1. Introduction
Despite recent advances, the pharmacological therapy of cystic fibrosis (CF), an autosomal recessive genetic disorder caused by the mutation of the cystic fibrosis transmembrane conductance regulator (CFTR or ABCC7) gene, is still a largely unmet medical need. Mutations related to the CFTR gene can result in the CFTR protein misfolding, decreased trafficking to and stability in the plasma membrane of epithelial cells and an impaired activity of this ATP-gated anion channel [
1]. This is the reason why CF is considered a disease of impaired protein folding and function, and rescue of the cells from stress due to chronic expression of misfolded proteins, including mutant CFTR, can improve the loss-of-function phenotype [
2]. Used in triple combination, small molecular CFTR modulators, like correctors, which act as pharmacological chaperones or proteostasis regulators, and potentiators, which directly improve CFTR channel functions, now offer a breakthrough therapy for the most common CFTR mutations [
3,
4]. Patients carrying the most common ΔF508-CFTR mutations can benefit from these new drugs, however these therapies are either not suitable for all patients, who might be unresponsive due to rare or unique CFTR mutations, or they cannot afford them due to their excessive cost [
5].
CF is a multi-organ disease with progressive decline in lung function, therefore the care of CF patients requires a multidisciplinary and multifaceted approach including pharmaco- and physiotherapy as well as psychosocial interventions [
6]. Adjuvant therapies also play an important role in the management of CF, especially to prevent the accumulation of viscous mucus and bacterial infection in the respiratory tract. CFTR and bicarbonate secretion are central elements in the regulation of mucus viscosity. CFTR not only functions as a cAMP/PKA-regulated epithelial anion channel, but also controls the activity of other ion channels and transporters, such as the epithelial Na
+ channel (ENaC) or the Cl
-/HCO
3- exchanger SLC26A4 [
7]. Gene mutations or exogenous noxae (i.e., cigarette smoke) can impair CFTR functions compromising transepithelial electrolyte and water transport in the respiratory tract [
8]. In recent years, a large number of evidence suggests that defective HCO
3- secretion plays a central role in the pathogenesis of CF- and non-CF-related airway diseases [
7,
8]. Importantly, HCO
3- controls the luminal pH in the airways. It has been demonstrated in a porcine model of CF that the acidic pH in the trachea reduced the bacterial-killing capacity of antimicrobial peptides, such as lysozyme and lactoferrin, which was corrected by administration of nebulized sodium bicarbonate [
9]. More recently, it has been reported that the antibacterial effects of defensins and LL-37 peptide are largely pH-dependent [
10]. Sodium bicarbonate not only restores the activity of antimicrobial peptides by regulating pH in the airway surface liquid, but has a direct bacteriostatic effects and inhibits biofilm formation of CF-related bacteria [
11]. It is also noteworthy, that HCO
3- is necessary for chelating protons and Ca
2+ for proper unfolding of secreted mucin molecules which helps to maintain the normal viscosity of the airway surface liquid [
12].
To better understand CF and develop new treatments, relevant model systems are needed. Immortalized airway epithelial cells have been proven to be particularly useful experimental tools [
13]. Several cell lines exist to study CF, especially CFTR mutations, the respective phenotypes and pharmacological therapies. Although in some studies non-respiratory cell lines (e.g., HeLa) transfected with wild-type (WT) or mutant CFTR are used to investigate the function of the CFTR channel, differences were found in both receptor trafficking and activation as compared to transfected respiratory cell lines [
14]. The human CF bronchial epithelial cell line CFBE41o
– (CFBE), developed by Kunzelmann et al. [
15], forms adherent, tight and polarized cell layers and expresses a transcriptome that is similar to that of primary airway epithelial cells [
16]. This cell line, which does not express endogenous CFTR at mRNA or protein level, has been transfected by several groups to generate cell lines expressing wild-type or ΔF508 CFTR [
14,
16,
17]. This human cell line model from a relevant tissue type maintains many features necessary for studies on CFTR function and treatment with small drug molecules at the cellular level. To increase the complexity and similarity of respiratory culture models to the lung tissue, in vitro co-culture models were introduced. Indeed, an increasing correlation was found between airway models and in vivo tissue by the combination of multiple lung-relevant cell types [
