Lemon Balm Extract ALS-L1023 Regulates Obesity and Improves Insulin Sensitivity via Activation of Hepatic PPARα in High-Fat Diet-Fed Obese C57BL/6J Mice


Abstract
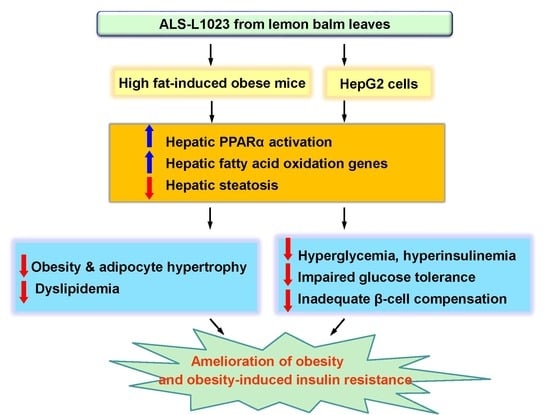
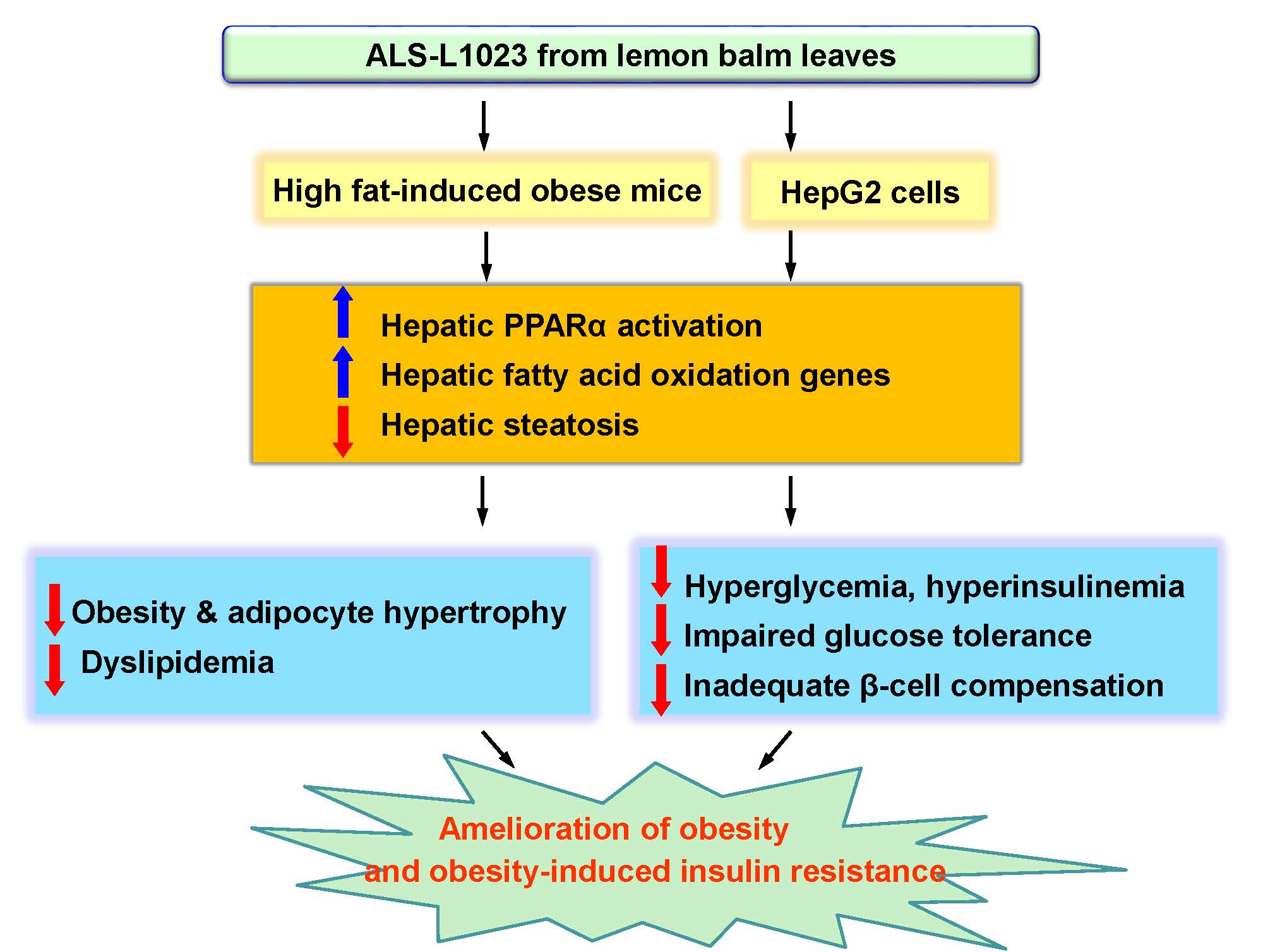
:
1. Introduction
2. Results
