N2A Titin: Signaling Hub and Mechanical Switch in Skeletal Muscle



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
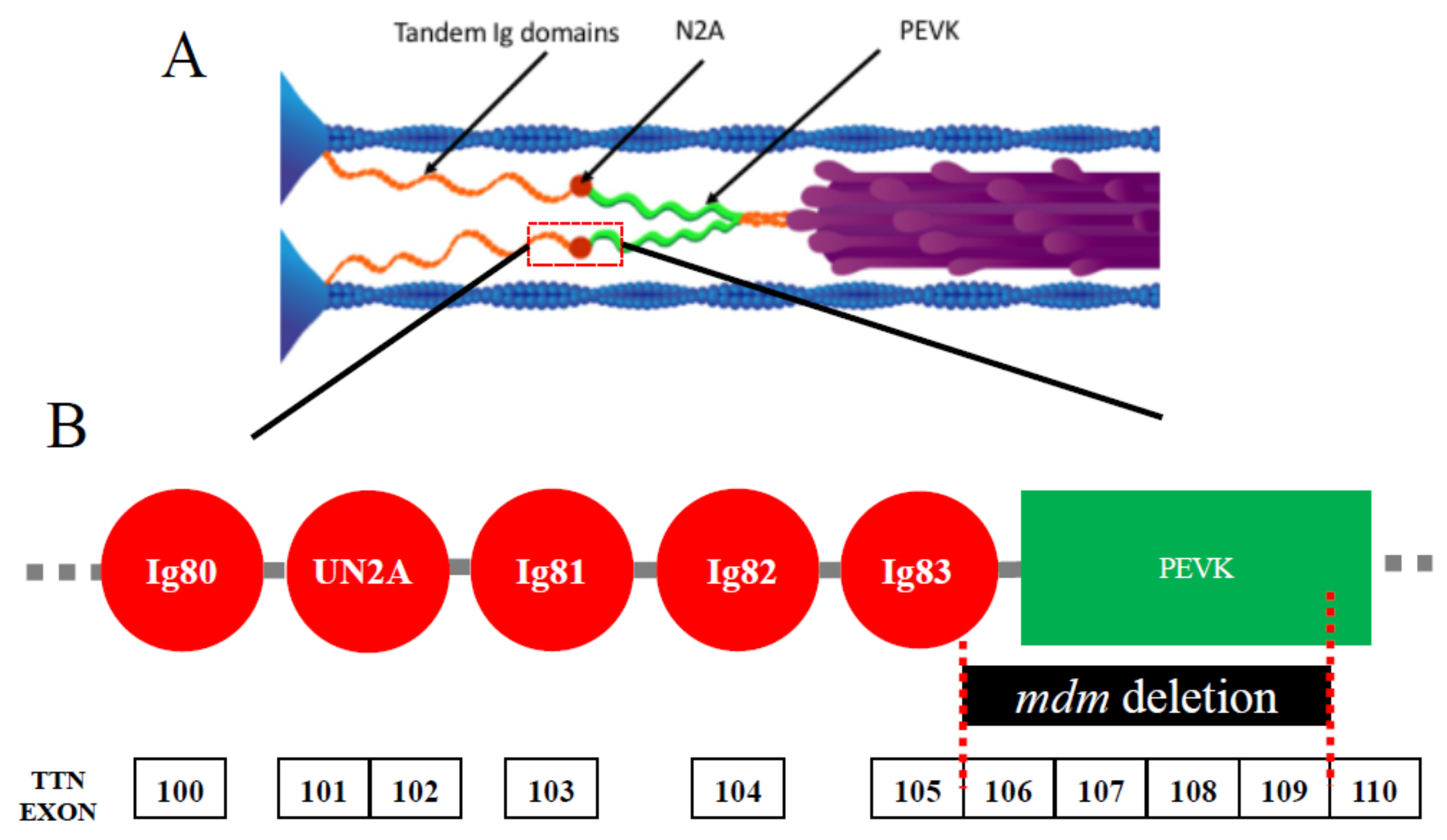
:1. Introduction: A Small Change in A Giant Protein Leads to Severe Titinopathy in mdm
2. Altered Gene Expression and Hypertrophic Signaling in mdm
2.1. Calpain-3/p94 Deficiency in mdm
2.2. MARP-Family Proteins
2.3. Mechano-Sensing in N2A Titin: Interactions between N2A Titin, MARPs, and CAPN3
3. N2A Titin: A Mechanical Switch in Skeletal Muscle
3.1. Titin: A Molecular Spring in the Resting Muscle
3.2. Titin: A Tunable Spring in Active Muscle
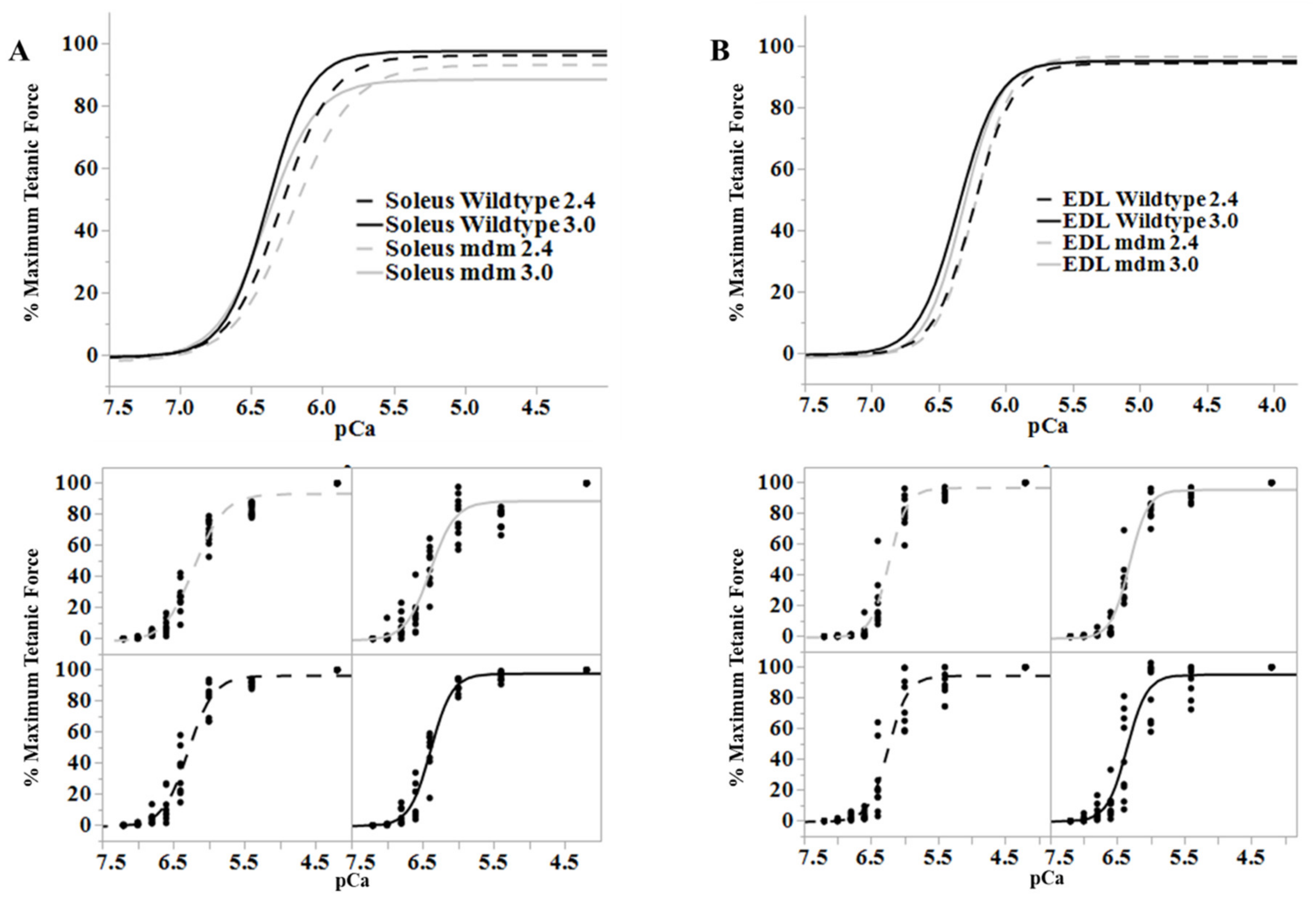
3.3. Altered Mechanics of mdm Muscles
3.3.1. Muscle Equilibrium Length Decreases upon Activation, but Not in mdm
3.3.2. Titin Force and Stiffness Increase upon Activation, but Not in mdm
3.3.3. Muscle Force Increases with Active Stretch and Decreases with Active Shortening, but Not in mdm
3.4. Conclusion: Mechano-Signaling Functions of N2A Titin and Mechanisms of Titinopathy
3.4.1. Effects of mdm on Hypertrophic Signaling
3.4.2. Effects of mdm on Muscle Mechanics
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| ANKRD | Ankyrin Repeat Domain |
| CAPN3 | Calcium activated neutral proteinase 3 |
| CARP | Cardiac ankyrin repeat protein |
| cDNA | Complementary DNA |
| EDL | Extensor digitorum longus |
| Ig | Immunoglobulin |
| KO | Knockout |
| L0 | Muscle optimal length |
| MARP | Muscle ankyrin-repeat protein |
| mdm | Muscular dystrophy with myositis |
| mdx | X chromosome-linked muscular dystrophy in mouse. |
| MLP | Muscle Lim protein |
| PEVK | Pro-Glu-Val-Lys |
| Rbm20 | RNA binding motif protein-20 |
| SOL | Soleus |
| Ttn | Titin gene |
| UDD | Unilateral diaphragm denervation |
References
- Hackman, P.; Vihola, A.; Haravuori, H.; Marchand, S.; Sarparanta, J.; De Seze, J.; Labeit, S.; Witt, C.; Peltonen, L.; Richard, I.; et al. Tibial muscular dystrophy is a titinopathy caused by mutations in TTN, the gene encoding the giant skeletal-muscle protein titin. Am. J. Hum. Genet. 2002, 71, 492–500. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bang, M.L.; Centner, T.; Fornoff, F.; Geach, A.J.; Gotthardt, M.; McNabb, M.; Witt, C.C.; Labeit, D.; Gregorio, C.C.; Granzier, H.; et al. The complete gene sequence of titin, expression of an unusual approximately 700-kDa titin isoform, and its interaction with obscurin identify a novel Z-line to I-band linking system. Circ. Res. 2001, 89, 1065–1072. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, K.; Ramirez-Mitchell, R.; Palter, D. Titin is an extraordinarily long, flexible, and slender myofibrillar protein. Sci. Proc. Natl. Acad. Sci. USA 1984, 81, 3685–3689. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gregorio, C.C.; Granzier, H.; Sorimachi, H.; Labeit, S. Muscle assembly: A titanic achievement? Curr. Opin. Cell Biol. 1999, 11, 18–25. [Google Scholar] [CrossRef]
- Linke, W.A.; Ivemeyer, M.; Mundel, P.; Stockmeier, M.R.; Kolmerer, B. Nature of PEVK-titin elasticity in skeletal muscle. Sci. Proc. Natl. Acad. Sci. USA 1998, 95, 8052–8057. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Horowits, R.; Podolsky, R.J. The positional stability of thick filaments in activated skeletal muscle depends on sarcomere length: Evidence for the role of titin filaments. J. Cell Biol. 1987, 105, 2217–2223. [Google Scholar] [CrossRef] [Green Version]
- Gautel, M.; Djinovic-Carugo, K. The sarcomeric cytoskeleton: From molecules to motion. J. Exp. Biol. 2016, 219, 135–145. [Google Scholar] [CrossRef] [Green Version]