18]. In these models in addition to alveolar epithelial cells, vascular endothelial cells and/or immune cells, like macrophages or mast cells are used [
18]. However, few co-culture models exist for bronchial epithelium. Recently a co-culture model of human bronchial epithelial cells with human microvascular endothelial cells was described and optimized for microscopical studies [
19]. However, no such co-culture models have been established for the study of CF.
Experimental and clinical results with inhaled sodium bicarbonate as an adjuvant therapy in CF are promising due to its mucolytic and bacteriostatic properties, but its direct effect has yet not been studied on respiratory epithelial cells. Our aim was to establish and characterize co-culture models of human CF bronchial epithelial cell lines expressing wild-type or ΔF508-CFTR channels with human vascular endothelial cells and investigate the effects of bicarbonate. Specifically, our study focused on the sodium-bicarbonate-treatment-induced changes in cell impedance, viability, morphology, barrier function, intracellular resting pH and cellular localization of the CFTR channel.
3. Discussion
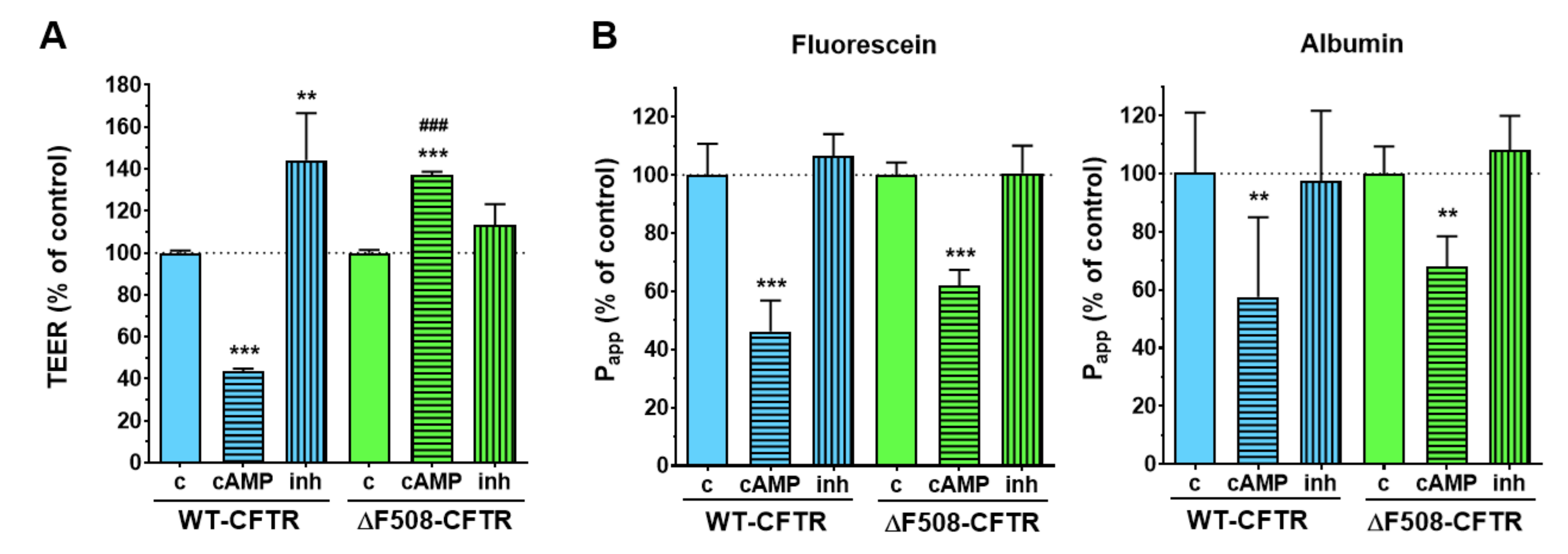
We have successfully established and characterized a new, physiologically more relevant co-culture model of cystic fibrosis using the CFBE cell line. This cell line, derived from samples from a CF patient, is widely used for studies on disease pathology and to test possible pharmacological treatments [
25,
26]. To better understand CF pathology, CFBE cells, which do not express endogenous CFTR, were transfected with WT and ΔF508-CFTR channels by several groups [
14,
17,
20]. Bebők et al. have characterized in detail a CFBE cell-line pair sharing the same genetic background but expressing either the WT or the mutant channel [
14], and this model was further developed in our experiments. Many studies prove that CFBE cells expressing WT and ΔF508-CFTR channels are a useful model system to study biogenesis, trafficking and regulation of this anion channel. The advantages of CFBE cells include easy culture of the cells, robust expression of the respective CFTR channels, epithelial characteristics, like a polarized phenotype and apical expression of CFTR in wild-type cells, and the expression of adenosine and adrenergic receptors which regulate CFTR activity [
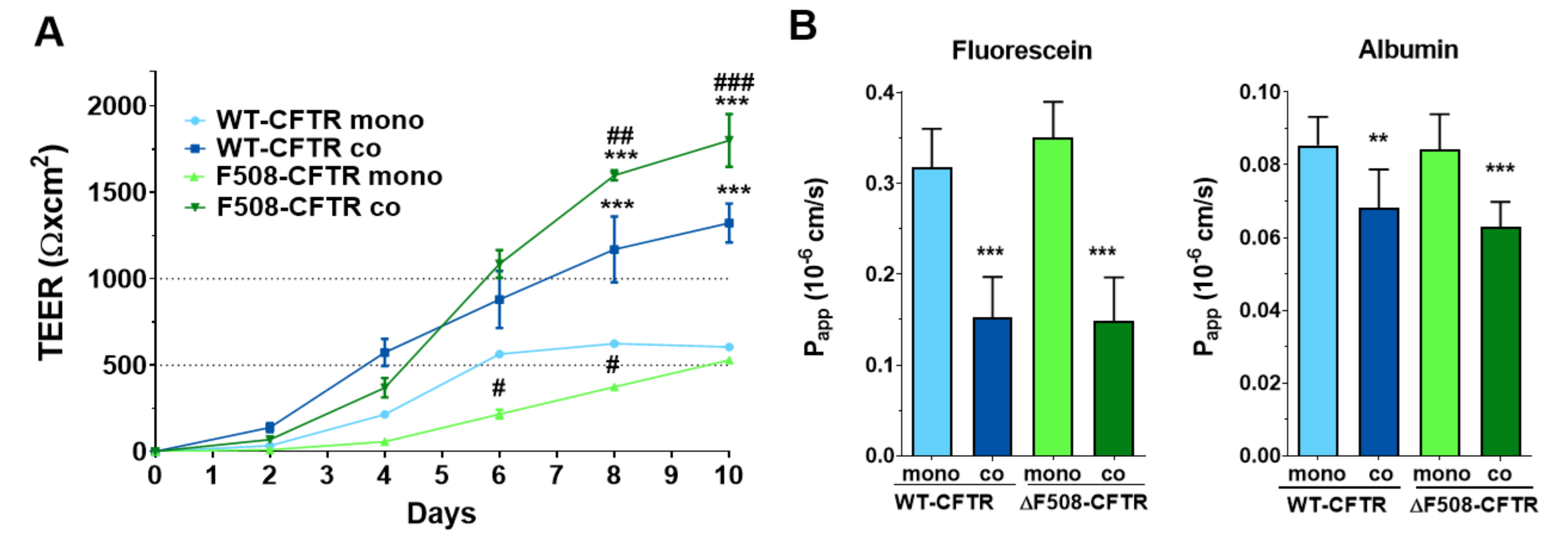
14]. We could confirm, that both CFBE cell lines formed tight barriers, showed high TEER values and well-established intercellular junctions in concordance with literature data [
20]. We also demonstrated the expression of functional CFTR channels in the WT-CFTR CFBE cells, but not in the ΔF508-CFTR CFBE cells. Intracellular cAMP signaling is linked to CFTR activation and increased ion flux, which was reflected in the WT-CFTR CFBE cells as a decrease in TEER values, while the opposite effect was observed after treatment with a CFTR inhibitor. In contrast, the TEER values of the ΔF508-CFTR CFBE cells increased, which is due to the well-known barrier-tightening effects of cAMP [
27] as indicated by the decreased paracellular permeability for tracer molecules of diffusion in both cell lines. These functional data are in accordance with the positive immunostaining for CFTR in the cells in our study, and functional TEER results from the literature [
20,
28], and indicate the validity of our models.
Co-culture models of airway epithelial cells are increasingly used as a more complex experimental setup [
18,
26]. However, these models are made either from non-bronchial, mostly alveolar epithelial cells [
18] or are used as 3D organoids, which are not suitable for permeability studies [
26]. The only study we could find presenting a human bronchial epithelial and vascular endothelial co-culture was a microfluidic model optimized for microscopy [
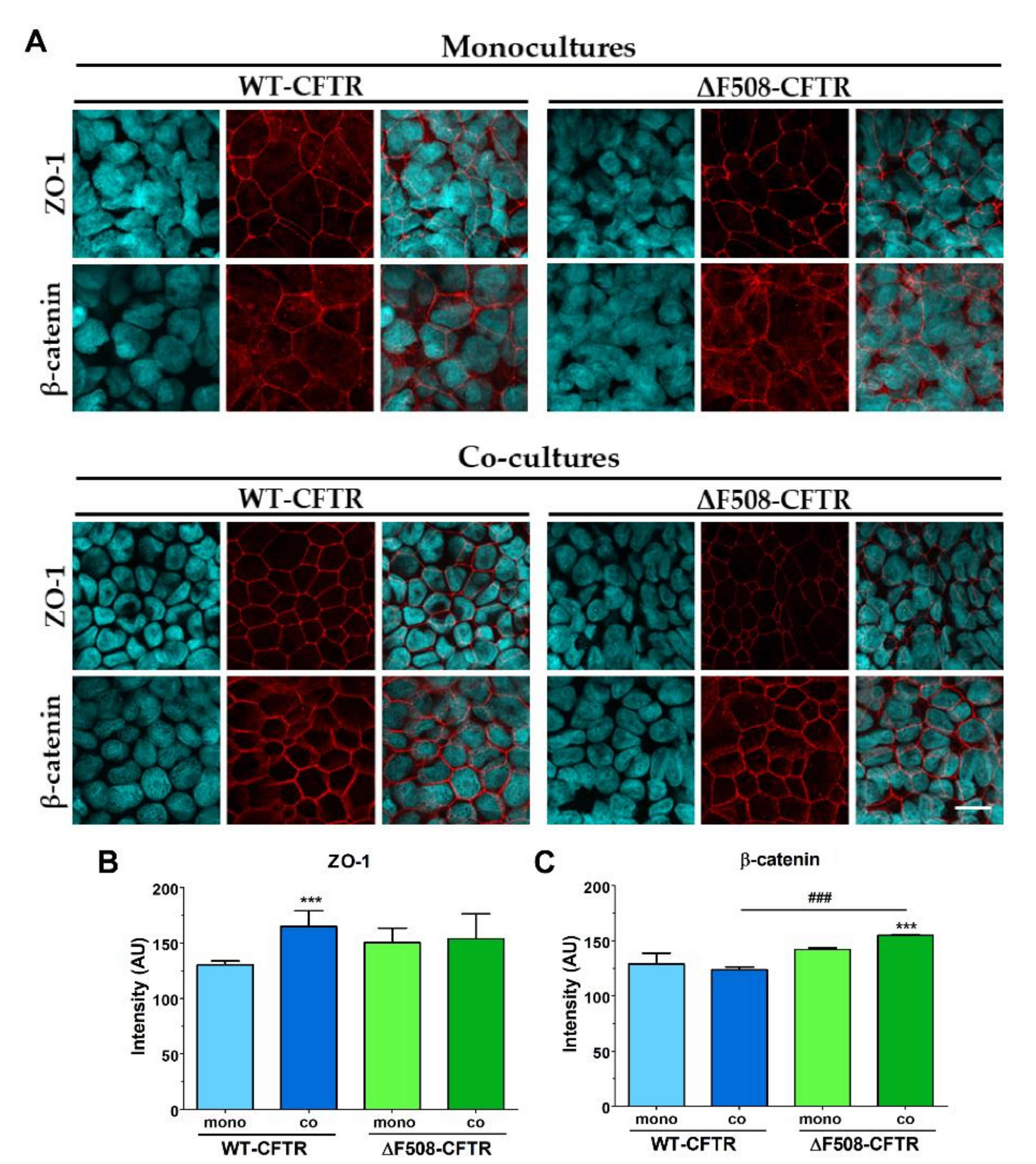
19]. Thus, our study is the first to establish and characterize a culture-insert-based co-culture model using a CFBE cell-line pair and human vascular endothelial cells. We found that the presence of endothelial cells induced better barrier properties in CFBE bronchial epithelial cells. The resistance values increased while the permeability values decreased for marker molecules. The co-culture conditions also increased the tightness of interepithelial junctions and made CFBE cells form a well-organized, tighter monolayer visualized by immunostaining. We observed, that the WT-CFBE model showed lower, while the ΔF508-CFTR CFBE model had significantly higher, TEER.
