Manipulation of Metabolic Pathways and Its Consequences for Anti-Tumor Immunity: A Clinical Perspective


Abstract
:1. The Tumor Microenvironment and Immune Checkpoint Inhibitors
2. The Implications of Metabolic Syndrome for Targeting Metabolic Pathways within the Tumor Microenvironment
3. The Metabolic Pathways in the Tumor Microenvironment: Glucose and Lipid
4. The Metabolic Pathways in the Tumor Microenvironment: Amino Acids and Metabolic Intermediates
5. The Effects of Exosomes and Immune Checkpoint Inhibitors on Metabolic Pathways
6. Conclusions
Funding
Conflicts of Interest
Abbreviations
| TME | tumor microenvironment |
| Treg | regulatory T |
| TGF | transforming growth factor |
| IL | interleukin |
| CTLA-4 | cytotoxic T-lymphocyte-associated antigen 4 |
| PD-1 | programmed death 1 |
| DC | dendritic cell |
| TAM | tumor-associated macrophage |
| ICI | immune checkpoint inhibitor |
| IGF | insulin-like growth factor |
| PI3K | phosphatidylinositol 3-kinase |
| APC | antigen-presenting cell |
| FAO | fatty acid oxidation |
| FAS | fatty acid synthesis |
| ACC1 | acetyl-CoA carboxylase 1 |
| ACAT | acetyl-CoA acetyltransferase |
| ASCT | alanine serine cysteine transporter |
| DON | 6-diazo-5-oxo-L-norleucine |
| IDO | indoleamine-2,3-dioxygenase |
| LDH | lactate dehydrogenase |
| OXPHOS | oxidative phosphorylation |
References
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.; Ertl, H.C. Starved and asphyxiated: How can CD8+ T cells within a tumor microenvironment prevent tumor progression. Front. Immunol. 2016, 7, 32. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gilkes, D.M.; Semenza, G.L.; Wirtz, D. Hypoxia and the extracellular matrix: Drivers of tumour metastasis. Nat. Rev. Cancer 2014, 14, 430–439. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meseure, D.; Alsibai, K.D.; Nicolas, A. Pivotal role of pervasive neoplastic and stromal cells reprogramming in circulating tumor cells dissemination and metastatic colonization. Cancer Microenviron. 2014, 7, 95–115. [Google Scholar] [CrossRef] [PubMed]
- Alkasalias, T.; Moyano-Galceran, L.; Arsenian-Henriksson, M.; Lehti, K. Fibroblasts in the tumor microenvironment: Shield or spear? Int. J. Mol. Sci. 2018, 19, 1532. [Google Scholar] [CrossRef] [Green Version]
- Poltavets, V.; Kochetkova, M.; Pitson, S.M.; Samuel, M.S. The role of the extracellular matrix and its molecular and cellular regulators in cancer cell plasticity. Front. Oncol. 2018, 8, 431. [Google Scholar] [CrossRef] [Green Version]
- Almendros, I.; Gileles-Hillel, A.; Khalyfa, A.; Wang, Y.; Zhang, S.X.; Carreras, A.; Farré, R.; Gozal, D. Adipose tissue macrophage polarization by intermittent hypoxia in a mouse model of OSA: Effect of tumor microenvironment. Cancer Lett. 2015, 361, 233–239. [Google Scholar] [CrossRef]
- Wang, M.; Zhao, J.; Zhang, L.; Wei, F.; Lian, Y.; Wu, Y.; Gong, Z.; Zhang, S.; Zhou, J.; Cao, K. Role of tumor microenvironment in tumorigenesis. J. Cancer 2017, 8, 761. [Google Scholar] [CrossRef]
- Ghajar, C.M.; Peinado, H.; Mori, H.; Matei, I.R.; Evason, K.J.; Brazier, H.; Almeida, D.; Koller, A.; Hajjar, K.A.; Stainier, D.Y. The perivascular niche regulates breast tumour dormancy. Nat. Cell Biol. 2013, 15, 807–817. [Google Scholar] [CrossRef]
- Dunn, G.P.; Bruce, A.T.; Ikeda, H.; Old, L.J.; Schreiber, R.D. Cancer immunoediting: From immunosurveillance to tumor escape. Nat. Immunol. 2002, 3, 991–998. [Google Scholar] [CrossRef] [PubMed]
- Fridman, W.H.; Pagès, F.; Sautes-Fridman, C.; Galon, J. The immune contexture in human tumours: Impact on clinical outcome. Nat. Rev. Cancer 2012, 12, 298–306. [Google Scholar] [CrossRef]
- Mellman, I.; Coukos, G.; Dranoff, G. Cancer immunotherapy comes of age. Nature 2011, 480, 480–489. [Google Scholar] [CrossRef] [PubMed]
- Balkwill, F.; Mantovani, A. Inflammation and cancer: Back to Virchow? Lancet 2001, 357, 539–545. [Google Scholar] [CrossRef]
- Nishikawa, H.; Sakaguchi, S. Regulatory T cells in cancer immunotherapy. Curr. Opin. Immunol. 2014, 27, 1–7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sakaguchi, S.; Yamaguchi, T.; Nomura, T.; Ono, M. Regulatory T cells and immune tolerance. Cell 2008, 133, 775–787. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- deLeeuw, R.J.; Kost, S.E.; Kakal, J.A.; Nelson, B.H. The prognostic value of FoxP3+ tumor-infiltrating lymphocytes in cancer: A critical review of the literature. Clin. Cancer Res. 2012, 18, 3022–3029. [Google Scholar] [CrossRef] [Green Version]
- Shevach, E.M. Mechanisms of foxp3+ T regulatory cell-mediated suppression. Immunity 2009, 30, 636–645. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schmidt, A.; Oberle, N.; Krammer, P.H. Molecular mechanisms of treg-mediated T cell suppression. Front. Immunol. 2012, 3, 51. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Engelhardt, J.J.; Boldajipour, B.; Beemiller, P.; Pandurangi, P.; Sorensen, C.; Werb, Z.; Egeblad, M.; Krummel, M.F. Marginating dendritic cells of the tumor microenvironment cross-present tumor antigens and stably engage tumor-specific T cells. Cancer Cell 2012, 21, 402–417. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cao, X.; Cai, S.F.; Fehniger, T.A.; Song, J.; Collins, L.I.; Piwnica-Worms, D.R.; Ley, T.J. Granzyme B and perforin are important for regulatory T cell-mediated suppression of tumor clearance. Immunity 2007, 27, 635–646. [Google Scholar] [CrossRef] [Green Version]
- Veldhoen, M.; Moncrieffe, H.; Hocking, R.J.; Atkins, C.J.; Stockinger, B. Modulation of dendritic cell function by naive and regulatory CD4+ T cells. J. Immunol. 2006, 176, 6202–6210. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grossman, W.J.; Verbsky, J.W.; Barchet, W.; Colonna, M.; Atkinson, J.P.; Ley, T.J. Human T regulatory cells can use the perforin pathway to cause autologous target cell death. Immunity 2004, 21, 589–601. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yan, Z.; Garg, S.K.; Banerjee, R. Regulatory T cells interfere with glutathione metabolism in dendritic cells and T cells. J. Biol. Chem. 2010, 285, 41525–41532. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vignali, D.A.; Collison, L.W.; Workman, C.J. How regulatory T cells work. Nat. Rev. Immunol. 2008, 8, 523–532. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, H.; Franco, F.; Ho, P.-C. Metabolic regulation of Tregs in cancer: Opportunities for immunotherapy. Trends Cancer 2017, 3, 583–592. [Google Scholar] [CrossRef]
- DeNardo, D.G.; Barreto, J.B.; Andreu, P.; Vasquez, L.; Tawfik, D.; Kolhatkar, N.; Coussens, L.M. CD4+ T cells regulate pulmonary metastasis of mammary carcinomas by enhancing protumor properties of macrophages. Cancer Cell 2009, 16, 91–102. [Google Scholar] [CrossRef] [Green Version]
- Shiao, S.L.; Ruffell, B.; DeNardo, D.G.; Faddegon, B.A.; Park, C.C.; Coussens, L.M. TH2-polarized CD4+ T cells and macrophages limit efficacy of radiotherapy. Cancer Immunol. Res. 2015, 3, 518–525. [Google Scholar] [CrossRef] [Green Version]
- De Monte, L.; Wörmann, S.; Brunetto, E.; Heltai, S.; Magliacane, G.; Reni, M.; Paganoni, A.M.; Recalde, H.; Mondino, A.; Falconi, M. Basophil recruitment into tumor-draining lymph nodes correlates with Th2 inflammation and reduced survival in pancreatic cancer patients. Cancer Res. 2016, 76, 1792–1803. [Google Scholar] [CrossRef] [Green Version]
- Kumar, V.; Cheng, P.; Condamine, T.; Mony, S.; Languino, L.R.; McCaffrey, J.C.; Hockstein, N.; Guarino, M.; Masters, G.; Penman, E. CD45 phosphatase inhibits STAT3 transcription factor activity in myeloid cells and promotes tumor-associated macrophage differentiation. Immunity 2016, 44, 303–315. [Google Scholar] [CrossRef] [Green Version]
- Franklin, R.A.; Liao, W.; Sarkar, A.; Kim, M.V.; Bivona, M.R.; Liu, K.; Pamer, E.G.; Li, M.O. The cellular and molecular origin of tumor-associated macrophages. Science 2014, 344, 921–925. [Google Scholar] [CrossRef] [Green Version]
- Sangaletti, S.; Di Carlo, E.; Gariboldi, S.; Miotti, S.; Cappetti, B.; Parenza, M.; Rumio, C.; Brekken, R.A.; Chiodoni, C.; Colombo, M.P. Macrophage-derived SPARC bridges tumor cell-extracellular matrix interactions toward metastasis. Cancer Res. 2008, 68, 9050–9059. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nucera, S.; Biziato, D.; De Palma, M. The interplay between macrophages and angiogenesis in development, tissue injury and regeneration. Int. J. Dev. Biol. 2011, 55, 495–503. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Noy, R.; Pollard, J.W. Tumor-associated macrophages: From mechanisms to therapy. Immunity 2014, 41, 49–61. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mantovani, A.; Marchesi, F.; Malesci, A.; Laghi, L.; Allavena, P. Tumour-associated macrophages as treatment targets in oncology. Nat. Rev. Clin. Oncol. 2017, 14, 399. [Google Scholar] [CrossRef] [PubMed]
- Johnstone, R.M.; Adam, M.; Hammond, J.; Orr, L.; Turbide, C. Vesicle formation during reticulocyte maturation. Association of plasma membrane activities with released vesicles (exosomes). J. Biol. Chem. 1987, 262, 9412–9420. [Google Scholar]
- Kim, H.K.; Song, K.; Park, Y.; Kang, Y.; Lee, Y.; Lee, K.; Ryu, K.; Bae, J.; Kim, S. Elevated levels of circulating platelet microparticles, VEGF, IL-6 and RANTES in patients with gastric cancer: Possible role of a metastasis predictor. Eur. J. Cancer 2003, 39, 184–191. [Google Scholar] [CrossRef]
- Kharaziha, P.; Ceder, S.; Li, Q.; Panaretakis, T. Tumor cell-derived exosomes: A message in a bottle. Biochim. Et Biophys. Acta (Bba)-Rev. Cancer 2012, 1826, 103–111. [Google Scholar] [CrossRef]
- Peinado, H.; Alečković, M.; Lavotshkin, S.; Matei, I.; Costa-Silva, B.; Moreno-Bueno, G.; Hergueta-Redondo, M.; Williams, C.; García-Santos, G.; Ghajar, C.M. Melanoma exosomes educate bone marrow progenitor cells toward a pro-metastatic phenotype through MET. Nat. Med. 2012, 18, 883. [Google Scholar] [CrossRef] [Green Version]
