdez, M.; Vanda, B.; Selman, M.; Pardo, A. Matrix metalloproteinases 2, 9, and 13, and tissue inhibitors of metalloproteinases 1 and 2 in experimental lung silicosis. Am. J. Respir. Crit. Care Med. 1999, 160, 1274–1282. [Google Scholar] [CrossRef]
- Yu, G.; Kovkarova-Naumovski, E.; Jara, P.; Parwani, A.; Kass, D.; Ruiz, V.; Lopez-Otín, C.; Rosas, I.O.; Gibson, K.F.; Cabrera, S. Matrix metalloproteinase-19 is a key regulator of lung fibrosis in mice and humans. Am. J. Respir. Crit. Care Med. 2012, 186, 752–762. [Google Scholar] [CrossRef] [Green Version]
- Summer, R.; Krishna, R.; Schriner, D.; Cuevas-Mora, K.; Sales, D.; Para, R.; Roman, J.; Nieweld, C.; Gochuico, B.R.; Romero, F. Matrix metalloproteinase activity in the lung is increased in Hermansky-Pudlak syndrome. Orphanet J. Rare Dis. 2019, 14, 162. [Google Scholar] [CrossRef] [Green Version]
- Hershko, A.; Ciechanover, A. The ubiquitin system. Annu. Rev. Biochem. 1998, 67, 425–479. [Google Scholar] [CrossRef] [PubMed]
- Budenholzer, L.; Cheng, C.L.; Li, Y.; Hochstrasser, M. Proteasome structure and assembly. J. Mol. Biol. 2017, 429, 3500–3524. [Google Scholar] [CrossRef]
- Pickart, C.M. Mechanisms underlying ubiquitination. Annu. Rev. Biochem. 2001, 70, 503–533. [Google Scholar] [CrossRef]
- Nam, T.; Han, J.H.; Devkota, S.; Lee, H.-W. Emerging paradigm of crosstalk between autophagy and the ubiquitin-proteasome system. Mol. Cells 2017, 40, 897. [Google Scholar]
- Radhakrishnan, S.K.; Lee, C.S.; Young, P.; Beskow, A.; Chan, J.Y.; Deshaies, R.J. Transcription factor Nrf1 mediates the proteasome recovery pathway after proteasome inhibition in mammalian cells. Mol. Cell 2010, 38, 17–28. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yun, Y.S.; Kim, K.H.; Tschida, B.; Sachs, Z.; Noble-Orcutt, K.E.; Moriarity, B.S.; Ai, T.; Ding, R.; Williams, J.; Chen, L. mTORC1 coordinates protein synthesis and immunoproteasome formation via PRAS40 to prevent accumulation of protein stress. Mol. Cell 2016, 61, 625–639. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, S.-J.; Cheresh, P.; Jablonski, R.P.; Williams, D.B.; Kamp, D.W. The role of mitochondrial DNA in mediating alveolar epithelial cell apoptosis and pulmonary fibrosis. Int. J. Mol. Sci. 2015, 16, 21486–21519. [Google Scholar] [CrossRef]
- López-Otín, C.; Blasco, M.A.; Partridge, L.; Serrano, M.; Kroemer, G. The hallmarks of aging. Cell 2013, 153, 1194–1217. [Google Scholar] [CrossRef] [Green Version]
- Powers, E.T.; Morimoto, R.I.; Dillin, A.; Kelly, J.W.; Balch, W.E. Biological and chemical approaches to diseases of proteostasis deficiency. Annu. Rev. Biochem. 2009, 78, 959–991. [Google Scholar] [CrossRef] [Green Version]
- Taylor, R.C.; Dillin, A. Aging as an event of proteostasis collapse. Cold Spring Harbor Perspect. Biol. 2011, 3, a004440. [Google Scholar] [CrossRef] [Green Version]
- Morimoto, R.I. Proteotoxic stress and inducible chaperone networks in neurodegenerative disease and aging. Genes & Dev. 2008, 22, 1427–1438. [Google Scholar]
- Vabulas, R.M.; Raychaudhuri, S.; Hayer-Hartl, M.; Hartl, F.U. Protein folding in the cytoplasm and the heat shock response. Cold Spring Harbor Perspect. Biol. 2010, 2, a004390. [Google Scholar] [CrossRef] [PubMed]
- Kopp, Y.; Lang, W.-H.; Schuster, T.B.; Martinez-Limon, A.; Hofbauer, H.F.; Ernst, R.; Calloni, G.; Vabulas, R.M. CHIP as a membrane-shuttling proteostasis sensor. Elife 2017, 6, e29388. [Google Scholar] [PubMed] [Green Version]
- Qian, S.-B.; McDonough, H.; Boellmann, F.; Cyr, D.M.; Patterson, C. CHIP-mediated stress recovery by sequential ubiquitination of substrates and Hsp70. Nature 2006, 440, 551–555. [Google Scholar] [PubMed] [Green Version]