- Gerull, B. The Rapidly Evolving Role of Titin in Cardiac Physiology and Cardiomyopathy. Can. J. Cardiol. 2015, 31, 1351–1359. [Google Scholar] [CrossRef]
- Herman, D.S.; Lam, L.; Taylor, M.R.; Wang, L.; Teekakirikul, P.; Christodoulou, D.; Conner, L.; DePalma, S.R.; McDonough, B.; Sparks, E.; et al. Truncations of titin causing dilated cardiomyopathy. N. Engl. J. Med. 2012, 366, 619–628. [Google Scholar] [CrossRef] [Green Version]
- Guo, W.; Bharmal, S.J.; Esbona, K.; Greaser, M.L. Titin diversity—Alternative splicing gone wild. J. Biomed. Biotechnol. 2010, 2010, 753675. [Google Scholar] [CrossRef] [Green Version]
- Savarese, M.; Sarparanta, J.; Vihola, A.; Udd, B.; Hackman, P. Increasing Role of Titin Mutations in Neuromuscular Disorders. J. Neuromuscul. Dis. 2016, 3, 293–308. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chauveau, C.; Rowell, J.; Ferreiro, A. A rising titan: TTN review and mutation update. Hum. Mutat. 2014, 35, 1046–1059. [Google Scholar] [CrossRef] [PubMed]
- Koser, F.; Loescher, C.; Linke, W.A. Posttranslational modifications of titin from cardiac muscle: How, where, and what for? FEBS J. 2019, 286, 2240–2260. [Google Scholar] [CrossRef]
- Kellermayer, D.; Smith, J.E.; Granzier, H. Titin mutations and muscle disease. Pflugers Arch. 2019, 471, 673–682. [Google Scholar] [CrossRef] [PubMed]
- Garvey, S.M.; Rajan, C.; Lerner, A.P.; Frankel, W.N.; Cox, G.A. The muscular dystrophy with myositis (mdm) mouse mutation disrupts a skeletal muscle-specific domain of titin. Genomics 2002, 79, 146–149. [Google Scholar] [CrossRef] [PubMed]
- Lane, P.W. Muscular dystrophy with myositis (mdm). Mouse News Lett. 1985, 73, 18. [Google Scholar]
- Gerull, B.; Gramlich, M.; Atherton, J.; McNabb, M.; Trombitas, K.; Sasse-Klaassen, S.; Seidman, J.G.; Seidman, C.; Granzier, H.; Labeit, S.; et al. Mutations of TTN, encoding the giant muscle filament titin, cause familial dilated cardiomyopathy. Nat. Genet. 2002, 30, 201–204. [Google Scholar] [CrossRef]
- Labeit, S.; Kolmerer, B. Titins: Giant proteins in charge of muscle ultrastructure and elasticity. Science 1995, 270, 293–296. [Google Scholar] [CrossRef] [PubMed]
- Heimann, P.; Menke, A.; Rothkegel, B.; Jockusch, H. Overshooting production of satellite cells in murine skeletal muscle affected by the mutation "muscular dystrophy with myositis" (mdm, Chr 2). Cell Tissue Res. 1996, 283, 435–441. [Google Scholar] [CrossRef] [PubMed]
- Brynnel, A.; Hernandez, Y.; Kiss, B.; Lindqvist, J.; Adler, M.; Kolb, J.; van der Pijl, R.; Gohlke, J.; Strom, J.; Smith, J.; et al. Downsizing the molecular spring of the giant protein titin reveals that skeletal muscle titin determines passive stiffness and drives longitudinal hypertrophy. eLife 2018, 7. [Google Scholar] [CrossRef]
- Nishikawa, K. Eccentric contraction: Unraveling mechanisms of force enhancement and energy conservation. J. Exp. Biol. 2016, 219, 189–196. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Muller-Seitz, M.; Kaupmann, K.; Labeit, S.; Jockusch, H. Chromosomal localization of the mouse titin gene and its relation to "muscular dystrophy with myositis" and nebulin genes on chromosome 2. Genomics 1993, 18, 559–561. [Google Scholar] [CrossRef]
- Nishikawa, K.C.; Monroy, J.A.; Tahir, U. Muscle Function from Organisms to Molecules. Integr. Comp. Biol. 2018, 58, 194–206. [Google Scholar] [CrossRef]