- Quail, D.F.; Joyce, J.A. Microenvironmental regulation of tumor progression and metastasis. Nat. Med. 2013, 19, 1423. [Google Scholar] [CrossRef]
- Zhang, H.-G.; Grizzle, W.E. Exosomes: A novel pathway of local and distant intercellular communication that facilitates the growth and metastasis of neoplastic lesions. Am. J. Pathol. 2014, 184, 28–41. [Google Scholar] [CrossRef] [Green Version]
- Gatalica, Z.; Snyder, C.; Maney, T.; Ghazalpour, A.; Holterman, D.A.; Xiao, N.; Overberg, P.; Rose, I.; Basu, G.D.; Vranic, S. Programmed cell death 1 (PD-1) and its ligand (PD-L1) in common cancers and their correlation with molecular cancer type. Cancer Epidemiol. Prev. Biomark. 2014, 23, 2965–2970. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, A.; Wang, H.; Liu, Y.; Zhao, M.; Zhang, H.; Lu, Z.; Fang, Y.; Chen, X.; Liu, G. The prognostic value of PD-L1 expression for non-small cell lung cancer patients: A meta-analysis. Eur. J. Surg. Oncol. (Ejso) 2015, 41, 450–456. [Google Scholar] [CrossRef] [PubMed]
- Muenst, S.; Schaerli, A.; Gao, F.; Däster, S.; Trella, E.; Droeser, R.; Muraro, M.; Zajac, P.; Zanetti, R.; Gillanders, W. Expression of programmed death ligand 1 (PD-L1) is associated with poor prognosis in human breast cancer. Breast Cancer Res. Treat. 2014, 146, 15–24. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kuang, D.-M.; Zhao, Q.; Peng, C.; Xu, J.; Zhang, J.-P.; Wu, C.; Zheng, L. Activated monocytes in peritumoral stroma of hepatocellular carcinoma foster immune privilege and disease progression through PD-L1. J. Exp. Med. 2009, 206, 1327–1337. [Google Scholar] [CrossRef] [PubMed]
- Topalian, S.L.; Drake, C.G.; Pardoll, D.M. Immune checkpoint blockade: A common denominator approach to cancer therapy. Cancer Cell 2015, 27, 450–461. [Google Scholar] [CrossRef] [Green Version]
- Curiel, T.J.; Coukos, G.; Zou, L.; Alvarez, X.; Cheng, P.; Mottram, P.; Evdemon-Hogan, M.; Conejo-Garcia, J.R.; Zhang, L.; Burow, M. Specific recruitment of regulatory T cells in ovarian carcinoma fosters immune privilege and predicts reduced survival. Nat. Med. 2004, 10, 942–949. [Google Scholar] [CrossRef]
- Han, Y.; Yang, Y.; Chen, Z.; Jiang, Z.; Gu, Y.; Liu, Y.; Xu, S.; Lin, C.; Pan, Z.; Zhou, W. Human hepatocellular carcinoma-infiltrating CD4+ CD69+ Foxp3− regulatory T cell suppresses T cell response via membrane-bound TGF-β1. J. Mol. Med. 2014, 92, 539–550. [Google Scholar] [CrossRef]
- Borghaei, H.; Paz-Ares, L.; Horn, L.; Spigel, D.R.; Steins, M.; Ready, N.E.; Chow, L.Q.; Vokes, E.E.; Felip, E.; Holgado, E. Nivolumab versus docetaxel in advanced nonsquamous non–small-cell lung cancer. New Engl. J. Med. 2015, 373, 1627–1639. [Google Scholar] [CrossRef]
- Garon, E.B.; Rizvi, N.A.; Hui, R.; Leighl, N.; Balmanoukian, A.S.; Eder, J.P.; Patnaik, A.; Aggarwal, C.; Gubens, M.; Horn, L. Pembrolizumab for the treatment of non–small-cell lung cancer. New Engl. J. Med. 2015, 372, 2018–2028. [Google Scholar] [CrossRef]
- Hodi, F.S.; O’Day, S.J.; McDermott, D.F.; Weber, R.W.; Sosman, J.A.; Haanen, J.B.; Gonzalez, R.; Robert, C.; Schadendorf, D.; Hassel, J.C. Improved survival with ipilimumab in patients with metastatic melanoma. New Engl. J. Med. 2010, 363, 711–723. [Google Scholar] [CrossRef]
- Larkin, J.; Chiarion-Sileni, V.; Gonzalez, R.; Grob, J.J.; Cowey, C.L.; Lao, C.D.; Schadendorf, D.; Dummer, R.; Smylie, M.; Rutkowski, P. Combined nivolumab and ipilimumab or monotherapy in untreated melanoma. New Engl. J. Med. 2015, 373, 23–34. [Google Scholar] [CrossRef] [Green Version]
- Schadendorf, D.; Hodi, F.S.; Robert, C.; Weber, J.S.; Margolin, K.; Hamid, O.; Patt, D.; Chen, T.-T.; Berman, D.M.; Wolchok, J.D. Pooled analysis of long-term survival data from phase II and phase III trials of ipilimumab in unresectable or metastatic melanoma. J. Clin. Oncol. 2015, 33, 1889. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Robert, C.; Thomas, L.; Bondarenko, I.; O’Day, S.; Weber, J.; Garbe, C.; Lebbe, C.; Baurain, J.-F.; Testori, A.; Grob, J.-J. Ipilimumab plus dacarbazine for previously untreated metastatic melanoma. New Engl. J. Med. 2011, 364, 2517–2526. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jenkins, R.W.; Barbie, D.A.; Flaherty, K.T. Mechanisms of resistance to immune checkpoint inhibitors. Br. J. Cancer 2018, 118, 9–16. [Google Scholar] [CrossRef] [Green Version]
- Darvin, P.; Toor, S.M.; Nair, V.S.; Elkord, E. Immune checkpoint inhibitors: Recent progress and potential biomarkers. Exp. Mol. Med. 2018, 50, 1–11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aguilar, M.; Bhuket, T.; Torres, S.; Liu, B.; Wong, R.J. Prevalence of the metabolic syndrome in the United States, 2003-2012. Jama 2015, 313, 1973–1974. [Google Scholar] [CrossRef]
- Ranasinghe, P.; Mathangasinghe, Y.; Jayawardena, R.; Hills, A.; Misra, A. Prevalence and trends of metabolic syndrome among adults in the asia-pacific region: A systematic review. Bmc Public Health 2017, 17, 101. [Google Scholar] [CrossRef] [Green Version]
- Dunkley, A.; Charles, K.; Gray, L.; Camosso-Stefinovic, J.; Davies, M.; Khunti, K. Effectiveness of interventions for reducing diabetes and cardiovascular disease risk in people with metabolic syndrome: Systematic review and mixed treatment comparison meta-analysis. Diabetesobesity Metab. 2012, 14, 616–625. [Google Scholar] [CrossRef]
- Lorenzo, C.; Okoloise, M.; Williams, K.; Stern, M.P.; Haffner, S.M. The metabolic syndrome as predictor of type 2 diabetes: The San Antonio heart study. Diabetes Care 2003, 26, 3153–3159. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grundy, S.M. Metabolic syndrome: Connecting and reconciling cardiovascular and diabetes worlds. J. Am. Coll. Cardiol. 2006, 47, 1093–1100. [Google Scholar] [CrossRef] [Green Version]
- Wilson, P.W.; D’Agostino, R.B.; Parise, H.; Sullivan, L.; Meigs, J.B. Metabolic syndrome as a precursor of cardiovascular disease and type 2 diabetes mellitus. Circulation 2005, 112, 3066–3072. [Google Scholar] [CrossRef] [PubMed]
- Arnold, M.; Pandeya, N.; Byrnes, G.; Renehan, A.G.; Stevens, G.A.; Ezzati, M.; Ferlay, J.; Miranda, J.J.; Romieu, I.; Dikshit, R. Global burden of cancer attributable to high body-mass index in 2012: A population-based study. Lancet Oncol. 2015, 16, 36–46. [Google Scholar] [CrossRef]
- Calle, E.E.; Kaaks, R. Overweight, obesity and cancer: Epidemiological evidence and proposed mechanisms. Nat. Rev. Cancer 2004, 4, 579–591. [Google Scholar] [CrossRef]
- Pearson-Stuttard, J.; Zhou, B.; Kontis, V.; Bentham, J.; Gunter, M.J.; Ezzati, M. Retracted: Worldwide Burden of Cancer Attributable to Diabetes and High Body-Mass Index: A Comparative Risk Assessment; Elsevier: Amsterdam, The Netherlands, 2018. [Google Scholar]
- Calle, E.E.; Rodriguez, C.; Walker-Thurmond, K.; Thun, M.J. Overweight, obesity, and mortality from cancer in a prospectively studied cohort of US adults. New Engl. J. Med. 2003, 348, 1625–1638. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barone, B.B.; Yeh, H.-C.; Snyder, C.F.; Peairs, K.S.; Stein, K.B.; Derr, R.L.; Wolff, A.C.; Brancati, F.L. Long-term all-cause mortality in cancer patients with preexisting diabetes mellitus: A systematic review and meta-analysis. Jama 2008, 300, 2754–2764. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lipscombe, L.L.; Goodwin, P.J.; Zinman, B.; McLaughlin, J.R.; Hux, J.E. The impact of diabetes on survival following breast cancer. Breast Cancer Res. Treat. 2008, 109, 389–395. [Google Scholar] [CrossRef]
- Chen, Y.; Wu, F.; Saito, E.; Lin, Y.; Song, M.; Luu, H.N.; Gupta, P.C.; Sawada, N.; Tamakoshi, A.; Shu, X.-O. Association between type 2 diabetes and risk of cancer mortality: A pooled analysis of over 771,000 individuals in the Asia Cohort Consortium. Diabetologia 2017, 60, 1022–1032. [Google Scholar] [CrossRef]
- Vigneri, P.; Frasca, F.; Sciacca, L.; Pandini, G.; Vigneri, R. Diabetes and cancer. Endocr. -Relat. Cancer 2009, 16, 1103–1123. [Google Scholar] [CrossRef] [Green Version]
- Warburg, O. On the origin of cancer cells. Science 1956, 123, 309–314. [Google Scholar] [CrossRef]
- Catrina, S.-B.; Okamoto, K.; Pereira, T.; Brismar, K.; Poellinger, L. Hyperglycemia regulates hypoxia-inducible factor-1α protein stability and function. Diabetes 2004, 53, 3226–3232. [Google Scholar] [CrossRef] [Green Version]
- Ma, J.; Li, H.; Giovannucci, E.; Mucci, L.; Qiu, W.; Nguyen, P.L.; Gaziano, J.M.; Pollak, M.; Stampfer, M.J. Prediagnostic body-mass index, plasma C-peptide concentration, and prostate cancer-specific mortality in men with prostate cancer: A long-term survival analysis. Lancet Oncol. 2008, 9, 1039–1047. [Google Scholar] [CrossRef] [Green Version]
- Wolpin, B.M.; Meyerhardt, J.A.; Chan, A.T.; Ng, K.; Chan, J.A.; Wu, K.; Pollak, M.N.; Giovannucci, E.L.; Fuchs, C.S. Insulin, the insulin-like growth factor axis, and mortality in patients with nonmetastatic colorectal cancer. J. Clin. Oncol. 2009, 27, 176. [Google Scholar] [CrossRef] [PubMed]
- Perseghin, G.; Calori, G.; Lattuada, G.; Ragogna, F.; Dugnani, E.; Garancini, M.P.; Crosignani, P.; Villa, M.; Bosi, E.; Ruotolo, G. Insulin resistance/hyperinsulinemia and cancer mortality: The Cremona study at the 15th year of follow-up. Acta Diabetol. 2012, 49, 421–428. [Google Scholar] [CrossRef] [PubMed]
- Tsujimoto, T.; Kajio, H.; Sugiyama, T. Association between hyperinsulinemia and increased risk of cancer death in nonobese and obese people: A population-based observational study. Int. J. Cancer 2017, 141, 102–111. [Google Scholar] [CrossRef] [PubMed]
- Coppack, S.W. Pro-inflammatory cytokines and adipose tissue. Proc. Nutr. Soc. 2001, 60, 349–356. [Google Scholar] [CrossRef] [Green Version]
- Weisberg, S.P.; McCann, D.; Desai, M.; Rosenbaum, M.; Leibel, R.L.; Ferrante, A.W. Obesity is associated with macrophage accumulation in adipose tissue. J. Clin. Investig. 2003, 112, 1796–1808. [Google Scholar] [CrossRef]