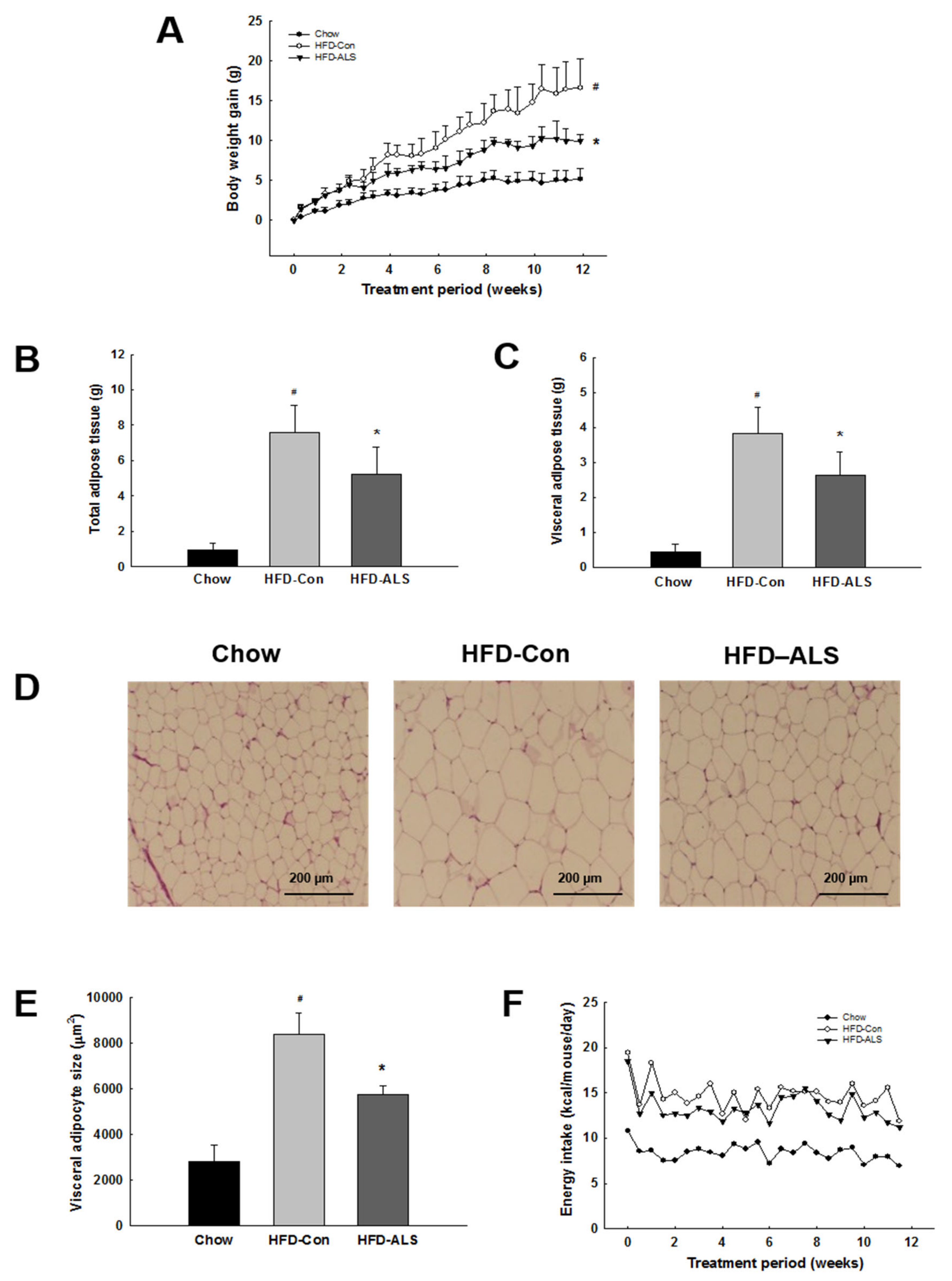
2.1. ALS-L1023 Reduces Weight Gain and Visceral Adipocyte Size in HFD-Fed Obese Mice
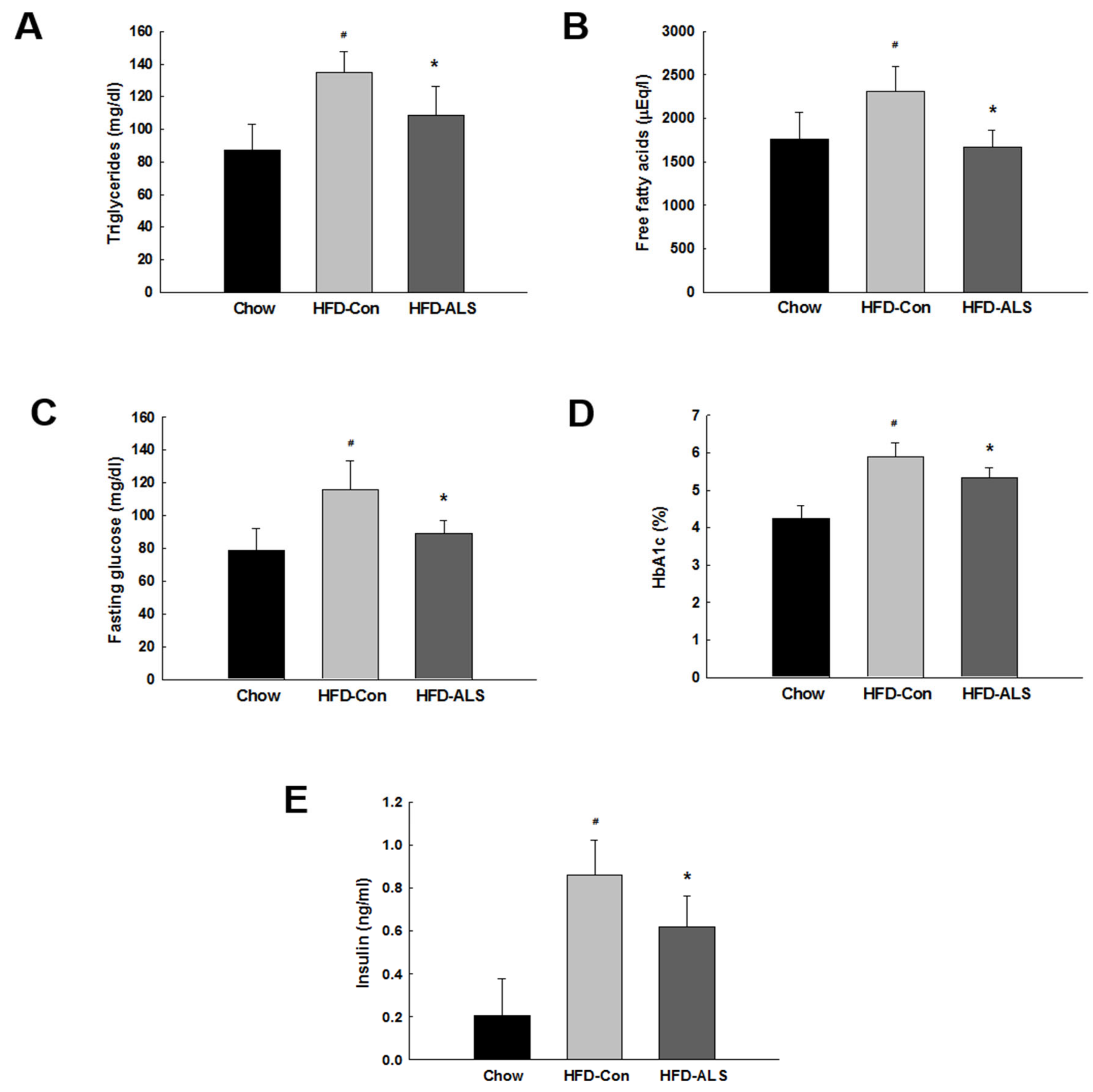