- Witt, C.C.; Ono, Y.; Puschmann, E.; McNabb, M.; Wu, Y.; Gotthardt, M.; Witt, S.H.; Haak, M.; Labeit, D.; Gregorio, C.C.; et al. Induction and myofibrillar targeting of CARP, and suppression of the Nkx2.5 pathway in the MDM mouse with impaired titin-based signaling. J. Mol. Biol. 2004, 336, 145–154. [Google Scholar] [CrossRef] [PubMed]
- Powers, K.; Nishikawa, K.; Joumaa, V.; Herzog, W. Decreased force enhancement in skeletal muscle sarcomeRes. with a deletion in titin. J. Exp. Biol. 2016. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hessel, A.L.; Joumaa, V.; Eck, S.; Herzog, W.; Nishikawa, K.C. Optimal length, calcium sensitivity and twitch characteristics of skeletal muscles from mdm mice with a deletion in N2A titin. J. Exp. Biol. 2019, 222. [Google Scholar] [CrossRef] [Green Version]
- Powers, K.; Joumaa, V.; Jinha, A.; Moo, E.K.; Smith, I.C.; Nishikawa, K.; Herzog, W. Titin force enhancement following active stretch of skinned skeletal muscle fibres. J. Exp. Biol. 2017, 220, 3110–3118. [Google Scholar] [CrossRef] [Green Version]
- Hessel, A.L.; Nishikawa, K.C. Effects of a titin mutation on negative work during stretch-shortening cycles in skeletal muscles. J. Exp. Biol. 2017, 220, 4177–4185. [Google Scholar] [CrossRef] [Green Version]
- Huebsch, K.A.; Kudryashova, E.; Wooley, C.M.; Sher, R.B.; Seburn, K.L.; Spencer, M.J.; Cox, G.A. Mdm muscular dystrophy: Interactions with calpain 3 and a novel functional role for titin’s N2A domain. Hum. Mol. Genet. 2005, 14, 2801–2811. [Google Scholar] [CrossRef]
- Pace, C.M.; Mortimer, S.; Monroy, J.A.; Nishikawa, K.C. The effects of a skeletal muscle titin mutation on walking in mice. J. Comp. Physiol. A Neuroethol. Sens. Neural Behav. Physiol. 2017, 203, 67–76. [Google Scholar] [CrossRef]
- Boulanger Piette, A.; Hamoudi, D.; Marcadet, L.; Morin, F.; Argaw, A.; Ward, L.; Frenette, J. Targeting the Muscle-Bone Unit: Filling Two Needs with One Deed in the Treatment of Duchenne Muscular Dystrophy. Curr. Osteoporos. Rep. 2018, 16, 541–553. [Google Scholar] [CrossRef]
- Taylor-Burt, K.R.; Monroy, J.; Pace, C.; Lindstedt, S.; Nishikawa, K.C. Shiver me titin! Elucidating titin’s role in shivering thermogenesis. J. Exp. Biol. 2015, 218, 694–702. [Google Scholar] [CrossRef] [Green Version]
- Miyano, C.A.; Orezzoli, S.F.; Buck, C.L.; Nishikawa, K.C. Severe thermoregulatory deficiencies in mice with a deletion in the titin gene TTN. J. Exp. Biol. 2019, 222. [Google Scholar] [CrossRef] [Green Version]
- Freundt, J.K.; Linke, W.A. Titin as a force-generating muscle protein under regulatory control. J. Appl. Physiol. 2019, 126, 1474–1482. [Google Scholar] [CrossRef]
- Kruger, M.; Kotter, S. Titin, a Central Mediator for Hypertrophic Signaling, Exercise-Induced Mechanosignaling and Skeletal Muscle Remodeling. Front. Physiol. 2016, 7, 76. [Google Scholar] [CrossRef] [Green Version]
- Linke, W.A. Titin Gene and Protein Functions in Passive and Active Muscle. Rev. Ann. Rev. Physiol. 2018, 80, 389–411. [Google Scholar] [CrossRef]
- Haravuori, H.; Vihola, A.; Straub, V.; Auranen, M.; Richard, I.; Marchand, S.; Voit, T.; Labeit, S.; Somer, H.; Peltonen, L.; et al. Secondary calpain3 deficiency in 2q-linked muscular dystrophy: Titin is the candidate gene. Neurology 2001, 56, 869–877. [Google Scholar] [CrossRef]
- Ono, Y.; Torii, F.; Ojima, K.; Doi, N.; Yoshioka, K.; Kawabata, Y.; Labeit, D.; Labeit, S.; Suzuki, K.; Abe, K.; et al. Suppressed disassembly of autolyzing p94/CAPN3 by N2A connectin/titin in a genetic reporter system. J. Biol. Chem. 2006, 281, 18519–18531. [Google Scholar] [CrossRef] [Green Version]