- Pollak, M. Insulin and insulin-like growth factor signalling in neoplasia. Nat. Rev. Cancer 2008, 8, 915–928. [Google Scholar] [CrossRef]
- Brahmkhatri, V.P.; Prasanna, C.; Atreya, H.S. Insulin-like growth factor system in cancer: Novel targeted therapies. Biomed Res. Int. 2015, 2015. [Google Scholar] [CrossRef] [Green Version]
- Le Roith, D. Insulin-like growth factors. New Engl. J. Med. 1997, 336, 633–640. [Google Scholar] [CrossRef]
- Rinderknecht, E.; Humbel, R.E. The amino acid sequence of human insulin-like growth factor I and its structural homology with proinsulin. J. Biol. Chem. 1978, 253, 2769–2776. [Google Scholar]
- LeRoith, D.; Werner, H.; Beitner-Johnson, D.; Roberts, C.T., Jr. Molecular and cellular aspects of the insulin-like growth factor I receptor. Endocr. Rev. 1995, 16, 143–163. [Google Scholar] [CrossRef]
- Kim, J.J.; Accili, D. Signalling through IGF-I and insulin receptors: Where is the specificity? Growth Horm. Igf Res. 2002, 12, 84–90. [Google Scholar] [CrossRef]
- Chitnis, M.M.; Yuen, J.S.; Protheroe, A.S.; Pollak, M.; Macaulay, V.M. The type 1 insulin-like growth factor receptor pathway. Clin. Cancer Res. 2008, 14, 6364–6370. [Google Scholar] [CrossRef] [Green Version]
- Hakuno, F.; Takahashi, S.-I. 40 years of IGF1: IGF1 receptor signaling pathways. J. Mol. Endocrinol. 2018, 61, T69–T86. [Google Scholar] [CrossRef] [Green Version]
- Robey, R.B.; Hay, N. Is Akt the “Warburg kinase”?—Akt-energy metabolism interactions and oncogenesis. Semin. Cancer Biol. 2009, 19, 25–31. [Google Scholar] [CrossRef] [Green Version]
- Cully, M.; You, H.; Levine, A.J.; Mak, T.W. Beyond PTEN mutations: The PI3K pathway as an integrator of multiple inputs during tumorigenesis. Nat. Rev. Cancer 2006, 6, 184–192. [Google Scholar] [CrossRef]
- Dai, N.; Ji, F.; Wright, J.; Minichiello, L.; Sadreyev, R.; Avruch, J. IGF2 mRNA binding protein-2 is a tumor promoter that drives cancer proliferation through its client mRNAs IGF2 and HMGA1. Elife 2017, 6, e27155. [Google Scholar] [CrossRef]
- Shukla, A.; Grisouard, J.; Ehemann, V.; Hermani, A.; Enzmann, H.; Mayer, D. Analysis of signaling pathways related to cell proliferation stimulated by insulin analogs in human mammary epithelial cell lines. Endocr. -Relat. Cancer 2009, 16, 429–441. [Google Scholar] [CrossRef] [Green Version]
- Shen, K.; Cui, D.; Sun, L.; Lu, Y.; Han, M.; Liu, J. Inhibition of IGF-IR increases chemosensitivity in human colorectal cancer cells through MRP-2 promoter suppression. J. Cell. Biochem. 2012, 113, 2086–2097. [Google Scholar] [CrossRef]
- Rochester, M.A.; Riedemann, J.; Hellawell, G.O.; Brewster, S.F.; Macaulay, V.M. Silencing of the IGF1R gene enhances sensitivity to DNA-damaging agents in both PTEN wild-type and mutant human prostate cancer. Cancer Gene Ther. 2005, 12, 90–100. [Google Scholar] [CrossRef]
- Chitnis, M.M.; Lodhia, K.A.; Aleksic, T.; Gao, S.; Protheroe, A.S.; Macaulay, V.M. IGF-1R inhibition enhances radiosensitivity and delays double-strand break repair by both non-homologous end-joining and homologous recombination. Oncogene 2014, 33, 5262–5273. [Google Scholar] [CrossRef] [Green Version]
- Ireland, L.; Santos, A.; Ahmed, M.S.; Rainer, C.; Nielsen, S.R.; Quaranta, V.; Weyer-Czernilofsky, U.; Engle, D.D.; Perez-Mancera, P.A.; Coupland, S.E. Chemoresistance in pancreatic cancer is driven by stroma-derived insulin-like growth factors. Cancer Res. 2016, 76, 6851–6863. [Google Scholar] [CrossRef] [Green Version]
- Nowak-Sliwinska, P.; van Beijnum, J.R.; Huijbers, E.J.; Gasull, P.C.; Mans, L.; Bex, A.; Griffioen, A.W. Oncofoetal insulin receptor isoform A marks the tumour endothelium; an underestimated pathway during tumour angiogenesis and angiostatic treatment. Br. J. Cancer 2019, 120, 218–228. [Google Scholar] [CrossRef] [Green Version]
- Ooi, G.T.; Tseng, L.; Tran, M.Q.; Rechler, M.M. Insulin rapidly decreases insulin-like growth factor-binding protein-1 gene transcription in streptozotocin-diabetic rats. Mol. Endocrinol. 1992, 6, 2219–2228. [Google Scholar]
- Powell, D.R.; Suwanichkul, A.; Cubbage, M.L.; DePaolis, L.A.; Snuggs, M.B.; Lee, P. Insulin inhibits transcription of the human gene for insulin-like growth factor-binding protein-1. J. Biol. Chem. 1991, 266, 18868–18876. [Google Scholar]
- Renehan, A.G.; Frystyk, J.; Flyvbjerg, A. Obesity and cancer risk: The role of the insulin–IGF axis. Trends Endocrinol. Metab. 2006, 17, 328–336. [Google Scholar] [CrossRef]
- Weinstein, D.; Simon, M.; Yehezkel, E.; Laron, Z.; Werner, H. Insulin analogues display IGF-I-like mitogenic and anti-apoptotic activities in cultured cancer cells. Diabetes/Metab. Res. Rev. 2009, 25, 41–49. [Google Scholar] [CrossRef]
- van Niekerk, G.; Christowitz, C.; Conradie, D.; Engelbrecht, A.-M. Insulin as an immunomodulatory hormone. Cytokine Growth Factor Rev. 2019. [Google Scholar] [CrossRef]
- Wensveen, F.M.; Šestan, M.; Turk Wensveen, T.; Polić, B. ‘Beauty and the beast’in infection: How immune–endocrine interactions regulate systemic metabolism in the context of infection. Eur. J. Immunol. 2019, 49, 982–995. [Google Scholar] [CrossRef] [Green Version]
- Bilbao, D.; Luciani, L.; Johannesson, B.; Piszczek, A.; Rosenthal, N. Insulin-like growth factor-1 stimulates regulatory T cells and suppresses autoimmune disease. Embo Mol. Med. 2014, 6, 1423–1435. [Google Scholar] [CrossRef]
- Johannesson, B.; Sattler, S.; Semenova, E.; Pastore, S.; Kennedy-Lydon, T.M.; Sampson, R.D.; Schneider, M.D.; Rosenthal, N.; Bilbao, D. Insulin-like growth factor-1 induces regulatory T cell-mediated suppression of allergic contact dermatitis in mice. Dis. Models Mech. 2014, 7, 977–985. [Google Scholar] [CrossRef] [Green Version]
- Miyagawa, I.; Nakayamada, S.; Nakano, K.; Yamagata, K.; Sakata, K.; Yamaoka, K.; Tanaka, Y. Induction of regulatory T cells and its regulation with insulin-like growth factor/insulin-like growth factor binding protein-4 by human mesenchymal stem cells. J. Immunol. 2017, 199, 1616–1625. [Google Scholar] [CrossRef] [Green Version]
- Kooijman, R.; Coppens, A. Insulin-like growth factor-I stimulates IL-10 production in human T cells. J. Leukoc. Biol. 2004, 76, 862–867. [Google Scholar] [CrossRef]
- Lu, M.C.; Yu, C.L.; Chen, H.C.; Yu, H.C.; Huang, H.B.; Lai, N.S. Increased miR-223 expression in T cells from patients with rheumatoid arthritis leads to decreased insulin-like growth factor-1-mediated interleukin-10 production. Clin. Exp. Immunol. 2014, 177, 641–651. [Google Scholar] [CrossRef]
- Barrett, J.P.; Minogue, A.M.; Falvey, A.; Lynch, M.A. Involvement of IGF-1 and Akt in M1/M2 activation state in bone marrow-derived macrophages. Exp. Cell Res. 2015, 335, 258–268. [Google Scholar] [CrossRef]
- Schabbauer, G.; Sharif, O.; Brunner, J.S.; Vogel, A. Macrophage rewiring by nutrient associated PI3K dependent pathways. Front. Immunol. 2019, 10, 2002. [Google Scholar]
- Ma, J.; Giovannucci, E.; Pollak, M.; Leavitt, A.; Tao, Y.; Gaziano, J.M.; Stampfer, M.J. A prospective study of plasma C-peptide and colorectal cancer risk in men. J. Natl. Cancer Inst. 2004, 96, 546–553. [Google Scholar] [CrossRef] [Green Version]
- Goodwin, P.J.; Ennis, M.; Pritchard, K.I.; Trudeau, M.E.; Koo, J.; Madarnas, Y.; Hartwick, W.; Hoffman, B.; Hood, N. Fasting insulin and outcome in early-stage breast cancer: Results of a prospective cohort study. J. Clin. Oncol. 2002, 20, 42–51. [Google Scholar] [CrossRef]
- Cummings, D.E.; Overduin, J.; Foster-Schubert, K.E. Gastric bypass for obesity: Mechanisms of weight loss and diabetes resolution. J. Clin. Endocrinol. Metab. 2004, 89, 2608–2615. [Google Scholar] [CrossRef]
- Lau, D.C.; Teoh, H. Benefits of modest weight loss on the management of type 2 diabetes mellitus. Can. J. Diabetes 2013, 37, 128–134. [Google Scholar] [CrossRef]
- Pendyala, S.; Neff, L.M.; Suarez-Farinas, M.; Holt, P.R. Diet-induced weight loss reduces colorectal inflammation: Implications for colorectal carcinogenesis. Am. J. Clin. Nutr. 2011, 93, 234–242. [Google Scholar] [CrossRef] [Green Version]
- Rapp, K.; Klenk, J.; Ulmer, H.; Concin, H.; Diem, G.; Oberaigner, W.; Schroeder, J. Weight change and cancer risk in a cohort of more than 65 000 adults in Austria. Ann. Oncol. 2008, 19, 641–648. [Google Scholar] [CrossRef] [PubMed]
- Sjöström, L.; Gummesson, A.; Sjöström, C.D.; Narbro, K.; Peltonen, M.; Wedel, H.; Bengtsson, C.; Bouchard, C.; Carlsson, B.; Dahlgren, S. Effects of bariatric surgery on cancer incidence in obese patients in Sweden (Swedish Obese Subjects Study): A prospective, controlled intervention trial. Lancet Oncol. 2009, 10, 653–662. [Google Scholar] [CrossRef]
- Bai, Y.; Sun, Q. Macrophage recruitment in obese adipose tissue. Obes. Rev. 2015, 16, 127–136. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Park, E.J.; Lee, J.H.; Yu, G.-Y.; He, G.; Ali, S.R.; Holzer, R.G.; Österreicher, C.H.; Takahashi, H.; Karin, M. Dietary and genetic obesity promote liver inflammation and tumorigenesis by enhancing IL-6 and TNF expression. Cell 2010, 140, 197–208. [Google Scholar] [CrossRef] [Green Version]
- Ceccarini, G.; Pelosini, C.; Ferrari, F.; Magno, S.; Vitti, J.; Salvetti, G.; Moretto, C.; Marioni, A.; Buccianti, P.; Piaggi, P. Serum IGF-binding protein 2 (IGFBP-2) concentrations change early after gastric bypass bariatric surgery revealing a possible marker of leptin sensitivity in obese subjects. Endocrine 2019, 65, 86–93. [Google Scholar] [CrossRef]
- Bailey, C.J. Metformin: Historical overview. Diabetologia 2017, 60, 1566–1576. [Google Scholar] [CrossRef] [Green Version]
- Kay, J.P.; Alemzadeh, R.; Langley, G.; D’angelo, L.; Smith, P.; Holshouser, S. Beneficial effects of metformin in normoglycemic morbidly obese adolescents. Metab. -Clin. Exp. 2001, 50, 1457–1461. [Google Scholar] [CrossRef]
- Fontbonne, A.; Charles, M.A.; Juhan-Vague, I.; Bard, J.M.; André, P.; Isnard, F.; Cohen, J.M.; Grandmottet, P.; Vague, P.; Safar, M.E. The effect of metformin on the metabolic abnormalities associated with upper-body fat distribution. BIGPRO Study Group. Diabetes Care 1996, 19, 920–926. [Google Scholar] [CrossRef]