- Min, J.-N.; Whaley, R.A.; Sharpless, N.E.; Lockyer, P.; Portbury, A.L.; Patterson, C. CHIP deficiency decreases longevity, with accelerated aging phenotypes accompanied by altered protein quality control. Mol. Cell. Biol. 2008, 28, 4018–4025. [Google Scholar] [PubMed] [Green Version]
- Xin, H.; Xu, X.; Li, L.; Ning, H.; Rong, Y.; Shang, Y.; Wang, Y.; Fu, X.-Y.; Chang, Z. CHIP controls the sensitivity of transforming growth factor-β signaling by modulating the basal level of Smad3 through ubiquitin-mediated degradation. J. Biol. Chem. 2005, 280, 20842–20850. [Google Scholar] [CrossRef] [Green Version]
- Geng, J.; Huang, X.; Li, Y.; Xu, X.; Li, S.; Jiang, D.; Liang, J.; Jiang, D.; Wang, C.; Dai, H. Down-regulation of USP13 mediates phenotype transformation of fibroblasts in idiopathic pulmonary fibrosis. Respir. Res. 2015, 16, 124. [Google Scholar]
- Nho, R.S.; Hergert, P.; Kahm, J.; Jessurun, J.; Henke, C. Pathological alteration of FoxO3a activity promotes idiopathic pulmonary fibrosis fibroblast proliferation on type i collagen matrix. Am. J. Pathol. 2011, 179, 2420–2430. [Google Scholar]
- Kral, J.B.; Kuttke, M.; Schrottmaier, W.C.; Birnecker, B.; Warszawska, J.; Wernig, C.; Paar, H.; Salzmann, M.; Sahin, E.; Brunner, J.S. Erratum: Sustained PI3K Activation exacerbates BLM-induced Lung Fibrosis via activation of pro-inflammatory and pro-fibrotic pathways. Sci. Rep. 2016, 6, 23034. [Google Scholar]
- Imamura, T.; Oshima, Y.; Hikita, A. Regulation of TGF-β family signalling by ubiquitination and deubiquitination. J. Biochem. 2013, 154, 481–489. [Google Scholar] [PubMed] [Green Version]
- Kavsak, P.; Rasmussen, R.K.; Causing, C.G.; Bonni, S.; Zhu, H.; Thomsen, G.H.; Wrana, J.L. Smad7 binds to Smurf2 to form an E3 ubiquitin ligase that targets the TGFβ receptor for degradation. Mol. Cell 2000, 6, 1365–1375. [Google Scholar] [PubMed]
- Semren, N.; Welk, V.; Korfei, M.; Keller, I.E.; Fernandez, I.E.; Adler, H.; Günther, A.; Eickelberg, O.; Meiners, S. Regulation of 26S proteasome activity in pulmonary fibrosis. Am. J. Respir. Crit. Care Med. 2015, 192, 1089–1101. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mutlu, G.M.; Budinger, G.S.; Wu, M.; Lam, A.P.; Zirk, A.; Rivera, S.; Urich, D.; Chiarella, S.E.; Go, L.H.; Ghosh, A.K. Proteasomal inhibition after injury prevents fibrosis by modulating TGF-β1 signalling. Thorax 2012, 67, 139–146. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roque, W.; Cuevas-Mora, K.; Romero, F. Mitochondrial Quality Control in Age-Related Pulmonary Fibrosis. Int. J. Mol. Sci. 2020, 21, 643. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bueno, M.; Lai, Y.-C.; Romero, Y.; Brands, J.; Croix, C.M.S.; Kamga, C.; Corey, C.; Herazo-Maya, J.D.; Sembrat, J.; Lee, J.S. PINK1 deficiency impairs mitochondrial homeostasis and promotes lung fibrosis. J. Clin. Investig. 2015, 125, 521–538. [Google Scholar] [PubMed] [Green Version]
- Zhang, Y.; Manning, B.D. mTORC1 signaling activates NRF1 to increase cellular proteasome levels. Cell Cycle 2015, 14, 2011–2017. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cherok, E.; Xu, S.; Li, S.; Das, S.; Meltzer, W.A.; Zalzman, M.; Wang, C.; Karbowski, M. Novel regulatory roles of Mff and Drp1 in E3 ubiquitin ligase MARCH5–dependent degradation of MiD49 and Mcl1 and control of mitochondrial dynamics. Mol. Biol. Cell 2017, 28, 396–410. [Google Scholar]
- Escobar-Henriques, M.; Westermann, B.; Langer, T. Regulation of mitochondrial fusion by the F-box protein Mdm30 involves proteasome-independent turnover of Fzo1. J. Cell Biol. 2006, 173, 645–650. [Google Scholar] [CrossRef]
- Tian, Y.; Li, H.; Qiu, T.; Dai, J.; Zhang, Y.; Chen, J.; Cai, H. Loss of PTEN induces lung fibrosis via alveolar epithelial cell senescence depending on NF-κB activation. Aging Cell 2019, 18, e12858. [Google Scholar]