- Porter, J.D.; Khanna, S.; Kaminski, H.J.; Rao, J.S.; Merriam, A.P.; Richmonds, C.R.; Leahy, P.; Li, J.; Guo, W.; Andrade, F.H. A chronic inflammatory response dominates the skeletal muscle molecular signature in dystrophin-deficient mdx mice. Hum. Mol. Genet. 2002, 11, 263–272. [Google Scholar] [CrossRef] [Green Version]
- Baker, P.E.; Kearney, J.A.; Gong, B.; Merriam, A.P.; Kuhn, D.E.; Porter, J.D.; Rafael-Fortney, J.A. Analysis of gene expression differences between utrophin/dystrophin-deficient vs mdx skeletal muscles reveals a specific upregulation of slow muscle genes in limb muscles. Neurogenetics 2006, 7, 81–91. [Google Scholar] [CrossRef]
- Miller, M.K.; Bang, M.L.; Witt, C.C.; Labeit, D.; Trombitas, C.; Watanabe, K.; Granzier, H.; McElhinny, A.S.; Gregorio, C.C.; Labeit, S. The muscle ankyrin repeat proteins: CARP, ankrd2/Arpp and DARP as a family of titin filament-based stress response molecules. J. Mol. Biol. 2003, 333, 951–964. [Google Scholar] [CrossRef] [PubMed]
- Kontrogianni-Konstantopoulos, A.; Ackermann, M.A.; Bowman, A.L.; Yap, S.V.; Bloch, R.J. Muscle giants: Molecular scaffolds in sarcomerogenesis. Physiol. Rev. 2009, 89, 1217–1267. [Google Scholar] [CrossRef] [PubMed]
- Hayashi, C.; Ono, Y.; Doi, N.; Kitamura, F.; Tagami, M.; Mineki, R.; Arai, T.; Taguchi, H.; Yanagida, M.; Hirner, S.; et al. Multiple molecular interactions implicate the connectin/titin N2A region as a modulating scaffold for p94/calpain 3 activity in skeletal muscle. J. Biol. Chem. 2008, 283, 14801–14814. [Google Scholar] [CrossRef] [Green Version]
- Sorimachi, H.; Kinbara, K.; Kimura, S.; Takahashi, M.; Ishiura, S.; Sasagawa, N.; Sorimachi, N.; Shimada, H.; Tagawa, K.; Maruyama, K.; et al. Muscle-specific calpain, p94, responsible for limb girdle muscular dystrophy type 2A, associates with connectin through IS2, a p94-specific sequence. J. Biol. Chem. 1995, 270, 31158–31162. [Google Scholar] [CrossRef] [Green Version]
- Ono, Y.; Ojima, K.; Shinkai-Ouchi, F.; Hata, S.; Sorimachi, H. An eccentric calpain, CAPN3/p94/calpain-3. Biochimie 2016, 122, 169–187. [Google Scholar] [CrossRef] [Green Version]
- Jones, S.W.; Parr, T.; Sensky, P.L.; Scothern, G.P.; Bardsley, R.G.; Buttery, P.J. Fibre type-specific expression of p94, a skeletal muscle-specific calpain. J. Muscle Res. Cell Motil. 1999, 20, 417–424. [Google Scholar] [CrossRef]
- Ojima, K.; Kawabata, Y.; Nakao, H.; Nakao, K.; Doi, N.; Kitamura, F.; Ono, Y.; Hata, S.; Suzuki, H.; Kawahara, H.; et al. Dynamic distribution of muscle-specific calpain in mice has a key role in physical-stress adaptation and is impaired in muscular dystrophy. J. Clin. Investig. 2010, 120, 2672–2683. [Google Scholar] [CrossRef] [Green Version]
- Richard, I.; Broux, O.; Allamand, V.; Fougerousse, F.; Chiannilkulchai, N.; Bourg, N.; Brenguier, L.; Devaud, C.; Pasturaud, P.; Roudaut, C.; et al. Mutations in the proteolytic enzyme calpain 3 cause limb-girdle muscular dystrophy type 2A. Cell 1995, 81, 27–40. [Google Scholar] [CrossRef] [Green Version]
- Richard, I.; Roudaut, C.; Marchand, S.; Baghdiguian, S.; Herasse, M.; Stockholm, D.; Ono, Y.; Suel, L.; Bourg, N.; Sorimachi, H.; et al. Loss of calpain 3 proteolytic activity leads to muscular dystrophy and to apoptosis-associated IkappaBalpha/nuclear factor kappaB pathway perturbation in mice. J. Cell Biol. 2000, 151, 1583–1590. [Google Scholar] [CrossRef]