- Zakikhani, M.; Dowling, R.; Fantus, I.G.; Sonenberg, N.; Pollak, M. Metformin is an AMP kinase–dependent growth inhibitor for breast cancer cells. Cancer Res. 2006, 66, 10269–10273. [Google Scholar] [CrossRef] [Green Version]
- Alimova, I.N.; Liu, B.; Fan, Z.; Edgerton, S.M.; Dillon, T.; Lind, S.E.; Thor, A.D. Metformin inhibits breast cancer cell growth, colony formation and induces cell cycle arrest in vitro. Cell Cycle 2009, 8, 909–915. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, B.; Fan, Z.; Edgerton, S.M.; Deng, X.-S.; Alimova, I.N.; Lind, S.E.; Thor, A.D. Metformin induces unique biological and molecular responses in triple negative breast cancer cells. Cell Cycle 2009, 8, 2031–2040. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dowling, R.J.; Zakikhani, M.; Fantus, I.G.; Pollak, M.; Sonenberg, N. Metformin inhibits mammalian target of rapamycin–dependent translation initiation in breast cancer cells. Cancer Res. 2007, 67, 10804–10812. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Algire, C.; Zakikhani, M.; Blouin, M.-J.; Shuai, J.H.; Pollak, M. Metformin attenuates the stimulatory effect of a high-energy diet on in vivo LLC1 carcinoma growth. Endocr. -Relat. Cancer 2008, 15, 833–839. [Google Scholar] [CrossRef]
- Algire, C.; Amrein, L.; Bazile, M.; David, S.; Zakikhani, M.; Pollak, M. Diet and tumor LKB1 expression interact to determine sensitivity to anti-neoplastic effects of metformin in vivo. Oncogene 2011, 30, 1174–1182. [Google Scholar] [CrossRef] [Green Version]
- Algire, C.; Amrein, L.; Zakikhani, M.; Panasci, L.; Pollak, M. Metformin blocks the stimulative effect of a high-energy diet on colon carcinoma growth in vivo and is associated with reduced expression of fatty acid synthase. Endocr. -Relat. Cancer 2010, 17, 351. [Google Scholar] [CrossRef] [Green Version]
- Gallagher, E.J.; LeRoith, D. Minireview: IGF, insulin, and cancer. Endocrinology 2011, 152, 2546–2551. [Google Scholar] [CrossRef] [Green Version]
- Tsugane, S.; Inoue, M. Insulin resistance and cancer: Epidemiological evidence. Cancer Sci. 2010, 101, 1073–1079. [Google Scholar] [CrossRef]
- Evans, J.M.; Donnelly, L.A.; Emslie-Smith, A.M.; Alessi, D.R.; Morris, A.D. Metformin and reduced risk of cancer in diabetic patients. Bmj 2005, 330, 1304–1305. [Google Scholar] [CrossRef] [Green Version]
- Bowker, S.L.; Majumdar, S.R.; Veugelers, P.; Johnson, J.A. Increased cancer-related mortality for patients with type 2 diabetes who use sulfonylureas or insulin. Diabetes Care 2006, 29, 254–258. [Google Scholar] [CrossRef] [Green Version]
- Currie, C.; Poole, C.; Gale, E. The influence of glucose-lowering therapies on cancer risk in type 2 diabetes. Diabetologia 2009, 52, 1766–1777. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Monami, M.; Lamanna, C.; Balzi, D.; Marchionni, N.; Mannucci, E. Sulphonylureas and cancer: A case–control study. Acta Diabetol. 2009, 46, 279. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wright, J.L.; Stanford, J.L. Metformin use and prostate cancer in Caucasian men: Results from a population-based case–control study. Cancer Causes Control 2009, 20, 1617. [Google Scholar] [CrossRef] [Green Version]
- Landman, G.W.; Kleefstra, N.; van Hateren, K.J.; Groenier, K.H.; Gans, R.O.; Bilo, H.J. Metformin associated with lower cancer mortality in type 2 diabetes: ZODIAC-16. Diabetes Care 2010, 33, 322–326. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jonasson, J.; Ljung, R.; Talbäck, M.; Haglund, B.; Gudbjörnsdòttir, S.; Steineck, G. Insulin glargine use and short-term incidence of malignancies—a population-based follow-up study in Sweden. Diabetologia 2009, 52, 1745–1754. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Colhoun, H. Use of insulin glargine and cancer incidence in Scotland: A study from the Scottish Diabetes Research Network Epidemiology Group. Diabetologia 2009, 52, 1755–1765. [Google Scholar] [CrossRef] [Green Version]
- Hemkens, L.G.; Grouven, U.; Bender, R.; Günster, C.; Gutschmidt, S.; Selke, G.W.; Sawicki, P.T. Risk of malignancies in patients with diabetes treated with human insulin or insulin analogues: A cohort study. Diabetologia 2009, 52, 1732–1744. [Google Scholar] [CrossRef] [Green Version]
- DeCensi, A.; Puntoni, M.; Goodwin, P.; Cazzaniga, M.; Gennari, A.; Bonanni, B.; Gandini, S. Metformin and cancer risk in diabetic patients: A systematic review and meta-analysis. Cancer Prev. Res. 2010, 3, 1451–1461. [Google Scholar] [CrossRef] [Green Version]
- Jabbour, M. Primary care physicians and insulin initiation: Multiple barriers, lack of knowledge or both? Int. J. Clin. Pract. 2008, 62, 845–847. [Google Scholar] [CrossRef] [Green Version]
- Kurtzhals, P.; Schäffer, L.; Sørensen, A.; Kristensen, C.; Jonassen, I.; Schmid, C.; Trüb, T. Correlations of receptor binding and metabolic and mitogenic potencies of insulin analogs designed for clinical use. Diabetes 2000, 49, 999–1005. [Google Scholar] [CrossRef] [Green Version]
- Liefvendahl, E.; Arnqvist, H. Mitogenic effect of the insulin analogue glargine in malignant cells in comparison with insulin and IGF-I. Horm. Metab. Res. 2008, 40, 369–374. [Google Scholar] [CrossRef] [PubMed]
- Cheong, J.-H.; Park, E.S.; Liang, J.; Dennison, J.B.; Tsavachidou, D.; Nguyen-Charles, C.; Cheng, K.W.; Hall, H.; Zhang, D.; Lu, Y. Dual inhibition of tumor energy pathway by 2-deoxyglucose and metformin is effective against a broad spectrum of preclinical cancer models. Mol. Cancer Ther. 2011, 10, 2350–2362. [Google Scholar] [CrossRef] [Green Version]
- Sahra, I.B.; Laurent, K.; Giuliano, S.; Larbret, F.; Ponzio, G.; Gounon, P.; Le Marchand-Brustel, Y.; Giorgetti-Peraldi, S.; Cormont, M.; Bertolotto, C. Targeting cancer cell metabolism: The combination of metformin and 2-deoxyglucose induces p53-dependent apoptosis in prostate cancer cells. Cancer Res. 2010, 70, 2465–2475. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Buzzai, M.; Jones, R.G.; Amaravadi, R.K.; Lum, J.J.; DeBerardinis, R.J.; Zhao, F.; Viollet, B.; Thompson, C.B. Systemic treatment with the antidiabetic drug metformin selectively impairs p53-deficient tumor cell growth. Cancer Res. 2007, 67, 6745–6752. [Google Scholar] [CrossRef] [Green Version]
- Yap, T.A.; Bjerke, L.; Clarke, P.A.; Workman, P. Drugging PI3K in cancer: Refining targets and therapeutic strategies. Curr. Opin. Pharmacol. 2015, 23, 98–107. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ma, C.X.; Sanchez, C.; Gao, F.; Crowder, R.; Naughton, M.; Pluard, T.; Creekmore, A.; Guo, Z.; Hoog, J.; Lockhart, A.C. A phase I study of the AKT inhibitor MK-2206 in combination with hormonal therapy in postmenopausal women with estrogen receptor–positive metastatic breast cancer. Clin. Cancer Res. 2016, 22, 2650–2658. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sprinzl, M.F.; Puschnik, A.; Schlitter, A.M.; Schad, A.; Ackermann, K.; Esposito, I.; Lang, H.; Galle, P.R.; Weinmann, A.; Heikenwälder, M. Sorafenib inhibits macrophage-induced growth of hepatoma cells by interference with insulin-like growth factor-1 secretion. J. Hepatol. 2015, 62, 863–870. [Google Scholar] [CrossRef]
- Bowers, L.W.; Rossi, E.L.; O’Flanagan, C.H.; deGraffenried, L.A.; Hursting, S.D. The role of the insulin/IGF system in cancer: Lessons learned from clinical trials and the energy balance-cancer link. Front. Endocrinol. 2015, 6, 77. [Google Scholar] [CrossRef] [Green Version]
- Simpson, A.; Petnga, W.; Macaulay, V.M.; Weyer-Czernilofsky, U.; Bogenrieder, T. Insulin-like growth factor (IGF) pathway targeting in cancer: Role of the IGF axis and opportunities for future combination studies. Target. Oncol. 2017, 12, 571–597. [Google Scholar] [CrossRef] [Green Version]
- Pollak, M. The insulin and insulin-like growth factor receptor family in neoplasia: An update. Nat. Rev. Cancer 2012, 12, 159–169. [Google Scholar] [CrossRef]
- Busaidy, N.L.; Farooki, A.; Dowlati, A.; Perentesis, J.P.; Dancey, J.E.; Doyle, L.A.; Brell, J.M.; Siu, L.L. Management of metabolic effects associated with anticancer agents targeting the PI3K-Akt-mTOR pathway. J. Clin. Oncol. 2012, 30, 2919. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fischer, H.J.; Sie, C.; Schumann, E.; Witte, A.-K.; Dressel, R.; van den Brandt, J.; Reichardt, H.M. The insulin receptor plays a critical role in T cell function and adaptive immunity. J. Immunol. 2017, 198, 1910–1920. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Quail, D.F.; Bowman, R.L.; Akkari, L.; Quick, M.L.; Schuhmacher, A.J.; Huse, J.T.; Holland, E.C.; Sutton, J.C.; Joyce, J.A. The tumor microenvironment underlies acquired resistance to CSF-1R inhibition in gliomas. Science 2016, 352, aad3018. [Google Scholar] [CrossRef] [Green Version]
- Andrews, D.W.; Resnicoff, M.; Flanders, A.E.; Kenyon, L.; Curtis, M.; Merli, G.; Baserga, R.; Iliakis, G.; Aiken, R.D. Results of a pilot study involving the use of an antisense oligodeoxynucleotide directed against the insulin-like growth factor type I receptor in malignant astrocytomas. J. Clin. Oncol. 2001, 19, 2189–2200. [Google Scholar] [CrossRef] [PubMed]
- Resnicoff, M.; Sell, C.; Rubini, M.; Coppola, D.; Ambrose, D.; Baserga, R.; Rubin, R. Rat glioblastoma cells expressing an antisense RNA to the insulin-like growth factor-1 (IGF-1) receptor are nontumorigenic and induce regression of wild-type tumors. Cancer Res. 1994, 54, 2218–2222. [Google Scholar] [PubMed]
- Trojan, J.; Johnson, T.R.; Rudin, S.D.; Ilan, J.; Tykocinski, M.L. Treatment and prevention of rat glioblastoma by immunogenic C6 cells expressing antisense insulin-like growth factor I RNA. Science 1993, 259, 94–97. [Google Scholar] [CrossRef] [PubMed]
- Somasundaram, R.; Zhang, G.; Wagner, S.N.; Fukunaga-Kalabis, M.; Herlyn, M. The role of tumor microenvironment in therapy resistance and melanoma progression. AACR 2015. [Google Scholar]