When the barrier integrity of the CFBE cell-line pair was compared, based on several independent experiments, we confirmed that monocultures of ΔF508-CFTR CFBE cells showed lower TEER and higher paracellular permeability values indicating weaker barrier properties. These findings are in agreement with literature data, in which bronchial epithelial cell monolayers expressing ΔF508-CFTR have a higher paracellular permeability [
26,
28]. These papers not only link CFTR dysfunction to the dysregulation of the paracellular transport route [
28], but also demonstrate that the expression of the WT-CFTR restores tight-junction localization and function in cystic fibrosis bronchial epithelial cells [
26]. Our novel finding is, that when ΔF508-CFTR CFBE cells are kept in co-culture with human vascular endothelial cells from a normal genetic background, this effect is rescued: the mutant cells show tighter barrier properties: increased resistance and decreased permeability for hydrophilic marker molecules.
Although air-liquid interface culture conditions are generally considered as better reflecting physiological conditions [
26], in our hands culturing CFBE cells in air-liquid interface showed weaker barrier properties compared to the traditional liquid submerged cultures. Our results are supported by a study, in which impaired barrier properties were also obtained at air-liquid-cultured CFBE cells [
17], indicating that this approach cannot necessarily be applied for all respiratory culture models.
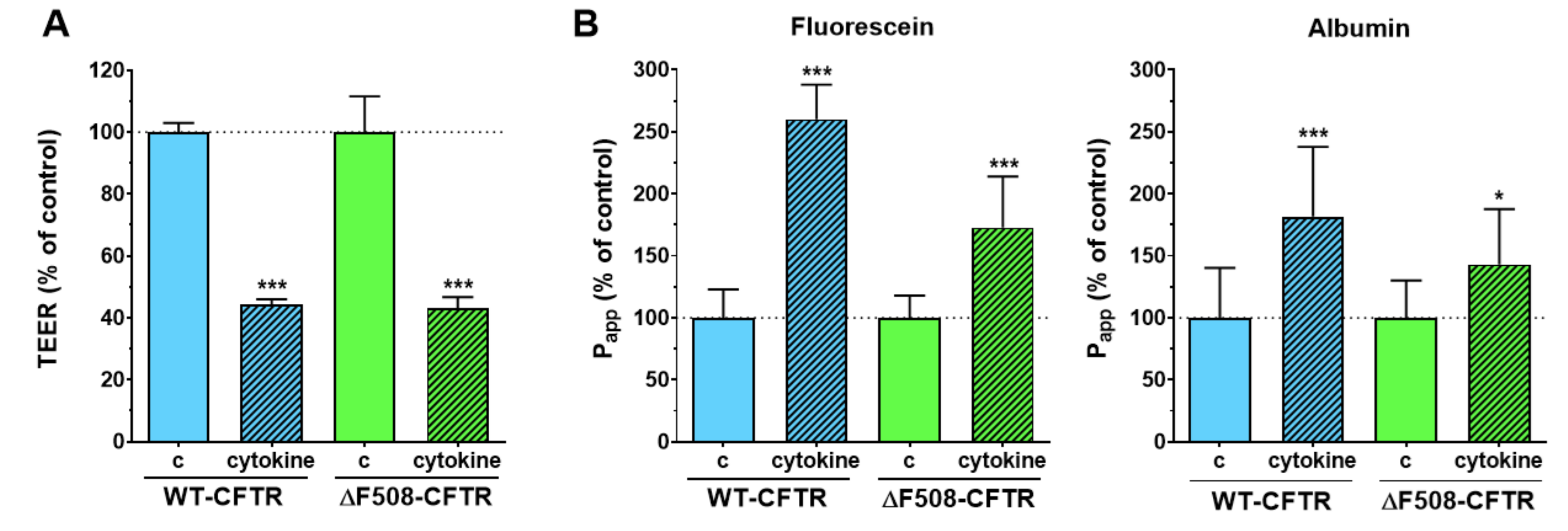
Inflammatory changes in the respiratory system are a hallmark of CF and have been investigated in culture models, where loss of barrier integrity was described in airway epithelial cultures treated with proinflammatory cytokines [
26,
29]. To test the reaction of the model to inflammatory challenge, co-cultures of CFBE epithelial and endothelial cells were treated with a combination of TNFα and IL-1β. This combination of cytokines caused NF-κB translocation and the loss of barrier integrity in our previous studies on other biological barrier models, namely on intestinal epithelial cells [
21] and brain endothelial cells [
30]. Treatment with proinflammatory cytokines caused barrier damage in both CFBE cell-based co-culture models as demonstrated by the lower TEER and higher permeability values. This effect was not observed in CFBE epithelial monocultures, where the response of the cells to cytokines was modest. The presence of endothelial cells contributed to the more robust response to proinflammatory cytokine treatment in epithelial cells and suggests that co-cultures can be more suitable models to investigate inflammatory conditions in vitro.
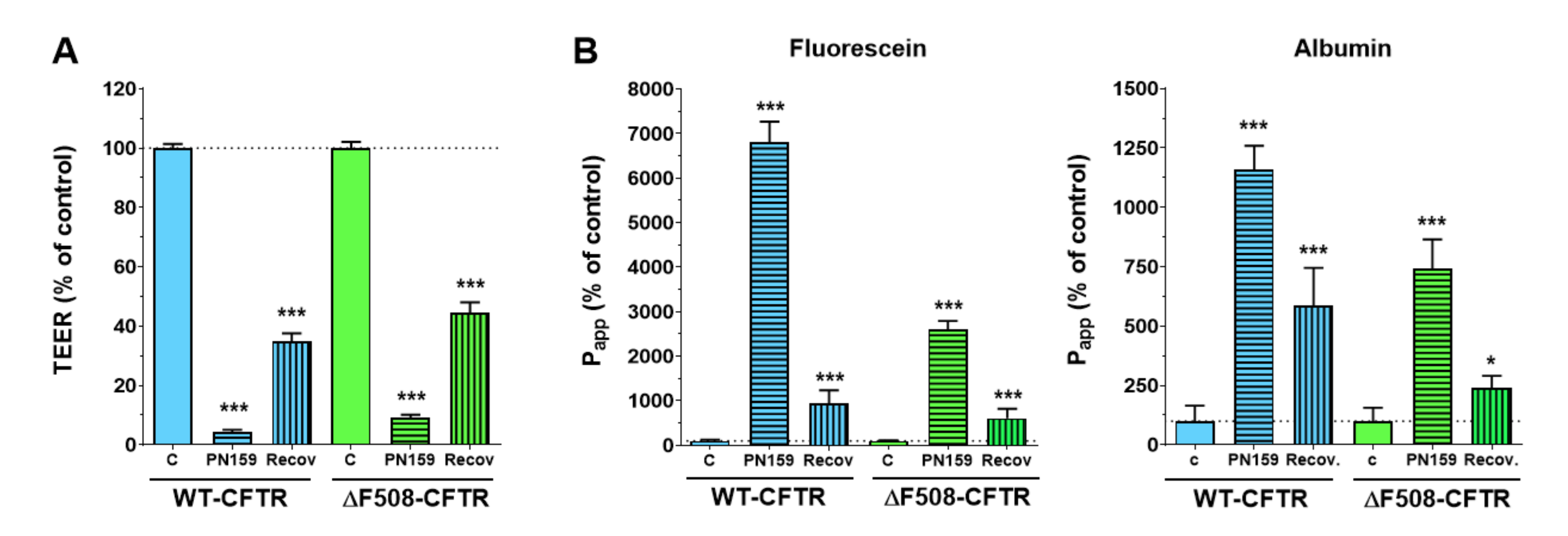
To further characterize the CFBE cell lines we tested their response to the PN159/KLAL peptide, which has a dual effect on barrier cells: it opens the intercellular-tight junctions and also acts as a cell-penetrating peptide [
23]. The junction-modulator effect of this synthetic cationic peptide was discovered on human bronchial epithelial cell cultures [
31], and we have described a similar effect on models of the intestinal epithelium and the blood-brain barrier [
22]. We found that the PN159/KLAL peptide efficiently and reversibly opened the junctions in CFBE bronchial epithelial cells and there was no difference between the reactions of the CFBE cell-line pair to the peptide.