- Tagawa, K.; Taya, C.; Hayashi, Y.; Nakagawa, M.; Ono, Y.; Fukuda, R.; Karasuyama, H.; Toyama-Sorimachi, N.; Katsui, Y.; Hata, S.; et al. Myopathy phenotype of transgenic mice expressing active site-mutated inactive p94 skeletal muscle-specific calpain, the gene product responsible for limb girdle muscular dystrophy type 2A. Hum. Mol. Genet. 2000, 9, 1393–1402. [Google Scholar] [CrossRef] [Green Version]
- Barash, I.A.; Mathew, L.; Ryan, A.F.; Chen, J.; Lieber, R.L. Rapid muscle-specific gene expression changes after a single bout of eccentric contractions in the mouse. Am. J. Physiol. Cell Physiol. 2004, 286, C355–C364. [Google Scholar] [CrossRef] [Green Version]
- Lun, A.S.; Chen, J.; Lange, S. Probing muscle ankyrin-repeat protein (MARP) structure and function. Anat. Rec. 2014, 297, 1615–1629. [Google Scholar] [CrossRef] [Green Version]
- van der Pijl, R.; Strom, J.; Conijn, S.; Lindqvist, J.; Labeit, S.; Granzier, H.; Ottenheijm, C. Titin-based mechanosensing modulates muscle hypertrophy. J. Cachexia Sarcopenia Muscle 2018, 9, 947–961. [Google Scholar] [CrossRef] [Green Version]
- Lange, S.; Gehmlich, K.; Lun, A.S.; Blondelle, J.; Hooper, C.; Dalton, N.D.; Alvarez, E.A.; Zhang, X.; Bang, M.L.; Abassi, Y.A.; et al. MLP and CARP are linked to chronic PKCalpha signalling in dilated cardiomyopathy. Nat. Commun. 2016, 7, 12120. [Google Scholar] [CrossRef]
- Ishiguro, N.; Baba, T.; Ishida, T.; Takeuchi, K.; Osaki, M.; Araki, N.; Okada, E.; Takahashi, S.; Saito, M.; Watanabe, M.; et al. Carp, a cardiac ankyrin-repeated protein, and its new homologue, Arpp, are differentially expressed in heart, skeletal muscle, and rhabdomyosarcomas. Am. J. Pathol. 2002, 160, 1767–1778. [Google Scholar] [CrossRef] [Green Version]
- Barash, I.A.; Bang, M.L.; Mathew, L.; Greaser, M.L.; Chen, J.; Lieber, R.L. Structural and regulatory roles of muscle ankyrin repeat protein family in skeletal muscle. Am. J. Physiol. Cell Physiol. 2007, 293, C218–C227. [Google Scholar] [CrossRef] [Green Version]
- Zhou, T.; Fleming, J.R.; Franke, B.; Bogomolovas, J.; Barsukov, I.; Rigden, D.J.; Labeit, S.; Mayans, O. CARP interacts with titin at a unique helical N2A sequence and at the domain Ig81 to form a structured complex. FEBS Lett. 2016, 590, 3098–3110. [Google Scholar] [CrossRef] [Green Version]
- Lanzicher, T.; Zhou, T.; Saripalli, C.; Keschrumrus, V.; Smith Iii, J.E.; Mayans, O.; Sbaizero, O.; Granzier, H. Single-Molecule Force Spectroscopy on the N2A Element of Titin: Effects of Phosphorylation and CARP. Front. Physiol. 2020, 11, 173. [Google Scholar] [CrossRef] [Green Version]
- Bianco, P.; Nagy, A.; Kengyel, A.; Szatmari, D.; Martonfalvi, Z.; Huber, T.; Kellermayer, M.S. Interaction forces between F-actin and titin PEVK domain measured with optical tweezers. Biophys. J. 2007, 93, 2102–2109. [Google Scholar] [CrossRef] [Green Version]
- Linke, W.A.; Granzier, H. A spring tale: New facts on titin elasticity. Biophys. J. 1998, 75, 2613–2614. [Google Scholar] [CrossRef] [Green Version]
- Trombitas, K.; Greaser, M.; French, G.; Granzier, H. PEVK extension of human soleus muscle titin revealed by immunolabeling with the anti-titin antibody 9D10. J. Struct. Biol. 1998, 122, 188–196. [Google Scholar] [CrossRef] [PubMed]
- Labeit, D.; Watanabe, K.; Witt, C.; Fujita, H.; Wu, Y.; Lahmers, S.; Funck, T.; Labeit, S.; Granzier, H. Calcium-dependent molecular spring elements in the giant protein titin. Sci. Proc. Natl. Acad. Sci. USA 2003, 100, 13716–13721. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cornachione, A.S.; Leite, F.; Bagni, M.A.; Rassier, D.E. The increase in non-cross-bridge forces after stretch of activated striated muscle is related to titin isoforms. Am. J. Physiol. Cell Physiol. 2016, 310, C19–C26. [Google Scholar] [CrossRef] [Green Version]