- Afzal, M.Z.; Mercado, R.R.; Shirai, K. Efficacy of metformin in combination with immune checkpoint inhibitors (anti-PD-1/anti-CTLA-4) in metastatic malignant melanoma. J. Immunother. Cancer 2018, 6, 64. [Google Scholar] [CrossRef] [Green Version]
- Pietras, R.; Xu, H.; Hu, X.; Matheny, C.; Sandler, A.; Patel, M. P1. 04-33 retrospective descriptive analysis of metformin with atezolizumab in advanced non-small cell lung cancer in the OAK trial. J. Thorac. Oncol. 2018, 13, S538–S539. [Google Scholar] [CrossRef] [Green Version]
- Bahrambeigi, S.; Shafiei-Irannejad, V. Immune-mediated anti-tumor effects of metformin; targeting metabolic reprogramming of T cells as a new possible mechanism for anti-cancer effects of metformin. Biochem. Pharmacol. 2020, 174, 113787. [Google Scholar] [CrossRef]
- Araki, K.; Turner, A.P.; Shaffer, V.O.; Gangappa, S.; Keller, S.A.; Bachmann, M.F.; Larsen, C.P.; Ahmed, R. mTOR regulates memory CD8 T-cell differentiation. Nature 2009, 460, 108–112. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Prlic, M.; Bevan, M.J. A metabolic switch to memory. Nature 2009, 460, 41–42. [Google Scholar] [CrossRef] [PubMed]
- Curry, J.M.; Johnson, J.; Mollaee, M.; Tassone, P.; Amin, D.; Knops, A.; Whitaker-Menezes, D.; Mahoney, M.G.; South, A.; Rodeck, U. Metformin Clinical Trial in HPV+ and HPV–Head and Neck Squamous Cell Carcinoma: Impact on Cancer Cell Apoptosis and Immune Infiltrate. Front. Oncol. 2018, 8, 436. [Google Scholar] [CrossRef] [PubMed]
- Ding, L.; Liang, G.; Yao, Z.; Zhang, J.; Liu, R.; Chen, H.; Zhou, Y.; Wu, H.; Yang, B.; He, Q. Metformin prevents cancer metastasis by inhibiting M2-like polarization of tumor associated macrophages. Oncotarget 2015, 6, 36441. [Google Scholar] [CrossRef] [Green Version]
- Uehara, T.; Eikawa, S.; Nishida, M.; Kunisada, Y.; Yoshida, A.; Fujiwara, T.; Kunisada, T.; Ozaki, T.; Udono, H. Metformin induces CD11b+-cell-mediated growth inhibition of an osteosarcoma: Implications for metabolic reprogramming of myeloid cells and anti-tumor effects. Int. Immunol. 2019, 31, 187–198. [Google Scholar] [CrossRef] [Green Version]
- Verdura, S.; Cuyàs, E.; Martin-Castillo, B.; Menendez, J.A. Metformin as an archetype immuno-metabolic adjuvant for cancer immunotherapy. OncoImmunology 2019, 8, e1633235. [Google Scholar] [CrossRef]
- Cha, J.-H.; Yang, W.-H.; Xia, W.; Wei, Y.; Chan, L.-C.; Lim, S.-O.; Li, C.-W.; Kim, T.; Chang, S.-S.; Lee, H.-H. Metformin promotes antitumor immunity via endoplasmic-reticulum-associated degradation of PD-L1. Mol. Cell 2018, 71, 606–620.e607. [Google Scholar] [CrossRef] [Green Version]
- Garner, H.; de Visser, K.E. Immune crosstalk in cancer progression and metastatic spread: A complex conversation. Nat. Rev. Immunol. 2020, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Chen, D.S.; Mellman, I. Oncology meets immunology: The cancer-immunity cycle. Immunity 2013, 39, 1–10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Palucka, A.K.; Coussens, L.M. The basis of oncoimmunology. Cell 2016, 164, 1233–1247. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Elgert, K.D.; Alleva, D.G.; Mullins, D.W. Tumor-induced immune dysfunction: The macrophage connection. J. Leukoc. Biol. 1998, 64, 275–290. [Google Scholar] [CrossRef] [PubMed]
- Fu, C.; Jiang, A. Dendritic cells and CD8 T cell immunity in tumor microenvironment. Front. Immunol. 2018, 9, 3059. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lewis, C.E.; Pollard, J.W. Distinct role of macrophages in different tumor microenvironments. Cancer Res. 2006, 66, 605–612. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Almand, B.; Resser, J.R.; Lindman, B.; Nadaf, S.; Clark, J.I.; Kwon, E.D.; Carbone, D.P.; Gabrilovich, D.I. Clinical significance of defective dendritic cell differentiation in cancer. Clin. Cancer Res. 2000, 6, 1755–1766. [Google Scholar] [PubMed]
- Tang, M.; Diao, J.; Cattral, M.S. Molecular mechanisms involved in dendritic cell dysfunction in cancer. Cell. Mol. Life Sci. 2017, 74, 761–776. [Google Scholar] [CrossRef]
- Xu, M.M.; Pu, Y.; Han, D.; Shi, Y.; Cao, X.; Liang, H.; Chen, X.; Li, X.-D.; Deng, L.; Chen, Z.J. Dendritic cells but not macrophages sense tumor mitochondrial DNA for cross-priming through signal regulatory protein α signaling. Immunity 2017, 47, 363–373.e365. [Google Scholar] [CrossRef]
- Vander Heiden, M.G.; Cantley, L.C.; Thompson, C.B. Understanding the Warburg effect: The metabolic requirements of cell proliferation. Science 2009, 324, 1029–1033. [Google Scholar] [CrossRef] [Green Version]
- Michalek, R.D.; Gerriets, V.A.; Jacobs, S.R.; Macintyre, A.N.; MacIver, N.J.; Mason, E.F.; Sullivan, S.A.; Nichols, A.G.; Rathmell, J.C. Cutting edge: Distinct glycolytic and lipid oxidative metabolic programs are essential for effector and regulatory CD4+ T cell subsets. J. Immunol. 2011, 186, 3299–3303. [Google Scholar] [CrossRef] [Green Version]
- Shi, L.Z.; Wang, R.; Huang, G.; Vogel, P.; Neale, G.; Green, D.R.; Chi, H. HIF1α–dependent glycolytic pathway orchestrates a metabolic checkpoint for the differentiation of TH17 and Treg cells. J. Exp. Med. 2011, 208, 1367–1376. [Google Scholar] [CrossRef] [Green Version]
- Gubser, P.M.; Bantug, G.R.; Razik, L.; Fischer, M.; Dimeloe, S.; Hoenger, G.; Durovic, B.; Jauch, A.; Hess, C. Rapid effector function of memory CD8+ T cells requires an immediate-early glycolytic switch. Nat. Immunol. 2013, 14, 1064. [Google Scholar] [CrossRef]
- Dang, E.V.; Barbi, J.; Yang, H.-Y.; Jinasena, D.; Yu, H.; Zheng, Y.; Bordman, Z.; Fu, J.; Kim, Y.; Yen, H.-R. Control of TH17/Treg balance by hypoxia-inducible factor 1. Cell 2011, 146, 772–784. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rodríguez-Prados, J.-C.; Través, P.G.; Cuenca, J.; Rico, D.; Aragonés, J.; Martín-Sanz, P.; Cascante, M.; Boscá, L. Substrate fate in activated macrophages: A comparison between innate, classic, and alternative activation. J. Immunol. 2010, 185, 605–614. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Krawczyk, C.M.; Holowka, T.; Sun, J.; Blagih, J.; Amiel, E.; DeBerardinis, R.J.; Cross, J.R.; Jung, E.; Thompson, C.B.; Jones, R.G. Toll-like receptor–induced changes in glycolytic metabolism regulate dendritic cell activation. Bloodthe J. Am. Soc. Hematol. 2010, 115, 4742–4749. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chang, C.-H.; Qiu, J.; O’Sullivan, D.; Buck, M.D.; Noguchi, T.; Curtis, J.D.; Chen, Q.; Gindin, M.; Gubin, M.M.; Van Der Windt, G.J. Metabolic competition in the tumor microenvironment is a driver of cancer progression. Cell 2015, 162, 1229–1241. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ho, P.-C.; Bihuniak, J.D.; Macintyre, A.N.; Staron, M.; Liu, X.; Amezquita, R.; Tsui, Y.-C.; Cui, G.; Micevic, G.; Perales, J.C. Phosphoenolpyruvate is a metabolic checkpoint of anti-tumor T cell responses. Cell 2015, 162, 1217–1228. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gottfried, E.; Kunz-Schughart, L.A.; Ebner, S.; Mueller-Klieser, W.; Hoves, S.; Andreesen, R.; Mackensen, A.; Kreutz, M. Tumor-derived lactic acid modulates dendritic cell activation and antigen expression. Blood 2006, 107, 2013–2021. [Google Scholar] [CrossRef]
- Dietl, K.; Renner, K.; Dettmer, K.; Timischl, B.; Eberhart, K.; Dorn, C.; Hellerbrand, C.; Kastenberger, M.; Kunz-Schughart, L.A.; Oefner, P.J. Lactic acid and acidification inhibit TNF secretion and glycolysis of human monocytes. J. Immunol. 2010, 184, 1200–1209. [Google Scholar] [CrossRef]
- Colegio, O.R.; Chu, N.-Q.; Szabo, A.L.; Chu, T.; Rhebergen, A.M.; Jairam, V.; Cyrus, N.; Brokowski, C.E.; Eisenbarth, S.C.; Phillips, G.M. Functional polarization of tumour-associated macrophages by tumour-derived lactic acid. Nature 2014, 513, 559–563. [Google Scholar] [CrossRef]
- Mukhopadhyay, R.; Jia, J.; Arif, A.; Ray, P.S.; Fox, P.L. The GAIT system: A gatekeeper of inflammatory gene expression. Trends Biochem. Sci. 2009, 34, 324–331. [Google Scholar] [CrossRef] [Green Version]
- Chang, C.-H.; Curtis, J.D.; Maggi Jr, L.B.; Faubert, B.; Villarino, A.V.; O’Sullivan, D.; Huang, S.C.-C.; Van Der Windt, G.J.; Blagih, J.; Qiu, J. Posttranscriptional control of T cell effector function by aerobic glycolysis. Cell 2013, 153, 1239–1251. [Google Scholar] [CrossRef] [Green Version]
- Sukumar, M.; Liu, J.; Ji, Y.; Subramanian, M.; Crompton, J.G.; Yu, Z.; Roychoudhuri, R.; Palmer, D.C.; Muranski, P.; Karoly, E.D. Inhibiting glycolytic metabolism enhances CD8+ T cell memory and antitumor function. J. Clin. Investig. 2013, 123, 4479–4488. [Google Scholar] [CrossRef] [PubMed]
- Pollizzi, K.N.; Patel, C.H.; Sun, I.-H.; Oh, M.-H.; Waickman, A.T.; Wen, J.; Delgoffe, G.M.; Powell, J.D. mTORC1 and mTORC2 selectively regulate CD8+ T cell differentiation. J. Clin. Investig. 2015, 125, 2090–2108. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jha, A.K.; Huang, S.C.-C.; Sergushichev, A.; Lampropoulou, V.; Ivanova, Y.; Loginicheva, E.; Chmielewski, K.; Stewart, K.M.; Ashall, J.; Everts, B. Network integration of parallel metabolic and transcriptional data reveals metabolic modules that regulate macrophage polarization. Immunity 2015, 42, 419–430. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gerriets, V.A.; Kishton, R.J.; Nichols, A.G.; Macintyre, A.N.; Inoue, M.; Ilkayeva, O.; Winter, P.S.; Liu, X.; Priyadharshini, B.; Slawinska, M.E. Metabolic programming and PDHK1 control CD4+ T cell subsets and inflammation. J. Clin. Investig. 2015, 125, 194–207. [Google Scholar] [CrossRef] [PubMed]
- Beier, U.H.; Angelin, A.; Akimova, T.; Wang, L.; Liu, Y.; Xiao, H.; Koike, M.A.; Hancock, S.A.; Bhatti, T.R.; Han, R. Essential role of mitochondrial energy metabolism in Foxp3+ T-regulatory cell function and allograft survival. Faseb J. 2015, 29, 2315–2326. [Google Scholar] [CrossRef] [Green Version]