By regulating mucus pH and viscosity, inhaled bicarbonate can be also crucial in the therapy of CF [
32]. Indeed, sodium bicarbonate decreased high viscosity of the CF bronchial secretion [
33] and a similar effect has been observed in a clinical investigation [
34]. This new clinical study on CF patients confirmed the safety and tolerability of nebulized sodium bicarbonate (4.2% and 8.4% solutions) and demonstrated an increased pH and decreased viscosity in bronchial mucus [
34]. In addition, sodium bicarbonate (100 mM) inhibited bacterial growth and biofilm formation of bacteria important in CF pathology, indicating it can have an additional beneficial effect in CF therapy [
11].
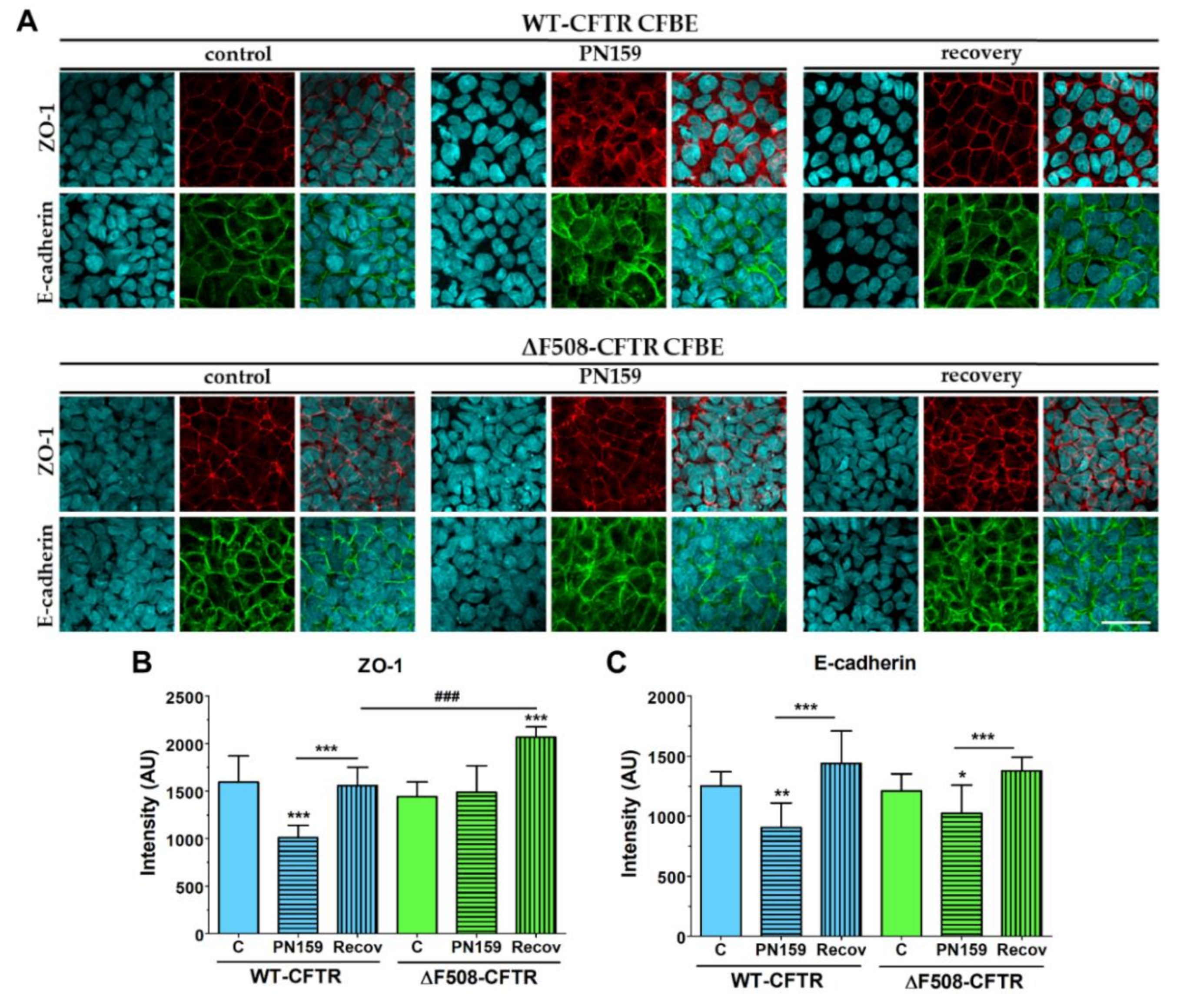
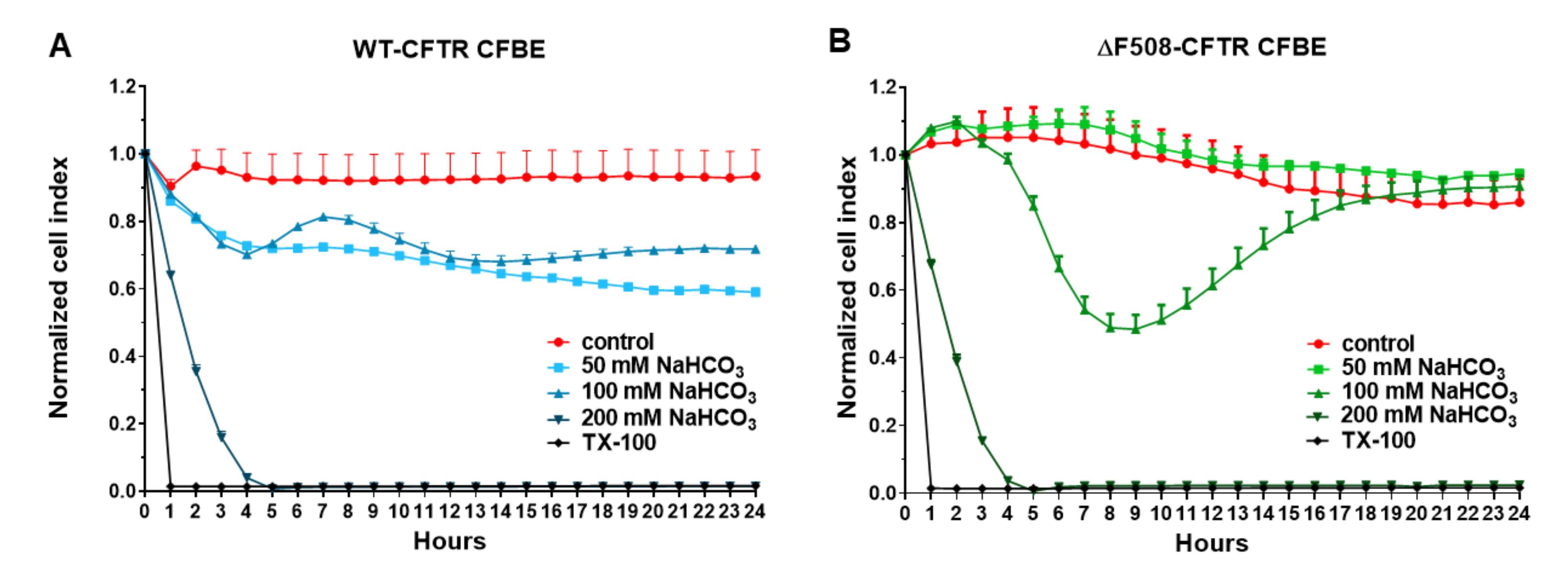
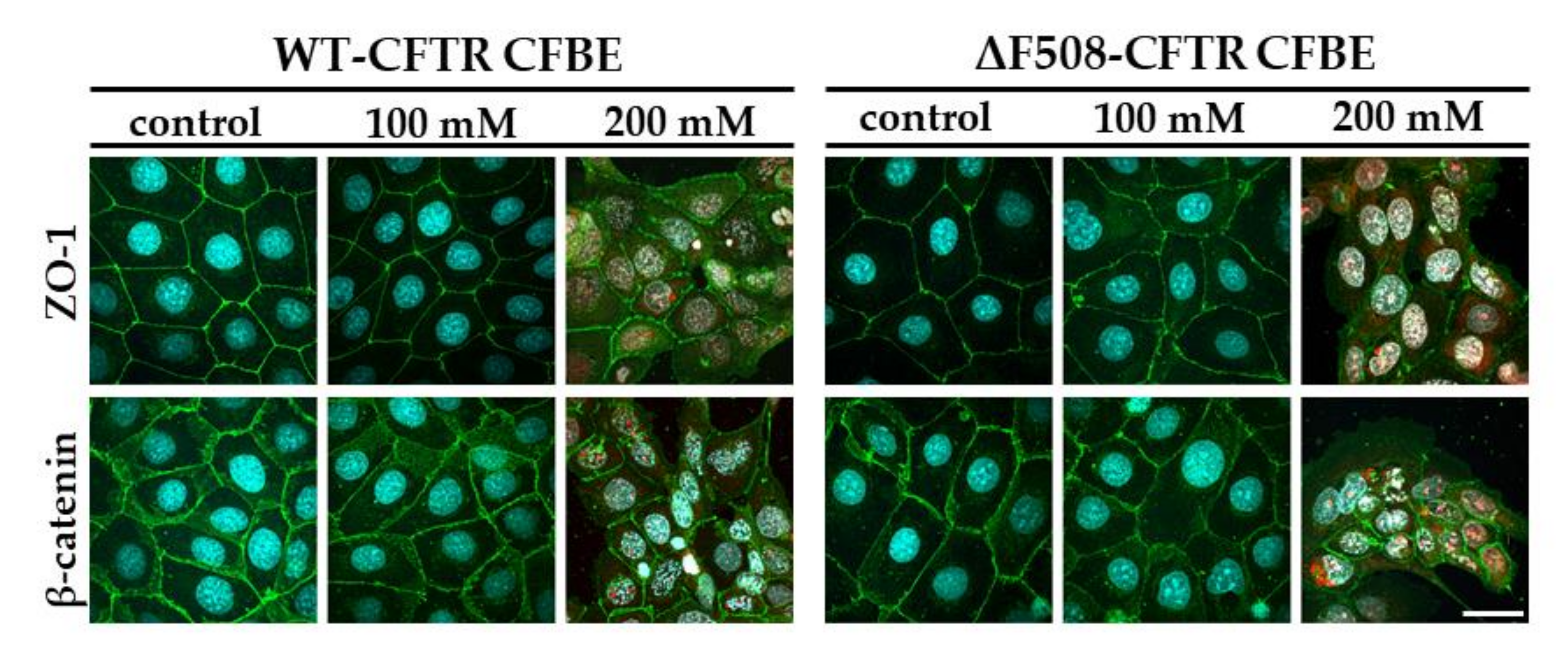
Our study aimed to investigate the direct effect of elevated sodium bicarbonate concentrations on bronchial epithelial cell viability, barrier properties and morphology, which have not been studied yet. Using impedance kinetics to monitor the cell-layer response we confirmed that CFBE epithelial cells tolerate well-elevated sodium bicarbonate concentrations. In our experiments 100 mM sodium bicarbonate (a four-times-higher concentration than in the basal culture medium) was the highest, but still a safe concentration based on viability and