- Fujita, H.; Labeit, D.; Gerull, B.; Labeit, S.; Granzier, H.L. Titin isoform-dependent effect of calcium on passive myocardial tension. Am. J. Physiol. Heart Circ. Physiol. 2004, 287, H2528–H2534. [Google Scholar] [CrossRef]
- Nishikawa, K. Titin: A Tunable Spring in Active Muscle. Physiology 2020, 35, 209–217. [Google Scholar] [CrossRef]
- Hessel, A.L.; Lindstedt, S.L.; Nishikawa, K.C. Physiological Mechanisms of Eccentric Contraction and Its Applications: A Role for the Giant Titin Protein. Front. Physiol. 2017, 8, 70. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lindstedt, S.L.; Nishikawa, K. Huxleys’ missing filament: Form and function of titin in vertebrate skelletal muscle. Ann. Rev. Physiol. 2017, 79, 145–166. [Google Scholar] [CrossRef]
- Nishikawa, K.; Dutta, S.; DuVall, M.; Nelson, B.; Gage, M.J.; Monroy, J.A. Calcium-dependent titin-thin filament interactions in muscle: Observations and theory. J. Muscle Res. Cell Motil. 2020, 41, 125–139. [Google Scholar] [CrossRef]
- Nishikawa, K.C.; Lindstedt, S.L.; LaStayo, P.C. Basic science and clinical use of eccentric contractions: History and uncertainties. J. Sport Health Sci. 2018, 7, 265–274. [Google Scholar] [CrossRef]
- Wang, K.; McCarter, R.; Wright, J.; Beverly, J.; Ramirez-Mitchell, R. Regulation of skeletal muscle stiffness and elasticity by titin isoforms: A test of the segmental extension model of resting tension. Sci. Proc. Natl. Acad. Sci. USA 1991, 88, 7101–7105. [Google Scholar] [CrossRef] [Green Version]
- Granzier, H.; Labeit, D.; Wu, Y.; Witt, C.; Watanabe, K.; Lahmers, S.; Gotthardt, M.; Labeit, S. Adaptations in titin’s spring elements in normal and cardiomyopathic hearts. Adv. Exp. Med. Biol. 2003, 538, 517–530. [Google Scholar]
- Gautel, M.; Goulding, D. A molecular map of titin/connectin elasticity reveals two different mechanisms acting in series. FEBS Lett. 1996, 385, 11–14. [Google Scholar] [CrossRef] [Green Version]
- Linke, W.A.; Ivemeyer, M.; Olivieri, N.; Kolmerer, B.; Ruegg, J.C.; Labeit, S. Towards a molecular understanding of the elasticity of titin. J. Mol. Biol. 1996, 261, 62–71. [Google Scholar] [CrossRef] [Green Version]
- Magid, A.; Law, D.J. Myofibrils bear most of the resting tension in frog skeletal muscle. Science 1985, 230, 1280–1282. [Google Scholar] [CrossRef]
- Opitz, C.A.; Kulke, M.; Leake, M.C.; Neagoe, C.; Hinssen, H.; Hajjar, R.J.; Linke, W.A. Damped elastic recoil of the titin spring in myofibrils of human myocardium. Sci. Proc. Natl. Acad. Sci. USA 2003, 100, 12688–12693. [Google Scholar] [CrossRef] [Green Version]
- Neagoe, C.; Opitz, C.A.; Makarenko, I.; Linke, W.A. Gigantic variety: Expression patterns of titin isoforms in striated muscles and consequences for myofibrillar passive stiffness. J. Muscle Res. Cell Motil. 2003, 24, 175–189. [Google Scholar] [CrossRef]
- Prado, L.G.; Makarenko, I.; Andresen, C.; Kruger, M.; Opitz, C.A.; Linke, W.A. Isoform diversity of giant proteins in relation to passive and active contractile properties of rabbit skeletal muscles. J. Gen. Physiol. 2005, 126, 461–480. [Google Scholar] [CrossRef] [Green Version]
- Lopez, M.A.; Pardo, P.S.; Cox, G.A.; Boriek, A.M. Early mechanical dysfunction of the diaphragm in the muscular dystrophy with myositis (Ttnmdm) model. Am. J. Physiol. Cell Physiol. 2008, 295, C1092–C1102. [Google Scholar] [CrossRef] [Green Version]