- Procaccini, C.; Carbone, F.; Di Silvestre, D.; Brambilla, F.; De Rosa, V.; Galgani, M.; Faicchia, D.; Marone, G.; Tramontano, D.; Corona, M. The proteomic landscape of human ex vivo regulatory and conventional T cells reveals specific metabolic requirements. Immunity 2016, 44, 406–421. [Google Scholar] [CrossRef] [Green Version]
- O’Sullivan, D.; van der Windt, G.J.; Huang, S.C.-C.; Curtis, J.D.; Chang, C.-H.; Buck, M.D.; Qiu, J.; Smith, A.M.; Lam, W.Y.; DiPlato, L.M. Memory CD8+ T cells use cell-intrinsic lipolysis to support the metabolic programming necessary for development. Immunity 2014, 41, 75–88. [Google Scholar] [CrossRef] [Green Version]
- O’Neill, L.A.; Kishton, R.J.; Rathmell, J. A guide to immunometabolism for immunologists. Nat. Rev. Immunol. 2016, 16, 553. [Google Scholar] [CrossRef] [Green Version]
- Molodtsov, A.; Turk, M.J. Tissue resident CD8 memory T cell responses in cancer and autoimmunity. Front. Immunol. 2018, 9, 2810. [Google Scholar] [CrossRef]
- Menares, E.; Gálvez-Cancino, F.; Cáceres-Morgado, P.; Ghorani, E.; López, E.; Díaz, X.; Saavedra-Almarza, J.; Figueroa, D.A.; Roa, E.; Quezada, S.A. Tissue-resident memory CD8+ T cells amplify anti-tumor immunity by triggering antigen spreading through dendritic cells. Nat. Commun. 2019, 10, 1–12. [Google Scholar] [CrossRef] [Green Version]
- van der Windt, G.J.; O’Sullivan, D.; Everts, B.; Huang, S.C.-C.; Buck, M.D.; Curtis, J.D.; Chang, C.-H.; Smith, A.M.; Ai, T.; Faubert, B. CD8 memory T cells have a bioenergetic advantage that underlies their rapid recall ability. Proc. Natl. Acad. Sci. USA 2013, 110, 14336–14341. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- van der Windt, G.J.; Everts, B.; Chang, C.-H.; Curtis, J.D.; Freitas, T.C.; Amiel, E.; Pearce, E.J.; Pearce, E.L. Mitochondrial respiratory capacity is a critical regulator of CD8+ T cell memory development. Immunity 2012, 36, 68–78. [Google Scholar] [CrossRef] [Green Version]
- Currie, E.; Schulze, A.; Zechner, R.; Walther, T.C.; Farese Jr, R.V. Cellular fatty acid metabolism and cancer. Cell Metab. 2013, 18, 153–161. [Google Scholar] [CrossRef] [Green Version]
- Svensson, R.U.; Parker, S.J.; Eichner, L.J.; Kolar, M.J.; Wallace, M.; Brun, S.N.; Lombardo, P.S.; Van Nostrand, J.L.; Hutchins, A.; Vera, L. Inhibition of acetyl-CoA carboxylase suppresses fatty acid synthesis and tumor growth of non-small-cell lung cancer in preclinical models. Nat. Med. 2016, 22, 1108. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Flaveny, C.A.; Griffett, K.; El-Gendy, B.E.-D.M.; Kazantzis, M.; Sengupta, M.; Amelio, A.L.; Chatterjee, A.; Walker, J.; Solt, L.A.; Kamenecka, T.M. Broad anti-tumor activity of a small molecule that selectively targets the Warburg effect and lipogenesis. Cancer Cell 2015, 28, 42–56. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Everts, B.; Amiel, E.; Huang, S.C.-C.; Smith, A.M.; Chang, C.-H.; Lam, W.Y.; Redmann, V.; Freitas, T.C.; Blagih, J.; Van Der Windt, G.J. TLR-driven early glycolytic reprogramming via the kinases TBK1-IKKɛ supports the anabolic demands of dendritic cell activation. Nat. Immunol. 2014, 15, 323. [Google Scholar] [CrossRef] [Green Version]
- Chen, H.W.; Heiniger, H.-J.; Kandutsch, A.A. Relationship between sterol synthesis and DNA synthesis in phytohemagglutinin-stimulated mouse lymphocytes. Proc. Natl. Acad. Sci. USA 1975, 72, 1950–1954. [Google Scholar] [CrossRef] [Green Version]
- Dufort, F.J.; Gumina, M.R.; Ta, N.L.; Tao, Y.; Heyse, S.A.; Scott, D.A.; Richardson, A.D.; Seyfried, T.N.; Chiles, T.C. Glucose-dependent de novo lipogenesis in B lymphocytes a requirement for ATP-citrate lyase in lipopolysaccharide-induced differentiation. J. Biol. Chem. 2014, 289, 7011–7024. [Google Scholar] [CrossRef] [Green Version]
- Lee, J.; Walsh, M.C.; Hoehn, K.L.; James, D.E.; Wherry, E.J.; Choi, Y. Regulator of fatty acid metabolism, acetyl coenzyme a carboxylase 1, controls T cell immunity. J. Immunol. 2014, 192, 3190–3199. [Google Scholar] [CrossRef] [Green Version]
- Berod, L.; Friedrich, C.; Nandan, A.; Freitag, J.; Hagemann, S.; Harmrolfs, K.; Sandouk, A.; Hesse, C.; Castro, C.N.; Bähre, H. De novo fatty acid synthesis controls the fate between regulatory T and T helper 17 cells. Nat. Med. 2014, 20, 1327. [Google Scholar] [CrossRef]
- Gullino, P.M.; Clark, S.H.; Grantham, F.H. The interstitial fluid of solid tumors. Cancer Res. 1964, 24, 780–797. [Google Scholar] [PubMed]
- Buck, M.D.; O’Sullivan, D.; Geltink, R.I.K.; Curtis, J.D.; Chang, C.-H.; Sanin, D.E.; Qiu, J.; Kretz, O.; Braas, D.; van der Windt, G.J. Mitochondrial dynamics controls T cell fate through metabolic programming. Cell 2016, 166, 63–76. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Menk, A.V.; Scharping, N.E.; Rivadeneira, D.B.; Calderon, M.J.; Watson, M.J.; Dunstane, D.; Watkins, S.C.; Delgoffe, G.M. 4-1BB costimulation induces T cell mitochondrial function and biogenesis enabling cancer immunotherapeutic responses. J. Exp. Med. 2018, 215, 1091–1100. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Teijeira, A.; Labiano, S.; Garasa, S.; Etxeberria, I.; Santamaría, E.; Rouzaut, A.; Enamorado, M.; Azpilikueta, A.; Inoges, S.; Bolaños, E. Mitochondrial morphological and functional reprogramming following CD137 (4-1BB) costimulation. Cancer Immunol. Res. 2018. [Google Scholar] [CrossRef] [Green Version]
- Cascone, T.; McKenzie, J.A.; Mbofung, R.M.; Punt, S.; Wang, Z.; Xu, C.; Williams, L.J.; Wang, Z.; Bristow, C.A.; Carugo, A. Increased tumor glycolysis characterizes immune resistance to adoptive T cell therapy. Cell Metab. 2018, 27, 977–987.e974. [Google Scholar] [CrossRef]
- Scharping, N.E.; Menk, A.V.; Whetstone, R.D.; Zeng, X.; Delgoffe, G.M. Efficacy of PD-1 blockade is potentiated by metformin-induced reduction of tumor hypoxia. Cancer Immunol. Res. 2017, 5, 9–16. [Google Scholar] [CrossRef] [Green Version]
- Yang, W.; Bai, Y.; Xiong, Y.; Zhang, J.; Chen, S.; Zheng, X.; Meng, X.; Li, L.; Wang, J.; Xu, C. Potentiating the antitumour response of CD8+ T cells by modulating cholesterol metabolism. Nature 2016, 531, 651–655. [Google Scholar] [CrossRef] [Green Version]
- Dean, E.J.; Falchook, G.S.; Patel, M.R.; Brenner, A.J.; Infante, J.R.; Arkenau, H.-T.; Borazanci, E.H.; Lopez, J.S.; Pant, S.; Schmid, P. Preliminary activity in the first in human study of the first-in-class fatty acid synthase (FASN) inhibitor, TVB-2640. Am. Soc. Clin. Oncol. 2016. [Google Scholar] [CrossRef]
- Hosios, A.M.; Hecht, V.C.; Danai, L.V.; Johnson, M.O.; Rathmell, J.C.; Steinhauser, M.L.; Manalis, S.R.; Vander Heiden, M.G. Amino acids rather than glucose account for the majority of cell mass in proliferating mammalian cells. Dev. Cell 2016, 36, 540–549. [Google Scholar] [CrossRef] [Green Version]
- Altman, B.J.; Stine, Z.E.; Dang, C.V. From Krebs to clinic: Glutamine metabolism to cancer therapy. Nat. Rev. Cancer 2016, 16, 619. [Google Scholar] [CrossRef]
- Wang, R.; Dillon, C.P.; Shi, L.Z.; Milasta, S.; Carter, R.; Finkelstein, D.; McCormick, L.L.; Fitzgerald, P.; Chi, H.; Munger, J. The transcription factor Myc controls metabolic reprogramming upon T lymphocyte activation. Immunity 2011, 35, 871–882. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carr, E.L.; Kelman, A.; Wu, G.S.; Gopaul, R.; Senkevitch, E.; Aghvanyan, A.; Turay, A.M.; Frauwirth, K.A. Glutamine uptake and metabolism are coordinately regulated by ERK/MAPK during T lymphocyte activation. J. Immunol. 2010, 185, 1037–1044. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Crawford, J.; Cohen, H.J. The essential role of L-glutamine in lymphocyte differentiation in vitro. J. Cell. Physiol. 1985, 124, 275–282. [Google Scholar] [CrossRef] [PubMed]
- Nakaya, M.; Xiao, Y.; Zhou, X.; Chang, J.-H.; Chang, M.; Cheng, X.; Blonska, M.; Lin, X.; Sun, S.-C. Inflammatory T cell responses rely on amino acid transporter ASCT2 facilitation of glutamine uptake and mTORC1 kinase activation. Immunity 2014, 40, 692–705. [Google Scholar] [CrossRef] [Green Version]
- Klysz, D.; Tai, X.; Robert, P.A.; Craveiro, M.; Cretenet, G.; Oburoglu, L.; Mongellaz, C.; Floess, S.; Fritz, V.; Matias, M.I. Glutamine-dependent α-ketoglutarate production regulates the balance between T helper 1 cell and regulatory T cell generation. Sci. Signal. 2015, 8, ra97. [Google Scholar] [CrossRef] [Green Version]
- Lemberg, K.M.; Vornov, J.J.; Rais, R.; Slusher, B.S. We’re not “DON” yet: Optimal dosing and prodrug delivery of 6-Diazo-5-oxo-L-norleucine. Mol. Cancer Ther. 2018, 17, 1824–1832. [Google Scholar] [CrossRef] [Green Version]
- Pinkus, L.M. [45] Glutamine binding sites. In Methods in Enzymology; Elsevier: Amsterdam, The Netherlands, 1977; Volume 46, pp. 414–427. [Google Scholar]
- Magill, G.; Myers, W.; Reilly, H.; Putnam, R.; Magill, J.; Sykes, M.; Escher, G.; Karnofsky, D.; Burchenal, J. Pharmacological and initial therapeutic observations on 6-Diazo-5-Oxo-L-Norleucine (Don) in human neoplastic disease. Cancer 1957, 10, 1138–1150. [Google Scholar] [CrossRef]
- Leone, R.D.; Zhao, L.; Englert, J.M.; Sun, I.-M.; Oh, M.-H.; Sun, I.-H.; Arwood, M.L.; Bettencourt, I.A.; Patel, C.H.; Wen, J. Glutamine blockade induces divergent metabolic programs to overcome tumor immune evasion. Science 2019, 366, 1013–1021. [Google Scholar] [CrossRef]
- Palmieri, E.M.; Menga, A.; Martín-Pérez, R.; Quinto, A.; Riera-Domingo, C.; De Tullio, G.; Hooper, D.C.; Lamers, W.H.; Ghesquière, B.; McVicar, D.W. Pharmacologic or genetic targeting of glutamine synthetase skews macrophages toward an M1-like phenotype and inhibits tumor metastasis. Cell Rep. 2017, 20, 1654–1666. [Google Scholar] [CrossRef] [Green Version]
- Oh, M.-H.; Sun, I.-H.; Zhao, L.; Leone, R.D.; Sun, I.-M.; Xu, W.; Collins, S.L.; Tam, A.J.; Blosser, R.L.; Patel, C.H. Targeting glutamine metabolism enhances tumor specific immunity by modulating suppressive myeloid cells. J. Clin. Investig. 2020. [Google Scholar] [CrossRef]