cell morphology data. Interestingly, we found differences between the reactions of the CFBE cell lines. CFBE cells expressing the wild-type CFTR channel were more sensitive to higher concentrations of sodium bicarbonate. In ΔF508-CFTR bronchial cells expressing the mutant channel the impedance values were the same after treatment with elevated concentrations of bicarbonate as in the control group. In addition, even a barrier-increasing effect was seen in the case of these ΔF508-CFTR epithelial cells based on the image analysis of junctional protein ZO-1. One of our most important findings is that the barrier function of the CFBE epithelial cells is preserved after sodium bicarbonate treatment based on resistance, permeability and morphological results.
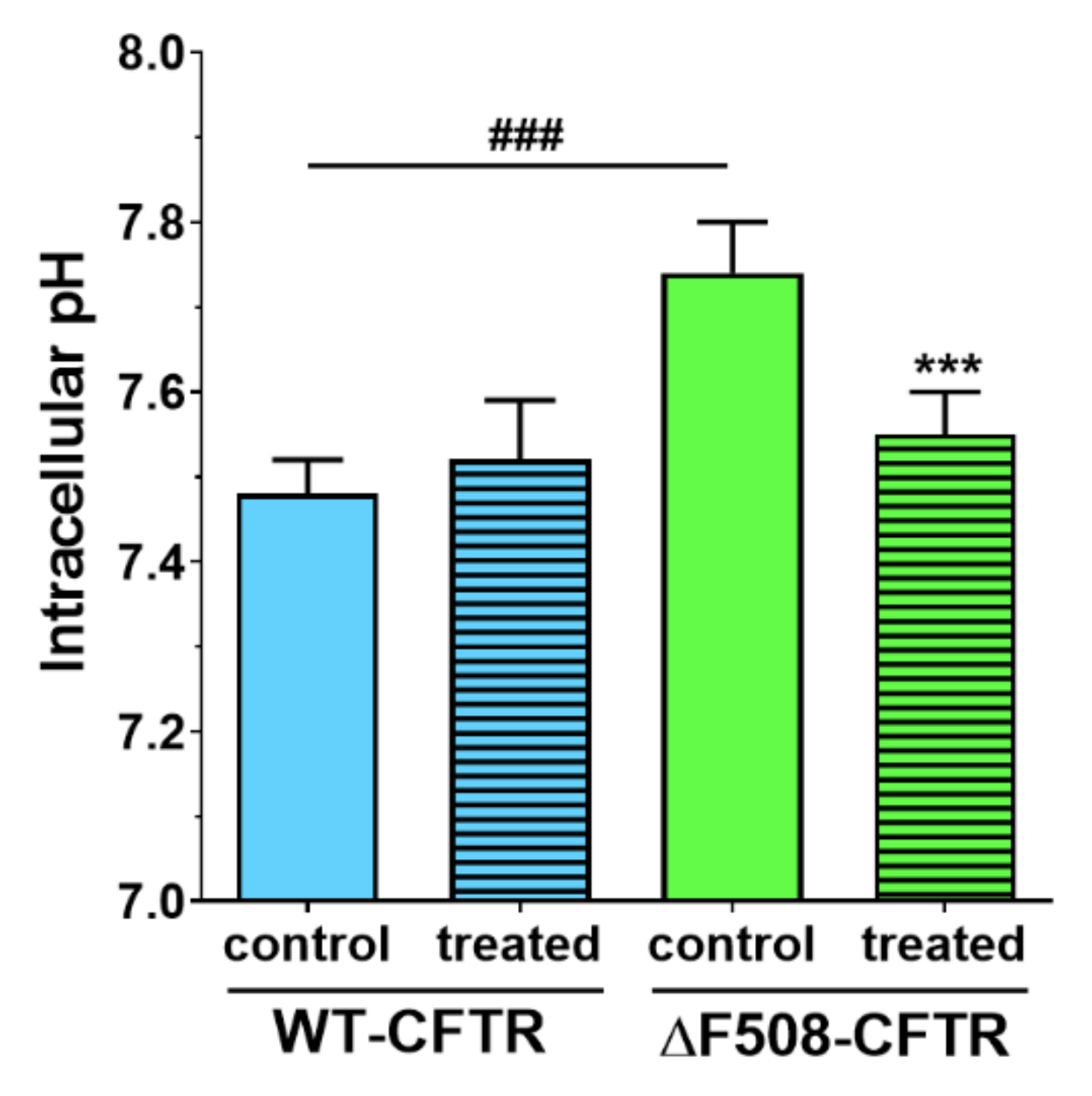
We not only demonstrated that higher concentrations of sodium bicarbonate can be safely used in CFBE cells, but we made observations that these might be also beneficial for cells expressing the mutant CFTR channel. We measured in ΔF508-CFTR bronchial epithelial cells a more alkaline-resting pH
i than in CFBE cells expressing wild-type CFTR. Similarly, in CFTR-KO crypt epithelial cells more alkaline pH
i was found [
24]. Treatment of the cells with 100 mM sodium bicarbonate normalized the pH
i of mutant cells to the level of the WT-CFTR CFBE cells.