- Monroy, J.A.; Powers, K.L.; Pace, C.M.; Uyeno, T.; Nishikawa, K.C. Effects of activation on the elastic properties of intact soleus muscles with a deletion in titin. J. Exp. Biol. 2017, 220, 828–836. [Google Scholar] [CrossRef] [Green Version]
- Tahir, U.; Monroy, J.A.; Rice, N.A.; Nishikawa, K.C. Effects of a titin mutation on force enhancement and force depression in mouse soleus muscles. J. Exp. Biol. 2020, 223. [Google Scholar] [CrossRef] [PubMed]
- Dutta, S.; Tsiros, C.; Sundar, S.L.; Athar, H.; Moore, J.; Nelson, B.; Gage, M.J.; Nishikawa, K. Calcium increases titin N2A binding to F-actin and regulated thin filaments. Sci. Rep. 2018, 8, 14575. [Google Scholar] [CrossRef] [PubMed]
- Linari, M.; Woledge, R.C.; Curtin, N.A. Energy storage during stretch of active single fibRes. from frog skeletal muscle. J. Physiol. 2003, 548, 461–474. [Google Scholar] [CrossRef] [PubMed]
- Julian, F.J.; Morgan, D.L. The effect on tension of non-uniform distribution of length changes applied to frog muscle fibres. J. Physiol. 1979, 293, 379–392. [Google Scholar] [CrossRef] [PubMed]
- Linke, W.A.; Stockmeier, M.R.; Ivemeyer, M.; Hosser, H.; Mundel, P. Characterizing titin’s I-band Ig domain region as an entropic spring. J. Cell Sci. 1998, 111 Pt 11, 1567–1574. [Google Scholar]
- Bagni, M.A.; Cecchi, G.; Colombini, B.; Colomo, F. A non-cross-bridge stiffness in activated frog muscle fibers. Biophys. J. 2002, 82, 3118–3127. [Google Scholar] [CrossRef] [Green Version]
- Bagni, M.A.; Cecchi, G.; Colomo, F.; Garzella, P. Development of stiffness precedes cross-bridge attachment during the early tension rise in single frog muscle fibres. J. Physiol. 1994, 481 Pt 2, 273–278. [Google Scholar] [CrossRef]
- Bagni, M.A.; Colombini, B.; Geiger, P.; Berlinguer Palmini, R.; Cecchi, G. Non-cross-bridge calcium-dependent stiffness in frog muscle fibers. Am. J. Physiol. Cell Physiol. 2004, 286, C1353–C1357. [Google Scholar] [CrossRef]
- Nocella, M.; Cecchi, G.; Bagni, M.A.; Colombini, B. Force enhancement after stretch in mammalian muscle fiber: No evidence of cross-bridge involvement. Am. J. Physiol. Cell Physiol. 2014, 307, C1123–C1129. [Google Scholar] [CrossRef] [Green Version]
- Cornachione, A.S.; Rassier, D.E. A non-cross-bridge, static tension is present in permeabilized skeletal muscle fibers after active force inhibition or actin extraction. Am. J. Physiol. Cell Physiol. 2012, 302, C566–C574. [Google Scholar] [CrossRef] [Green Version]
- Herzog, W.; Leonard, T.R. Force enhancement following stretching of skeletal muscle: A new mechanism. J. Exp. Biol. 2002, 205, 1275–1283. [Google Scholar]
- Joumaa, V.; Rassier, D.E.; Leonard, T.R.; Herzog, W. Passive force enhancement in single myofibrils. Pflugers Arch. 2007, 455, 367–371. [Google Scholar] [CrossRef]
- Joumaa, V.; Rassier, D.E.; Leonard, T.R.; Herzog, W. The origin of passive force enhancement in skeletal muscle. Am. J. Physiol. Cell Physiol. 2008, 294, C74–C78. [Google Scholar] [CrossRef] [Green Version]
- Minozzo, F.C.; Lira, C.A. Muscle residual force enhancement: A brief review. Clinics 2013, 68, 269–274. [Google Scholar] [CrossRef]
- Shalabi, N.; Cornachione, A.; de Souza Leite, F.; Vengallatore, S.; Rassier, D.E. Residual force enhancement is regulated by titin in skeletal and cardiac myofibrils. J. Physiol. 2017, 595, 2085–2098. [Google Scholar] [CrossRef]