- Guerra, V.; Dinardo, C.D.; Konopleva, M.; Burger, J.A.; Borthakur, G.; Jabbour, E.; Pemmaraju, N.; Sheppard, K.; Garcia-Manero, G.; Kantarjian, H.M. Interim results from a phase Ib/II clinical study of the glutaminase inhibitor telaglenastat (CB-839) in combination with azacitidine in patients with advanced myelodysplastic syndrome (MDS). Am. Soc. Clin. Oncol. 2019. [Google Scholar] [CrossRef]
- Kizilbash, S.H.; McBrayer, S.; Port, J.; Reid, J.M.; Lanza, I.; Allred, J.B.; Chakravarti, A.; Kunos, C.; Adjei, A.A. A phase Ib trial of CB-839 (telaglenastat) in combination with radiation therapy and temozolomide in patients with IDH-mutated diffuse astrocytoma and anaplastic astrocytoma (NCT03528642). Am. Soc. Clin. Oncol. 2019. [Google Scholar] [CrossRef]
- Wang, E.S.; Frankfurt, O.; Orford, K.W.; Bennett, M.; Flinn, I.W.; Maris, M.; Konopleva, M. Phase 1 study of CB-839, a first-in-class, orally administered small molecule inhibitor of glutaminase in patients with relapsed/refractory leukemia. Blood 2015, 126, 2566. [Google Scholar] [CrossRef]
- Lee, G.K.; Park, H.J.; Macleod, M.; Chandler, P.; Munn, D.H.; Mellor, A.L. Tryptophan deprivation sensitizes activated T cells to apoptosis prior to cell division. Immunology 2002, 107, 452–460. [Google Scholar] [CrossRef] [PubMed]
- Munn, D.H.; Shafizadeh, E.; Attwood, J.T.; Bondarev, I.; Pashine, A.; Mellor, A.L. Inhibition of T cell proliferation by macrophage tryptophan catabolism. J. Exp. Med. 1999, 189, 1363–1372. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.; Huang, L.; Bradley, J.; Liu, K.; Bardhan, K.; Ron, D.; Mellor, A.L.; Munn, D.H.; McGaha, T.L. GCN2-dependent metabolic stress is essential for endotoxemic cytokine induction and pathology. Mol. Cell. Biol. 2014, 34, 428–438. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bessede, A.; Gargaro, M.; Pallotta, M.T.; Matino, D.; Servillo, G.; Brunacci, C.; Bicciato, S.; Mazza, E.M.; Macchiarulo, A.; Vacca, C. Aryl hydrocarbon receptor control of a disease tolerance defence pathway. Nature 2014, 511, 184–190. [Google Scholar] [CrossRef] [Green Version]
- Uyttenhove, C.; Pilotte, L.; Théate, I.; Stroobant, V.; Colau, D.; Parmentier, N.; Boon, T.; Van den Eynde, B.J. Evidence for a tumoral immune resistance mechanism based on tryptophan degradation by indoleamine 2, 3-dioxygenase. Nat. Med. 2003, 9, 1269–1274. [Google Scholar] [CrossRef]
- Okamoto, A.; Nikaido, T.; Ochiai, K.; Takakura, S.; Saito, M.; Aoki, Y.; Ishii, N.; Yanaihara, N.; Yamada, K.; Takikawa, O. Indoleamine 2, 3-dioxygenase serves as a marker of poor prognosis in gene expression profiles of serous ovarian cancer cells. Clin. Cancer Res. 2005, 11, 6030–6039. [Google Scholar] [CrossRef] [Green Version]
- Minhas, P.S.; Liu, L.; Moon, P.K.; Joshi, A.U.; Dove, C.; Mhatre, S.; Contrepois, K.; Wang, Q.; Lee, B.A.; Coronado, M. Macrophage de novo NAD+ synthesis specifies immune function in aging and inflammation. Nat. Immunol. 2019, 20, 50–63. [Google Scholar] [CrossRef]
- Munn, D.H.; Sharma, M.D.; Hou, D.; Baban, B.; Lee, J.R.; Antonia, S.J.; Messina, J.L.; Chandler, P.; Koni, P.A.; Mellor, A.L. Expression of indoleamine 2, 3-dioxygenase by plasmacytoid dendritic cells in tumor-draining lymph nodes. J. Clin. Investig. 2004, 114, 280–290. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weinlich, G.; Murr, C.; Richardsen, L.; Winkler, C.; Fuchs, D. Decreased serum tryptophan concentration predicts poor prognosis in malignant melanoma patients. Dermatology 2007, 214, 8–14. [Google Scholar] [CrossRef] [PubMed]
- Holmgaard, R.B.; Zamarin, D.; Munn, D.H.; Wolchok, J.D.; Allison, J.P. Indoleamine 2, 3-dioxygenase is a critical resistance mechanism in antitumor T cell immunotherapy targeting CTLA-4. J. Exp. Med. 2013, 210, 1389–1402. [Google Scholar] [CrossRef] [PubMed]
- Cronin, S.J.; Seehus, C.; Weidinger, A.; Talbot, S.; Reissig, S.; Seifert, M.; Pierson, Y.; McNeill, E.; Longhi, M.S.; Turnes, B.L. The metabolite BH4 controls T cell proliferation in autoimmunity and cancer. Nature 2018, 563, 564–568. [Google Scholar] [CrossRef] [PubMed]
- Gu, T.; Rowswell-Turner, R.B.; Kilinc, M.O.; Egilmez, N.K. Central role of IFNγ–indoleamine 2, 3-dioxygenase axis in regulation of interleukin-12–mediated antitumor immunity. Cancer Res. 2010, 70, 129–138. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Friberg, M.; Jennings, R.; Alsarraj, M.; Dessureault, S.; Cantor, A.; Extermann, M.; Mellor, A.L.; Munn, D.H.; Antonia, S.J. Indoleamine 2, 3-dioxygenase contributes to tumor cell evasion of T cell-mediated rejection. Int. J. Cancer 2002, 101, 151–155. [Google Scholar] [CrossRef] [PubMed]
- Labadie, B.W.; Bao, R.; Luke, J.J. Reimagining IDO pathway inhibition in cancer immunotherapy via downstream focus on the tryptophan–kynurenine–aryl hydrocarbon axis. Clin. Cancer Res. 2019, 25, 1462–1471. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Geiger, R.; Rieckmann, J.C.; Wolf, T.; Basso, C.; Feng, Y.; Fuhrer, T.; Kogadeeva, M.; Picotti, P.; Meissner, F.; Mann, M. L-arginine modulates T cell metabolism and enhances survival and anti-tumor activity. Cell 2016, 167, 829–842.e813. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rodriguez, P.C.; Zea, A.H.; Culotta, K.S.; Zabaleta, J.; Ochoa, J.B.; Ochoa, A.C. Regulation of t cell receptor cd3ζ chain expression byl-arginine. J. Biol. Chem. 2002, 277, 21123–21129. [Google Scholar] [CrossRef] [Green Version]
- Rodriguez, P.C.; Quiceno, D.G.; Ochoa, A.C. L-arginine availability regulates T-lymphocyte cell-cycle progression. Blood 2007, 109, 1568–1573. [Google Scholar] [CrossRef] [Green Version]
- El-Gayar, S.; Thüring-Nahler, H.; Pfeilschifter, J.; Röllinghoff, M.; Bogdan, C. Translational control of inducible nitric oxide synthase by IL-13 and arginine availability in inflammatory macrophages. J. Immunol. 2003, 171, 4561–4568. [Google Scholar] [CrossRef] [Green Version]
- Pesce, J.T.; Ramalingam, T.R.; Mentink-Kane, M.M.; Wilson, M.S.; El Kasmi, K.C.; Smith, A.M.; Thompson, R.W.; Cheever, A.W.; Murray, P.J.; Wynn, T.A. Arginase-1–expressing macrophages suppress Th2 cytokine–driven inflammation and fibrosis. Plos Pathog. 2009, 5. [Google Scholar] [CrossRef] [Green Version]
- Murray, P.J.; Allen, J.E.; Biswas, S.K.; Fisher, E.A.; Gilroy, D.W.; Goerdt, S.; Gordon, S.; Hamilton, J.A.; Ivashkiv, L.B.; Lawrence, T. Macrophage activation and polarization: Nomenclature and experimental guidelines. Immunity 2014, 41, 14–20. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cobbold, S.P.; Adams, E.; Farquhar, C.A.; Nolan, K.F.; Howie, D.; Lui, K.O.; Fairchild, P.J.; Mellor, A.L.; Ron, D.; Waldmann, H. Infectious tolerance via the consumption of essential amino acids and mTOR signaling. Proc. Natl. Acad. Sci. USA 2009, 106, 12055–12060. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Doedens, A.L.; Stockmann, C.; Rubinstein, M.P.; Liao, D.; Zhang, N.; DeNardo, D.G.; Coussens, L.M.; Karin, M.; Goldrath, A.W.; Johnson, R.S. Macrophage expression of hypoxia-inducible factor-1α suppresses T-cell function and promotes tumor progression. Cancer Res. 2010, 70, 7465–7475. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sharda, D.R.; Yu, S.; Ray, M.; Squadrito, M.L.; De Palma, M.; Wynn, T.A.; Morris, S.M.; Hankey, P.A. Regulation of macrophage arginase expression and tumor growth by the Ron receptor tyrosine kinase. J. Immunol. 2011, 187, 2181–2192. [Google Scholar] [CrossRef]
- Rodriguez, P.C.; Quiceno, D.G.; Zabaleta, J.; Ortiz, B.; Zea, A.H.; Piazuelo, M.B.; Delgado, A.; Correa, P.; Brayer, J.; Sotomayor, E.M. Arginase I production in the tumor microenvironment by mature myeloid cells inhibits T-cell receptor expression and antigen-specific T-cell responses. Cancer Res. 2004, 64, 5839–5849. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Steggerda, S.M.; Bennett, M.K.; Chen, J.; Emberley, E.; Huang, T.; Janes, J.R.; Li, W.; MacKinnon, A.L.; Makkouk, A.; Marguier, G. Inhibition of arginase by CB-1158 blocks myeloid cell-mediated immune suppression in the tumor microenvironment. J. Immunother. Cancer 2017, 5, 101. [Google Scholar] [CrossRef]
- Papadopoulos, K.P.; Tsai, F.Y.-C.; Bauer, T.M.; Muigai, L.; Liang, Y.; Bennett, M.K.; Orford, K.W.; Fu, S. CX-1158-101: A first-in-human phase 1 study of CB-1158, a small molecule inhibitor of arginase, as monotherapy and in combination with an anti-PD-1 checkpoint inhibitor in patients (pts) with solid tumors. Am. Soc. Clin. Oncol. 2017. [Google Scholar] [CrossRef]
- Naing, A.; Bauer, T.; Papadopoulos, K.; Rahma, O.; Tsai, F.; Garralda, E.; Naidoo, J.; Pai, S.; Gibson, M.; Rybkin, I. Phase I study of the arginase inhibitor INCB001158 (1158) alone and in combination with pembrolizumab (PEM) in patients (Pts) with advanced/metastatic (adv/met) solid tumours. Ann. Oncol. 2019, 30, v160. [Google Scholar] [CrossRef]
- Hui, S.; Ghergurovich, J.M.; Morscher, R.J.; Jang, C.; Teng, X.; Lu, W.; Esparza, L.A.; Reya, T.; Zhan, L.; Guo, J.Y. Glucose feeds the TCA cycle via circulating lactate. Nature 2017, 551, 115–118. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Faubert, B.; Li, K.Y.; Cai, L.; Hensley, C.T.; Kim, J.; Zacharias, L.G.; Yang, C.; Do, Q.N.; Doucette, S.; Burguete, D. Lactate metabolism in human lung tumors. Cell 2017, 171, 358–371.e359. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ippolito, L.; Morandi, A.; Giannoni, E.; Chiarugi, P. Lactate: A metabolic driver in the tumour landscape. Trends Biochem. Sci. 2019, 44, 153–166. [Google Scholar] [CrossRef] [PubMed]
- Husain, Z.; Huang, Y.; Seth, P.; Sukhatme, V.P. Tumor-derived lactate modifies antitumor immune response: Effect on myeloid-derived suppressor cells and NK cells. J. Immunol. 2013, 191, 1486–1495. [Google Scholar] [CrossRef]