- Wang, H.X.; Wang, H.M.; Lin, H.Y.; Yang, Q.; Zhang, H.; Tsang, B.K.; Zhu, C. Proteasome subunit LMP2 is required for matrix metalloproteinase-2 and-9 expression and activities in human invasive extravillous trophoblast cell line. J. Cell. Physiol. 2006, 206, 616–623. [Google Scholar]
- De Carvalho, J.E.R.; Verwoert, M.T.; Vogels, I.M.; Reits, E.A.; Van Noorden, C.J.; Klaassen, I.; Schlingemann, R.O. Involvement of the ubiquitin-proteasome system in the expression of extracellular matrix genes in retinal pigment epithelial cells. Biochem. Biophys. Rep. 2018, 13, 83–92. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.M.; Zhang, X.; Qian, D.; Lin, H.Y.; Li, Q.L.; Liu, D.L.; Liu, G.Y.; Yu, X.D.; Zhu, C. Effect of ubiquitin-proteasome pathway on mouse blastocyst implantation and expression of matrix metalloproteinases-2 and-9. Biol. Reprod. 2004, 70, 481–487. [Google Scholar] [PubMed]
- Meiners, S.; Hocher, B.; Weller, A.; Laule, M.; Stangl, V.; Guenther, C.; Godes, M.; Mrozikiewicz, A.; Baumann, G.; Stangl, K. Downregulation of matrix metalloproteinases and collagens and suppression of cardiac fibrosis by inhibition of the proteasome. Hypertension 2004, 44, 471–477. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ortiz-Lazareno, P.C.; Hernandez-Flores, G.; Dominguez-Rodriguez, J.R.; Lerma-Diaz, J.M.; Jave-Suarez, L.F.; Aguilar-Lemarroy, A.; Gomez-Contreras, P.C.; Scott-Algara, D.; Bravo-Cuellar, A. MG132 proteasome inhibitor modulates proinflammatory cytokines production and expression of their receptors in U937 cells: Involvement of nuclear factor-κB and activator protein-1. Immunology 2008, 124, 534–541. [Google Scholar]
- Sales, D.; Shaghaghi, H.; Summer, R.S.; Romero, F. The Differential Effects of Hermansky-Pudlak Syndrome 1 and 2 on Lung Fibroblast Behavior. In C59. Genetic and Epigenetic Mechanisms in Pulmonary Fibrosis; American Thoracic Society: New York, NY, USA, 2019; p. 5266. [Google Scholar]
- Hara, K.; Suzuyama, Y. Future of antibiotic therapy in various medical fields. 1. Internal medicine. b. Respiratory tract infections. Nihon Rinsho. Jpn. J. Clin. Med. 1984, 42, A566. [Google Scholar]
- De Langhe, S.P.; Reynolds, S.D. Wnt signaling in lung organogenesis. Organogenesis 2008, 4, 100–108. [Google Scholar]
- Dominguez-Brauer, C.; Khatun, R.; Elia, A.J.; Thu, K.L.; Ramachandran, P.; Baniasadi, S.P.; Hao, Z.; Jones, L.D.; Haight, J.; Sheng, Y. E3 ubiquitin ligase Mule targets β-catenin under conditions of hyperactive Wnt signaling. Proc. Natl. Acad. Sci. USA 2017, 114, E1148–E1157. [Google Scholar] [CrossRef] [Green Version]
- Königshoff, M.; Balsara, N.; Pfaff, E.-M.; Kramer, M.; Chrobak, I.; Seeger, W.; Eickelberg, O. Functional Wnt signaling is increased in idiopathic pulmonary fibrosis. PLoS ONE 2008, 3, e2142. [Google Scholar] [CrossRef] [Green Version]
- Kim, J.Y.; Choeng, H.C.; Ahn, C.; Cho, S.-H. Early and late changes of MMP-2 and MMP-9 in bleomycin-induced pulmonary fibrosis. Yonsei Med J. 2009, 50, 68–77. [Google Scholar] [CrossRef] [Green Version]
- Kim, T.H.; Kim, S.-H.; Seo, J.-Y.; Chung, H.; Kwak, H.J.; Lee, S.-K.; Yoon, H.J.; Shin, D.H.; Park, S.S.; Sohn, J.W. Blockade of the Wnt/β-catenin pathway attenuates bleomycin-induced pulmonary fibrosis. Tohoku J. Exp. Med. 2011, 223, 45–54. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, C.; Zhu, H.; Sun, Z.; Xiang, Z.; Ge, Y.; Ni, C.; Luo, Z.; Qian, W.; Han, X. Inhibition of Wnt/β-catenin signaling promotes epithelial differentiation of mesenchymal stem cells and repairs bleomycin-induced lung injury. Am. J. Physiol. Cell Physiol. 2014, 307, C234–C244. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, V.S.; Ng, S.S.; Boersema, P.J.; Low, T.Y.; Karthaus, W.R.; Gerlach, J.P.; Mohammed, S.; Heck, A.J.; Maurice, M.M.; Mahmoudi, T. Wnt signaling through inhibition of β-catenin degradation in an intact Axin1 complex. Cell 2012, 149, 1245–1256. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koizumi, S.; Hamazaki, J.; Murata, S. Transcriptional regulation of the 26S proteasome by Nrf1. Proc. Jpn. Acad. Ser. B 2018, 94, 325–336. [Google Scholar] [CrossRef] [Green Version]