- Leonard, T.R.; Herzog, W. Regulation of muscle force in the absence of actin-myosin-based cross-bridge interaction. Am. J. Physiol. Cell Physiol. 2010, 299, C14–C20. [Google Scholar] [CrossRef] [Green Version]
- Nishikawa, K.C.; Monroy, J.A.; Uyeno, T.E.; Yeo, S.H.; Pai, D.K.; Lindstedt, S.L. Is titin a ‘winding filament’? A new twist on muscle contraction. Proc. Biol. Sci. 2012, 279, 981–990. [Google Scholar] [CrossRef] [Green Version]
- Lappin, A.K.; Monroy, J.A.; Pilarski, J.Q.; Zepnewski, E.D.; Pierotti, D.J.; Nishikawa, K.C. Storage and recovery of elastic potential energy powers ballistic prey capture in toads. J. Exp. Biol. 2006, 209, 2535–2553. [Google Scholar] [CrossRef] [Green Version]
- Sugi, H.; Akimoto, T.; Kobayashi, T.; Suzuki, S.; Shimada, M. Possible contribution of titin filaments to the compliant series elastic component in horseshoe crab skeletal muscle fibers. Adv. Exp. Med. Biol. 2000, 481, 371–380. [Google Scholar]
- Powers, K.; Schappacher-Tilp, G.; Jinha, A.; Leonard, T.; Nishikawa, K.; Herzog, W. Titin force is enhanced in actively stretched skeletal muscle. J. Exp. Biol. 2014, 217, 3629–3636. [Google Scholar] [CrossRef] [Green Version]
- Schappacher-Tilp, G.; Leonard, T.; Desch, G.; Herzog, W. A novel three-filament model of force generation in eccentric contraction of skeletal muscles. PLoS ONE 2015, 10, e0117634. [Google Scholar] [CrossRef] [Green Version]
- Filomena, M.C.; Yamamoto, D.L.; Caremani, M.; Kadarla, V.K.; Mastrototaro, G.; Serio, S.; Vydyanath, A.; Mutarelli, M.; Garofalo, A.; Pertici, I.; et al. Myopalladin promotes muscle growth through modulation of the serum response factor pathway. J. Cachexia Sarcopenia Muscle 2020, 11, 169–194. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gutierrez-Cruz, G.; Van Heerden, A.H.; Wang, K. Modular motif, structural folds and affinity profiles of the PEVK segment of human fetal skeletal muscle titin. J. Biol. Chem. 2001, 276, 7442–7449. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ma, K.; Wang, K. Interaction of nebulin SH3 domain with titin PEVK and myopalladin: Implications for the signaling and assembly role of titin and nebulin. FEBS Lett. 2002, 532, 273–278. [Google Scholar] [CrossRef] [Green Version]
- Ma, K.; Forbes, J.G.; Gutierrez-Cruz, G.; Wang, K. Titin as a giant scaffold for integrating stress and Src homology domain 3-mediated signaling pathways: The clustering of novel overlap ligand motifs in the elastic PEVK segment. J. Biol. Chem. 2006, 281, 27539–27556. [Google Scholar] [CrossRef] [PubMed] [Green Version]






© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nishikawa, K.; Lindstedt, S.L.; Hessel, A.; Mishra, D. N2A Titin: Signaling Hub and Mechanical Switch in Skeletal Muscle. Int. J. Mol. Sci. 2020, 21, 3974. https://doi.org/10.3390/ijms21113974
Nishikawa K, Lindstedt SL, Hessel A, Mishra D. N2A Titin: Signaling Hub and Mechanical Switch in Skeletal Muscle. International Journal of Molecular Sciences. 2020; 21(11):3974. https://doi.org/10.3390/ijms21113974
Chicago/Turabian StyleNishikawa, Kiisa, Stan L. Lindstedt, Anthony Hessel, and Dhruv Mishra. 2020. "N2A Titin: Signaling Hub and Mechanical Switch in Skeletal Muscle" International Journal of Molecular Sciences 21, no. 11: 3974. https://doi.org/10.3390/ijms21113974
APA StyleNishikawa, K., Lindstedt, S. L., Hessel, A., & Mishra, D. (2020). N2A Titin: Signaling Hub and Mechanical Switch in Skeletal Muscle. International Journal of Molecular Sciences, 21(11), 3974. https://doi.org/10.3390/ijms21113974