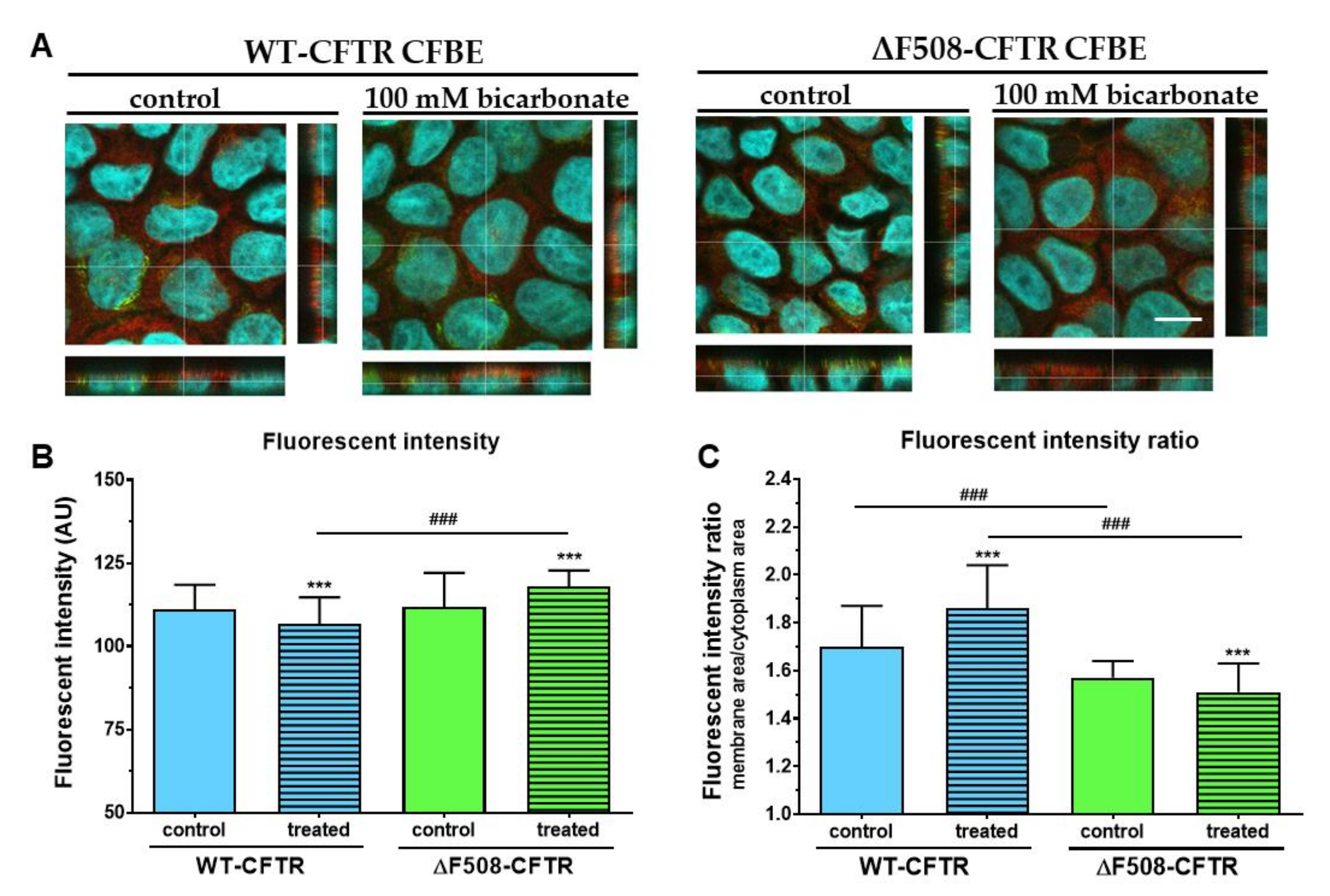
Although regulation of CFTR expression and targeting is not fully understood, transcription factors and miRNAs are known to regulate these processes [
35]. Our results show that administration of 100 mM sodium bicarbonate modifies the apical expression of CFTR in both cell lines. Extracellular HCO
3- acting as a signal molecule, stimulates the activity of soluble adenylyl cyclase increasing intracellular cAMP concentrations, which in turn activates protein kinase A [
36]. Protein kinase A could trigger NF-κB, a transcription factor known to mediate up-regulation of the CFTR gene expression in pulmonary epithelial cells [
37]. Thus, it is conceivable that higher HCO
3--permeability of WT-CFTR expressing cells causes up-regulation of adenylyl cyclase activity increasing apical expression of CFTR. Further investigations are needed to explain the reduced CFTR immunofluorescence ratio in ΔF508-CFTR-expressing cells.
Our morphological analysis indicates that mutant CFBE cells have an increased average height suggesting larger cell volume than WT-CFTR-expressing cells. The important role of CFTR in cell-volume regulation has already been demonstrated by other research groups. For instance, it has been shown that cell-volume regulation is defective of small intestinal crypts isolated from CFTR
-/- mutant mice [
38]. The authors claimed that this was due to impaired CFTR-dependent regulation of volume sensitive K
+ channels. In another study, CFTR potentiated and accelerated regulatory-volume decrease following hypotonic challenge by an autocrine mechanism involving ATP release and signaling [
39]. Consequently, lack of functional CFTR could lead to impaired ATP secretion and depletion of this autocrine, purinergic signaling system under both basal and hypotonic conditions. Furthermore, altered intracellular Cl
- concentrations of CF cells may also contribute to defective cell-volume regulation [
40].
In conclusion, we characterized a new co-culture model of bronchial epithelial cells and demonstrated that vascular endothelial cells induced better barrier properties in CFBE cells as reflected by the higher resistance and lower permeability values. Activation of CFTR by cAMP decreased the electrical resistance in wild-type but not in mutant CFBE cell layers confirming the presence and absence of functional channels, respectively. Elevated concentrations of sodium bicarbonate were well-tolerated by CFBE cells: it slightly reduced the impedance of wild-type but not that of the mutant CFBE cells. Sodium bicarbonate significantly increased the junctional staining intensity of ZO-1 protein and decreased the more-alkaline intracellular pH of the mutant CFBE cells, while the functional barrier properties of the models were preserved. Our observations on the direct effects of sodium bicarbonate on bronchial epithelial cells confirm the safe therapeutic use of inhaled sodium bicarbonate.
4. Materials and Methods
4.1. Materials
Unless specified otherwise, all reagents were purchased from Sigma-Aldrich Ltd. (Budapest, Hungary).