- Chen, J.; Wang, M.; Xiang, Y.; Ru, X.; Ren, Y.; Liu, X.; Qiu, L.; Zhang, Y. Nrf1 Is Endowed with a Dominant Tumor-Repressing Effect onto the Wnt/β-Catenin-Dependent and Wnt/β-Catenin-Independent Signaling Networks in the Human Liver Cancer. Oxidative Med. Cell. Longev. 2020, 2020, 5138539. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
| Pro/Anti-Fibrotic Action | Studies in Transgenic Animal Models | Cellular Source | Mechanism of Action | IPF Biomarker | Outcome | |
|---|---|---|---|---|---|---|
| MMP-3 | Pro | Yes. Bleomycin | AEC, fibroblasts, alveolar macrophages | EMT [33] Wnt/B-catenin pathway activation [33] Endostatin release | No | Aberrant cellular repair. Fibrocytes extravasation. CD103+ dendritic cells.Myofibroblast proliferation. Re-epithelization by alpha(2) beta(1) integrin activation [36]. |
| MMP-7 | Pro | Yes. Bleomycin | AEC | Osteopontin production [37] E-cadherin cleavage [38] Syndecan-1 shedding [39] | Yes | |
| MMP-9 | Pro | Yes. Bleomycin | AEC, fibroblasts | Abnormal alveolar broncholization. [40] Induced by TGF-B in Thy (-) fibroblasts [41] | No | |
| MMP-28 | Pro | Yes. Bleomycin | Macrophages | Switch M1-M2 phenotype and TGF-B production [42] | No | Fibroblast stimulation by M2 macrophages. |
| MMP-8 | Pro | Yes. Bleomycin | Fibrocytes, macrophages, AEC and several other immune cells | Collagenase activity [43] | No | Fibrocyte migration through dense collagen fibers and posterior maturation to fibroblast |
| MMP-19 | Anti | Yes. Bleomycin | AEC in spared areas in IPF lung areas. | COX-2 stimulation and PGE32 production exert antifibrotic effects [44] Regulation of adaptive immune response [45] | No | Spared lung tissue around fibrotic zones. |
| MMP-1 | Anti | No | AEC | Collagenase Inhibits apoptosis [30] Promotes normal re-epithelization of wounds | No | Inhibited by TGF-B and osteopontin in IPF lungs [37]. |
| MMP-13 | Pro and Anti | Yes. Bleomycin and Radiation | Fibroblasts | Increased lung inflammation with Bleomycin [46] Decreased inflammation with radiation [47] | No | Not clear role in IPF lungs. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Roque, W.; Boni, A.; Martinez-Manzano, J.; Romero, F. A Tale of Two Proteolytic Machines: Matrix Metalloproteinases and the Ubiquitin–Proteasome System in Pulmonary Fibrosis. Int. J. Mol. Sci. 2020, 21, 3878. https://doi.org/10.3390/ijms21113878
Roque W, Boni A, Martinez-Manzano J, Romero F. A Tale of Two Proteolytic Machines: Matrix Metalloproteinases and the Ubiquitin–Proteasome System in Pulmonary Fibrosis. International Journal of Molecular Sciences. 2020; 21(11):3878. https://doi.org/10.3390/ijms21113878
Chicago/Turabian StyleRoque, Willy, Alexandra Boni, Jose Martinez-Manzano, and Freddy Romero. 2020. "A Tale of Two Proteolytic Machines: Matrix Metalloproteinases and the Ubiquitin–Proteasome System in Pulmonary Fibrosis" International Journal of Molecular Sciences 21, no. 11: 3878. https://doi.org/10.3390/ijms21113878
APA StyleRoque, W., Boni, A., Martinez-Manzano, J., & Romero, F. (2020). A Tale of Two Proteolytic Machines: Matrix Metalloproteinases and the Ubiquitin–Proteasome System in Pulmonary Fibrosis. International Journal of Molecular Sciences, 21(11), 3878. https://doi.org/10.3390/ijms21113878