- Angelin, A.; Gil-de-Gómez, L.; Dahiya, S.; Jiao, J.; Guo, L.; Levine, M.H.; Wang, Z.; Quinn III, W.J.; Kopinski, P.K.; Wang, L. Foxp3 reprograms T cell metabolism to function in low-glucose, high-lactate environments. Cell Metab. 2017, 25, 1282–1293.e1287. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brand, A.; Singer, K.; Koehl, G.E.; Kolitzus, M.; Schoenhammer, G.; Thiel, A.; Matos, C.; Bruss, C.; Klobuch, S.; Peter, K. LDHA-associated lactic acid production blunts tumor immunosurveillance by T and NK cells. Cell Metab. 2016, 24, 657–671. [Google Scholar] [CrossRef] [Green Version]
- Fischer, K.; Hoffmann, P.; Voelkl, S.; Meidenbauer, N.; Ammer, J.; Edinger, M.; Gottfried, E.; Schwarz, S.; Rothe, G.; Hoves, S. Inhibitory effect of tumor cell–derived lactic acid on human T cells. Blood 2007, 109, 3812–3819. [Google Scholar] [CrossRef]
- Huber, V.; Camisaschi, C.; Berzi, A.; Ferro, S.; Lugini, L.; Triulzi, T.; Tuccitto, A.; Tagliabue, E.; Castelli, C.; Rivoltini, L. Cancer acidity: An ultimate frontier of tumor immune escape and a novel target of immunomodulation. Semin. Cancer Biol. 2017, 43, 74–89. [Google Scholar] [CrossRef]
- Lacroix, R.; Rozeman, E.A.; Kreutz, M.; Renner, K.; Blank, C.U. Targeting tumor-associated acidity in cancer immunotherapy. Cancer Immunol. Immunother. 2018, 67, 1331–1348. [Google Scholar] [CrossRef]
- Peng, M.; Yin, N.; Chhangawala, S.; Xu, K.; Leslie, C.S.; Li, M.O. Aerobic glycolysis promotes T helper 1 cell differentiation through an epigenetic mechanism. Science 2016, 354, 481–484. [Google Scholar] [CrossRef]
- Farabegoli, F.; Vettraino, M.; Manerba, M.; Fiume, L.; Roberti, M.; Di Stefano, G. Galloflavin, a new lactate dehydrogenase inhibitor, induces the death of human breast cancer cells with different glycolytic attitude by affecting distinct signaling pathways. Eur. J. Pharm. Sci. 2012, 47, 729–738. [Google Scholar] [CrossRef]
- Patel, C.H.; Leone, R.D.; Horton, M.R.; Powell, J.D. Targeting metabolism to regulate immune responses in autoimmunity and cancer. Nat. Rev. Drug Discov. 2019, 18, 669–688. [Google Scholar] [CrossRef]
- Miller, W.L.; Thomas, R.A.; Berne, R.M.; Rubio, R. Adenosine production in the ischemic kidney. Circ. Res. 1978, 43, 390–397. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sorrentino, C.; Miele, L.; Porta, A.; Pinto, A.; Morello, S. Myeloid-derived suppressor cells contribute to A2B adenosine receptor-induced VEGF production and angiogenesis in a mouse melanoma model. Oncotarget 2015, 6, 27478. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, L.; Huang, L.; Ye, H.; Song, S.P.; Bajwa, A.; Lee, S.J.; Moser, E.K.; Jaworska, K.; Kinsey, G.R.; Day, Y.J. Dendritic cells tolerized with adenosine A 2A R agonist attenuate acute kidney injury. J. Clin. Investig. 2012, 122, 3931–3942. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vijayan, D.; Young, A.; Teng, M.W.; Smyth, M.J. Targeting immunosuppressive adenosine in cancer. Nat. Rev. Cancer 2017, 17, 709. [Google Scholar] [CrossRef]
- Cai, X.Y.; Wang, X.F.; Li, J.; Dong, J.N.; Liu, J.Q.; Li, N.P.; Yun, B.; Xia, R.L.; Qin, J.; Sun, Y.H. High expression of CD39 in gastric cancer reduces patient outcome following radical resection. Oncol. Lett. 2016, 12, 4080–4086. [Google Scholar] [CrossRef] [Green Version]
- Turcotte, M.; Spring, K.; Pommey, S.; Chouinard, G.; Cousineau, I.; George, J.; Chen, G.M.; Gendoo, D.M.; Haibe-Kains, B.; Karn, T. CD73 is associated with poor prognosis in high-grade serous ovarian cancer. Cancer Res. 2015, 75, 4494–4503. [Google Scholar] [CrossRef] [Green Version]
- Inoue, Y.; Yoshimura, K.; Kurabe, N.; Kahyo, T.; Kawase, A.; Tanahashi, M.; Ogawa, H.; Inui, N.; Funai, K.; Shinmura, K. Prognostic impact of CD73 and A2A adenosine receptor expression in non-small-cell lung cancer. Oncotarget 2017, 8, 8738. [Google Scholar] [CrossRef] [Green Version]
- Maj, T.; Wang, W.; Crespo, J.; Zhang, H.; Wang, W.; Wei, S.; Zhao, L.; Vatan, L.; Shao, I.; Szeliga, W. Oxidative stress controls regulatory T cell apoptosis and suppressor activity and PD-L1-blockade resistance in tumor. Nat. Immunol. 2017, 18, 1332. [Google Scholar] [CrossRef]
- Sun, X.; Wu, Y.; Gao, W.; Enjyoji, K.; Csizmadia, E.; Müller, C.E.; Murakami, T.; Robson, S.C. CD39/ENTPD1 expression by CD4+ Foxp3+ regulatory T cells promotes hepatic metastatic tumor growth in mice. Gastroenterology 2010, 139, 1030–1040. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, S.; Apasov, S.; Koshiba, M.; Sitkovsky, M. Role of A2a extracellular adenosine receptor-mediated signaling in adenosine-mediated inhibition of T-cell activation and expansion. Bloodthe J. Am. Soc. Hematol. 1997, 90, 1600–1610. [Google Scholar]
- Ohta, A.; Kini, R.; Ohta, A.; Subramanian, M.; Madasu, M.; Sitkovsky, M. The development and immunosuppressive functions of CD4+ CD25+ FoxP3+ regulatory T cells are under influence of the adenosine-A2A adenosine receptor pathway. Front. Immunol. 2012, 3, 190. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Allard, B.; Pommey, S.; Smyth, M.J.; Stagg, J. Targeting CD73 enhances the antitumor activity of anti-PD-1 and anti-CTLA-4 mAbs. Clin. Cancer Res. 2013, 19, 5626–5635. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Iannone, R.; Miele, L.; Maiolino, P.; Pinto, A.; Morello, S. Adenosine limits the therapeutic effectiveness of anti-CTLA4 mAb in a mouse melanoma model. Am. J. Cancer Res. 2014, 4, 172. [Google Scholar]
- Beavis, P.A.; Milenkovski, N.; Henderson, M.A.; John, L.B.; Allard, B.; Loi, S.; Kershaw, M.H.; Stagg, J.; Darcy, P.K. Adenosine receptor 2A blockade increases the efficacy of anti–PD-1 through enhanced antitumor T-cell responses. Cancer Immunol. Res. 2015, 3, 506–517. [Google Scholar] [CrossRef] [Green Version]
- Fong, L.; Forde, P.M.; Powderly, J.D.; Goldman, J.W.; Nemunaitis, J.J.; Luke, J.J.; Hellmann, M.D.; Kummar, S.; Doebele, R.C.; Mahadevan, D. Safety and clinical activity of adenosine A2a receptor (A2aR) antagonist, CPI-444, in anti-PD1/PDL1 treatment-refractory renal cell (RCC) and non-small cell lung cancer (NSCLC) patients. Am. Soc. Clin. Oncol. 2017. [Google Scholar] [CrossRef]
- Hammami, A.; Allard, D.; Allard, B.; Stagg, J. Targeting the adenosine pathway for cancer immunotherapy. Semin. Immunol. 2019, 42, 101304. [Google Scholar] [CrossRef]
- Parolini, I.; Federici, C.; Raggi, C.; Lugini, L.; Palleschi, S.; De Milito, A.; Coscia, C.; Iessi, E.; Logozzi, M.; Molinari, A. Microenvironmental pH is a key factor for exosome traffic in tumor cells. J. Biol. Chem. 2009, 284, 34211–34222. [Google Scholar] [CrossRef] [Green Version]
- Qu, J.-L.; Qu, X.-J.; Zhao, M.-F.; Teng, Y.-E.; Zhang, Y.; Hou, K.-Z.; Jiang, Y.-H.; Yang, X.-H.; Liu, Y.-P. Gastric cancer exosomes promote tumour cell proliferation through PI3K/Akt and MAPK/ERK activation. Dig. Liver Dis. 2009, 41, 875–880. [Google Scholar] [CrossRef]
- Zhao, H.; Yang, L.; Baddour, J.; Achreja, A.; Bernard, V.; Moss, T.; Marini, J.C.; Tudawe, T.; Seviour, E.G.; San Lucas, F.A. Tumor microenvironment derived exosomes pleiotropically modulate cancer cell metabolism. elife 2016, 5, e10250. [Google Scholar] [CrossRef] [PubMed]
- Achreja, A.; Zhao, H.; Yang, L.; Yun, T.H.; Marini, J.; Nagrath, D. Exo-MFA–a 13C metabolic flux analysis framework to dissect tumor microenvironment-secreted exosome contributions towards cancer cell metabolism. Metab. Eng. 2017, 43, 156–172. [Google Scholar] [CrossRef] [PubMed]
- Lazar, I.; Clement, E.; Dauvillier, S.; Milhas, D.; Ducoux-Petit, M.; Legonidec, S.; Moro, C.; Soldan, V.; Dalle, S.; Balor, S. Adipocyte exosomes promote melanoma aggressiveness through fatty acid oxidation: A novel mechanism linking obesity and cancer. Cancer Res. 2016, 76, 4051–4057. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Patsoukis, N.; Bardhan, K.; Chatterjee, P.; Sari, D.; Liu, B.; Bell, L.N.; Karoly, E.D.; Freeman, G.J.; Petkova, V.; Seth, P. PD-1 alters T-cell metabolic reprogramming by inhibiting glycolysis and promoting lipolysis and fatty acid oxidation. Nat. Commun. 2015, 6, 1–13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Patsoukis, N.; Brown, J.; Petkova, V.; Liu, F.; Li, L.; Boussiotis, V.A. Selective effects of PD-1 on Akt and Ras pathways regulate molecular components of the cell cycle and inhibit T cell proliferation. Sci. Signal. 2012, 5, ra46. [Google Scholar] [CrossRef] [Green Version]
- Parry, R.V.; Chemnitz, J.M.; Frauwirth, K.A.; Lanfranco, A.R.; Braunstein, I.; Kobayashi, S.V.; Linsley, P.S.; Thompson, C.B.; Riley, J.L. CTLA-4 and PD-1 receptors inhibit T-cell activation by distinct mechanisms. Mol. Cell. Biol. 2005, 25, 9543–9553. [Google Scholar] [CrossRef] [Green Version]
- Li, X.; Wenes, M.; Romero, P.; Huang, S.C.-C.; Fendt, S.-M.; Ho, P.-C. Navigating metabolic pathways to enhance antitumour immunity and immunotherapy. Nat. Rev. Clin. Oncol. 2019, 16, 425–441. [Google Scholar] [CrossRef]


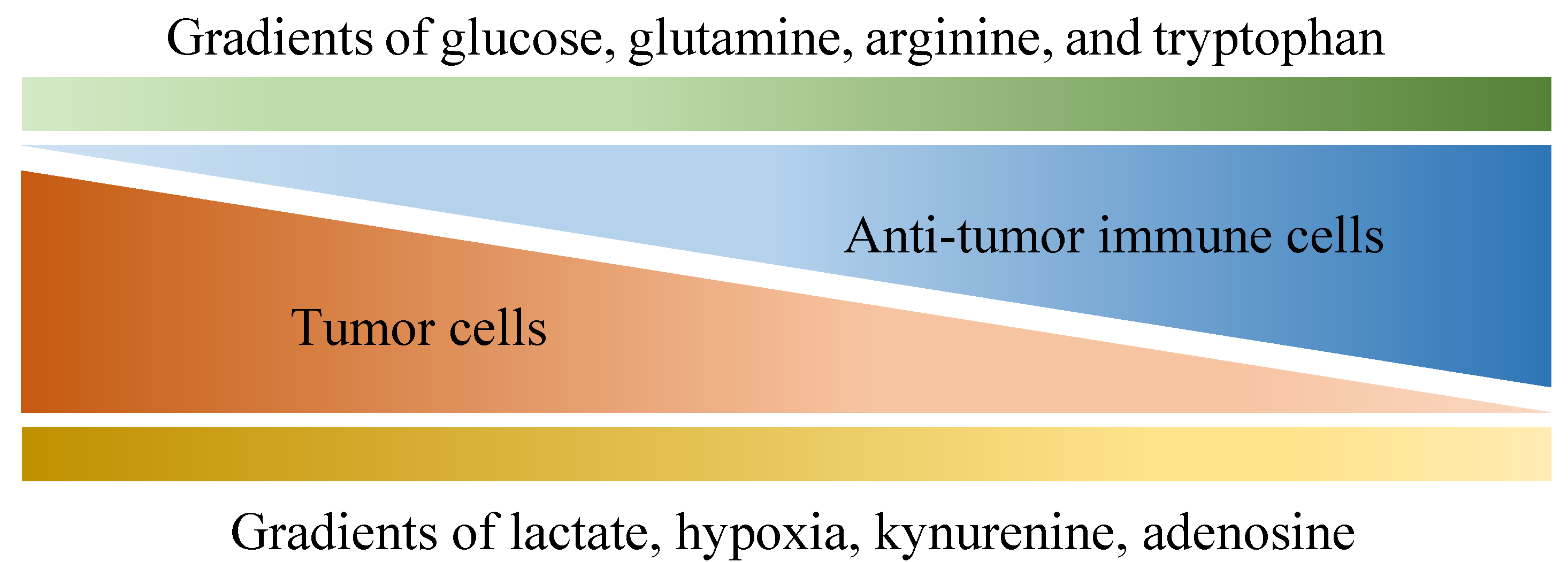{kind=link}
| Glycolysis | OXPHOS | FAO | FAS | |
|---|---|---|---|---|
| Naïve T cell | ○ | ○ | ||
| Effector T cell | ○ | ○ | ○ | |
| Memory T cell | ○ | ○ | ○ | |
| Regulatory T cell | ○ | ○ |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yang, H.-Y.; Wu, C.-Y.; Powell, J.D.; Lu, K.-L. Manipulation of Metabolic Pathways and Its Consequences for Anti-Tumor Immunity: A Clinical Perspective. Int. J. Mol. Sci. 2020, 21, 4030. https://doi.org/10.3390/ijms21114030
Yang H-Y, Wu C-Y, Powell JD, Lu K-L. Manipulation of Metabolic Pathways and Its Consequences for Anti-Tumor Immunity: A Clinical Perspective. International Journal of Molecular Sciences. 2020; 21(11):4030. https://doi.org/10.3390/ijms21114030
Chicago/Turabian StyleYang, Huang-Yu, Chao-Yi Wu, Jonathan D. Powell, and Kun-Lin Lu. 2020. "Manipulation of Metabolic Pathways and Its Consequences for Anti-Tumor Immunity: A Clinical Perspective" International Journal of Molecular Sciences 21, no. 11: 4030. https://doi.org/10.3390/ijms21114030
APA StyleYang, H. -Y., Wu, C. -Y., Powell, J. D., & Lu, K. -L. (2020). Manipulation of Metabolic Pathways and Its Consequences for Anti-Tumor Immunity: A Clinical Perspective. International Journal of Molecular Sciences, 21(11), 4030. https://doi.org/10.3390/ijms21114030