4.2. Cell Cultures
The human cystic fibrosis bronchial epithelial (CFBE) cell-line pair was kindly provided by Dr. Zsuzsanna Bebők. The cells expressing the wild-type human CFTR (WT-CFTR CFBE) and the CFTR with the most common ΔF508 mutation (ΔF508-CFTR CFBE) were generated by lentiviral transformation of the human CFBE41o
- cell line, which does not express CFTR [
14]. CFBE human bronchial epithelial cells were grown under selective pressure in puromycin-containing medium to express the transgenes and used for the experiments until passage 18. The cells were grown in Minimum Eagle Medium (MEM) (Gibco, Life Technologies, Carlsbad, CA, USA) supplemented with 10% fetal bovine serum (Pan-Biotech GmbH, Aidenbach, Germany), stable glutamine (Glutamax, 2 mM), puromycin (2 µg/mL) and 50 μg/mL gentamycin in a humidified incubator with 5% CO
2 at 37 °C. All plastic surfaces were coated with 0.05% rat tail collagen in sterile, distilled water before cell seeding in culture dishes and the medium was changed every 2 days.
Bronchial epithelial cells were cultured in the presence of human vascular endothelial cells [
41,
42] to create the co-culture model. The endothelial culture media (ECM-NG, Sciencell, Carlsbad, CA, USA) was supplemented with 5% FBS, 1% endothelial growth supplement (ECGS, Sciencell, Carlsbad, CA, USA) and 0.5% gentamicin. For the permeability measurements the epithelial cells were cultured on inserts (Transwell, polycarbonate membrane, 0.4 μm pore size, 1.12 cm
2, Corning Costar Co., MA, USA) placed in 12-well plates as mono- or co-cultures for 10 days. To prepare the co-culture model, endothelial cells (≤ P9) were passaged (8 × 10
4 cells/cm
2) to the collagen-type-IV- and fibronectin- (both at 100 μg/mL) coated bottom side of tissue culture inserts and bronchial epithelial cells were seeded (1 × 10
5 cells/cm
2) to the upper side of the membranes which were coated with rat tail collagen. CFBE cells were cultured for 10 days in the presence or absence of endothelial cells and were treated when the cell layer had reached steady resistance values. In the liquid-liquid interface (LLI) culture mode the volume of the medium in the upper compartment was 0.5 mL, in the lower compartment 1.5 mL. For culturing the cells at air-liquid interface (ALI) the medium was removed from the confluent bronchial epithelial cell layer after three days, while the endothelial cells in the bottom compartment received 1 mL fresh culture medium.
4.3. Treatments
To functionally validate the presence of wild-type and mutant CFTR channels in the respective cell lines an activator and an inhibitor of CFTR channels were tested on the monocultures. The stock solution (25 mM) of the CFTR channel activator 8-(4-chlorophenylthio)-cAMP (cAMP) was prepared in sterile distilled water and it was further diluted in culture medium at 250 µM final concentration for treatments. To avoid the degradation of cAMP RO 20 1724 (17.5 µM), a cAMP-specific phosphodiesterase 4 inhibitor was added to the treatment solution. For the stock solution (100 mM) the CFTR inhibitor 3-[(3-trifluoromethyl) phenyl]-5-[(4-carboxyphenyl) methylene]-2-thioxo-4-thiazolidinone (CFTRinh-172) was dissolved in dimethyl sulfoxide (DMSO), further diluted in culture medium and tested at 10 µM concentration also for 1 h. For both reagents, cell-layer integrity was monitored by electrical resistance and permeability measurements after 1 h of treatment.
To model inflammatory conditions, often observed in CF lung, the mono- and co-cultures were treated with a combination of TNF-α (50 ng/mL) and IL-1β (25 ng/mL) in both compartments of the inserts [
21]. After the 6-h cytokine treatment, cell-layer integrity was investigated by electrical resistance and permeability measurement.
To test the similarities and differences between the response of the CFBE cell lines to different treatments we also tested the effect of the PN159/KLAL peptide (NH
2-KLALKLALKALKAALKLA-amide). This peptide, which shows both cell-penetrating effects and opens intercellular junctions, has been characterized in detail by our research group [
22,
23]. The stock solution of the peptide (5 mM) was prepared in DMSO, the final concentration of the peptide in cell culture medium was 10 µM, as used in our previous studies. We treated the CFBE monoculture with the peptide for 30 min (only in upper compartments, where the apical junctional complex is situated). After the treatment medium was changed, TEER and permeability measurements were performed immediately and at the end of the 24 h recovery period.
To test the effect of sodium bicarbonate we prepared 50, 100 and 200 mM treatment solutions. The MEM culture medium already contains 26 mM sodium bicarbonate. To achieve a 500 mM stock solution we dissolved 200.5 mg NaHCO3 in 5 mL cell culture medium, then sterilized with a 0.2 µm syringe filter. The treatment solutions were diluted from the stock solution in culture medium and were freshly prepared before the experiments. For the viability assay 50, 100 and 200 mM concentrations, for the cell-layer integrity and intracellular pH measurements 100 mM concentration of bicarbonate were used (only upper, apical compartments were treated to mimic inhaled sodium bicarbonate treatment).
4.4. Measurement of the Electrical Resistance of CFBE Cell Layers
Transepithelial electrical resistance (TEER) indicates the integrity and paracellular permeability of cell layers for ions. TEER reflects the tightness of the intercellular junctions closing the paracellular cleft, therefore the overall tightness of cell layers of biological barriers. Before measuring the transepithelial electrical resistance (TEER) of the cell layers 500 µl of fresh medium was added to the upper and lower compartments. The measurement was performed after 15 min of equilibration time in the CO2 incubator. We used an EVOM volt-ohmmeter (World Precision Instruments, Sarasota, FL, USA) combined with STX-2 electrodes to determine TEER values which were expressed relative to the surface area of the monolayers as Ω × cm2. TEER of cell-free inserts (90–100 Ω × cm2) was subtracted from the measured data. The culture medium was changed and TEER was checked every second day. TEER values were measured before and right after the experiments.
4.5. Permeability Study on CFBE Cell Culture Model
To determine the tightness of the bronchial epithelial culture model two passive permeability marker molecules, fluorescein (10 μg/mL; Mw: 376 Da) and Evans-blue labeled albumin (167.5 μg/mL Evans-blue dye and 10 mg/mL bovine serum albumin; MW: 67.5 kDa) were tested [
22]. The inserts were transferred to 12-well plates containing 1.5 mL Ringer buffer in the acceptor (lower/basal) compartments. In the donor (upper/apical) compartments 0.5 mL buffer was pipetted containing both markers. To avoid the unstirred-water-layer effect, the plates were kept on a horizontal shaker (120 rpm) during the assay. The assay lasted for 30 or 60 min. The concentrations of the marker molecules in the samples from both compartments were determined by a fluorescence multiwell plate reader (Fluostar Optima, BMG Labtechnologies, Germany; fluorescein: 485 nm excitation wavelength, 520 nm emission wavelength; Evans-blue labeled albumin: 584 nm excitation wavelength, 680 nm emission wavelength).
The apparent permeability coefficients (P
app) were calculated as previously described [
23]. Briefly, cleared volume was calculated from the concentration difference of the tracer in the acceptor compartment (Δ[C]
A) after 30 or 60 min and in donor compartments at 0 h ([C]
D), the volume of the acceptor compartment (V
A; 1.5 mL) and the surface area available for permeability (A; 1.12 cm
2) using this equation:
4.6. Cell Viability Measurements
The kinetics of the epithelial cell reaction to the sodium bicarbonate treatment was monitored by impedance measurement at 10 kHz (RTCA-SP instrument, ACEA Biosciences, San Diego, CA, USA). Real-time cell electronic sensing is a non-invasive, label-free, impedance-based technique to quantify the kinetics of proliferation, viability and cellular reaction of adherent cells. This method to follow cell damage and/or protection in living barrier forming cells has been successfully used by our team in the last seven years [
23,
30]. The 96-well E-plates with built-in gold electrodes were coated with collagen type I (50 μg/mL) and dried for 30 min under sterile airflow. For background measurements 50 μL cell culture medium was added to the wells, then cells were seeded at a density of 1 × 10
4 cells/well to the coated 96-well plates with integrated gold electrodes (E-plate 96, ACEA Biosciences). Cells were cultured for 3–4 days in a CO
2 incubator at 37 °C and the impedance of cell layers was monitored every 10 min until the end of experiments by an RTCA-SP instrument (ACEA Biosciences). Cell index was defined as (Rn-Rb)/15, where Rn is the cell-electrode impedance of the well when it contains cells and Rb is the background impedance of the well with the medium alone. Cells were treated with culture medium containing sodium bicarbonate at 50, 100 and 200 mM concentrations at the beginning of the plateau phase of growth and the effects were followed for 24 h. Triton X-100 detergent (1 mg/mL) was used as a reference compound to induce cell death.
4.7. Immunohistochemistry
Immunostaining was used to evaluate morphological differences between the two CFBE cell lines when cultured in different conditions (mono- vs. co-culture, LLI vs. ALI) or treated by different concentrations of sodium bicarbonate. Cells were grown on culture inserts and after the treatments they were washed with phosphate buffered saline (PBS) and fixed with 3% paraformaldehyde solution for 15 min at room temperature. The cells were permeabilized by 0.2% TX-100 solution for 10 min and the nonspecific binding sites were blocked with 3% bovine serum albumin in PBS. Primary antibodies rabbit anti-ZO-1 (AB_138452, 1:400; Life Technologies, Carlsbad, CA, USA), rabbit anti-β-catenin (AB_476831, 1:400) and mouse anti-E-cadherin (AB_397580, 1:400; Life Technologies, Carlsbad, CA, USA) were applied as an overnight treatment. Incubation with secondary antibodies Alexa Fluor-488-labeled anti-mouse (AB_2534088, 1:400; Life Technologies, Invitrogen, USA) and anti-rabbit IgG conjugated with Cy3 (AB_258792, 1:400) lasted for 1 h. Hoechst dye 33342 was used to stain cell nuclei. After mounting the samples (Fluoromount-G; Southern Biotech, Birmingham, USA) staining was visualized by confocal laser scanning microscope (Olympus Fluoview FV1000, Olympus Life Science Europa GmbH, Hamburg, Germany).
We used double nuclei staining to evaluate cell-membrane damage. The blue Hoechst dye stains every cell nuclei and the red ethidium homodimer-1 dye stains only the nuclei of damaged cells [
23]. CFBE cells were cultured on collagen-coated glass coverslips (Menzel-Glaser, Braunschweig, Germany) in a 12-well plate. Cells were seeded at a 10
5 cells/well density and cultured for 3 days. Cell layers after reaching confluency were treated with culture medium containing 50, 100 and 200 mM bicarbonate for 24 h. Ethidium homodimer-1 dye (1 μM) was added to the cells in the last 30 min of the 24 h bicarbonate treatment. After a washing step with PBS the cells were fixed with aceton:methanol (1:1) solution for 3 min. Then the samples were processed for immunostaining for junctional proteins ZO-1 and β-catenin as described above.
The localization of CFTR channels in the CFBE cells was studied by concomitant staining of the samples with a rabbit polyclonal anti-CFTR antibody (Alomone Labs, Jerusalem, Israel; 1:400) and wheat germ agglutinin lectin (WGA) conjugated with Alexa fluor 488 (Invitrogen, W11261) which binds to the
n-acetyl-
d-glucosamine and sialic acid residues of the glycocalyx covering the cells’ surface. The cells were fixed with 3% paraformaldehyde, washed with PBS and incubated with WGA (5 µg/mL) in PBS for 10 min at room temperature [
43]. This step was followed by permeabilization with 0.2% TX-100 solution for 10 min, blocking with 3% bovine serum albumin in PBS for 1 h and incubation with the CFTR antibody overnight. After the washing steps cells were incubated with secondary antibody Cy3-conjugated anti-rabbit IgG for 1 h. Cell nuclei were stained with Hoechst dye 33342. Samples were mounted and the staining was visualized by a Leica TCS SP5 confocal laser scanning microscope (Leica Microsystems GmbH, Wetzlar, Germany).
The cellular localization of the CFTR channels was determined by image analysis (MATLAB, The Mathworks Inc, Natick, Ma, USA) performed on the immunostained CFBE image stacks. First, the image stacks of the WGA staining were used to determine the apical region of the imaged cells. For this the WGA image stacks were laterally smoothed by a 10-pixel-wide 2D Gaussian filter. Within each column (z-direction) of a smoothed WGA stack the cells’ apical membrane position was determined by finding the pixel position where the intensity first rises above 30% of the maximum value in the column, going from the top of the image stack (apical side). Next, based on the obtained apical contour of the cells, spatial masks were calculated for the cell membrane and cytoplasm. The membrane mask was calculated as a 12-pixels- (2.4 µm) wide stripe starting from the top edge of the cells. The cytoplasm mask was taken as the area between the bottom of the membrane mask and the bottom of the image stack (basal side). Finally, the spatial masks were used to calculate the mean pixel intensities of the CFTR staining in the membrane and cytoplasm regions (Video S1–S4). A total of 9 images were analyzed in each group consisting of 512 image stacks (4018 image stacks/group).
The intensity of junctional stainings (ZO-1, E-cadherin, β-catenin) were evaluated by image analysis (MATLAB, The Mathworks Inc, Natick, Ma, USA) performed on the immunostained images. A K-means clustering-based image segmentation function was used to segment the images into areas corresponding to the cytoplasm and the junctional regions. Based on the segmentation, spatial masks were obtained and used for calculating the mean pixel intensity in the junctional areas.
4.8. Measurement of Resting Intracellular pH
CFBE cells were cultured on rat tail collagen-coated glass coverslips (24 mm) in 35 mm cell culture dishes (Orange Scientific, Braine-l’Alleud, Belgium). The cells were seeded at 2 × 10
5 cells/dish density and cultured until confluency. Cells treated with 100 mM NaHCO
3 in cell culture medium were kept for 1 h in a CO
2 incubator at 37 °C. The cells were washed once with PBS and were loaded with the pH-sensitive fluorescent dye 2’,7’-bis-(2-carboxyethyl)-5-(and-6-) carboxyfluorescein-acetoxymethyl ester (BCECF-AM; Biotium Inc., Fremont, CA, USA) at 2 µM concentration in HEPES-buffered solution (HEPES-BS; 140 mM NaCl, 5 mM KCl, 1 mM CaCl
2, 1 mM MgCl
2, 10 mM D-glucose and 10 mM HEPES acid; pH = 7.4) at 37 °C in a wet chamber for 20–30 min. Thereafter, the cells on coverslips were continuously perfused at a rate of 1–1.5 mL/minute in a perfusion chamber (QE-1, Warner Instruments, Hamden, CT, USA) mounted on a Zeiss Axio Observer 7 microscope (Carl Zeiss Microscopy GmbH, Jena, Germany). The cells were excited with light at wavelengths of 495 (±3) nm and 436 (±10) nm, and the 495/436 fluorescence emission ratio was measured at 540 ± 20 nm to determine intracellular pH (pH
i). Images from fourteen to seventeen randomly selected cells (regions of interests, ROIs) were obtained by using the ZEN software (Carl Zeiss Microscopy GmbH). One measurement was recorded every two seconds. The 495/436 emission ratio of cells perfused with HEPES-BS was determined at the beginning of the measurements. To convert the emission ratio to resting pH
i, cells were perfused with a high K
+ HEPES-BS (K-HEPES-BS; 15 mM NaCl, 130 mM KCl, 1 mM CaCl
2, 1 mM MgCl
2, 10 mM
d-glucose, 10 mM HEPES acid; pH set between 6.0 and 8.4, and 10 µM nigericin sodium salt (Tocris Bioscience, Bristol, UK)) calibration solutions with known pH values [
44].
4.9. Statistical Analysis
All data presented are means ± SD. The values were compared using the analysis of variance followed by Dunett’s test or two-way ANOVA and Bonferroni post-hoc test posttest using GraphPad Prism 5.0 software (GraphPad Software Inc., San Diego, CA, USA). Changes were considered statistically significant at p < 0.05.

 ,
, 


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}