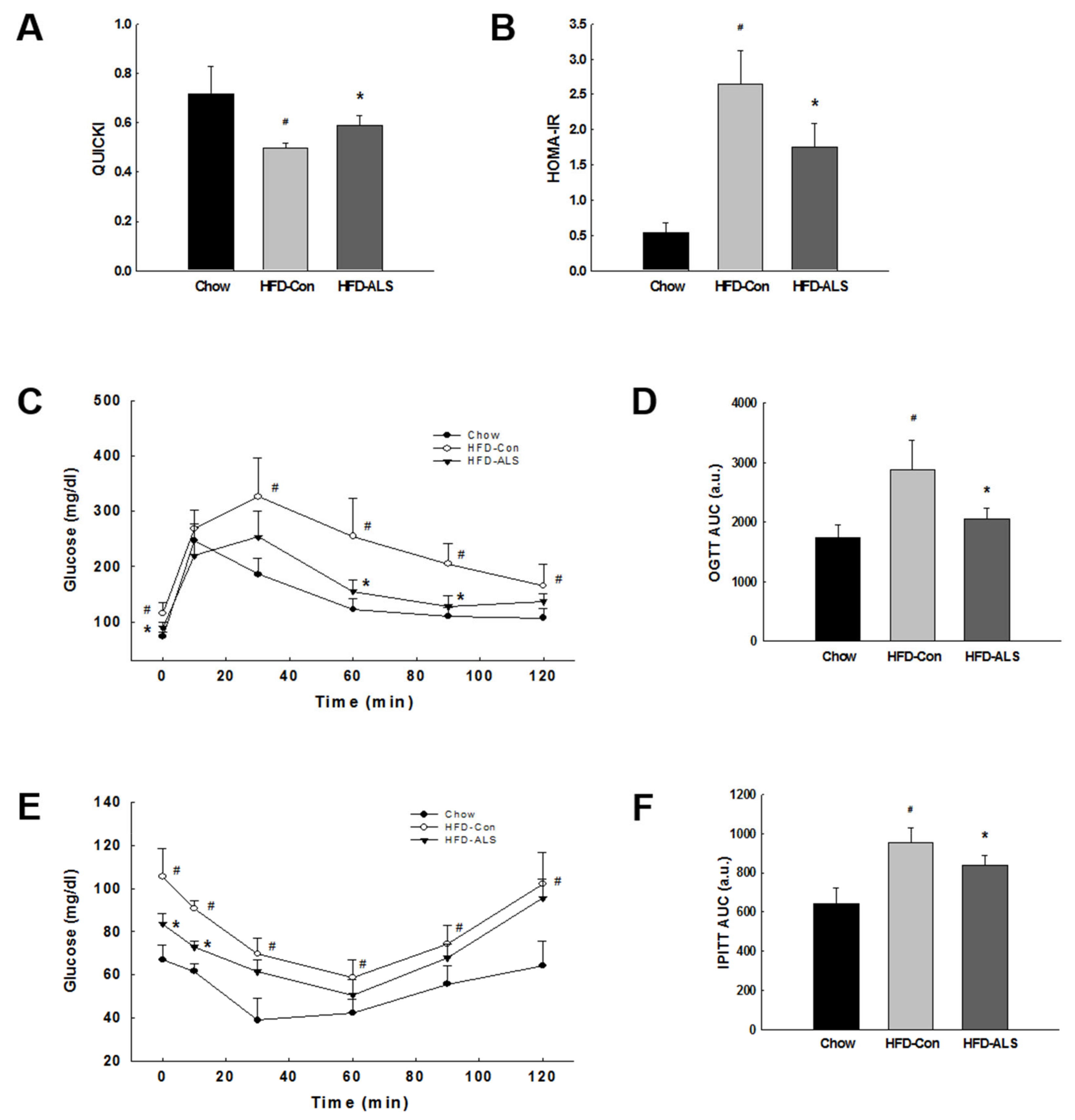
2.2. ALS-L1023 Lowers Elevated Glucose Levels and Increases Insulin Sensitivity in HFD-Fed Obese Mice
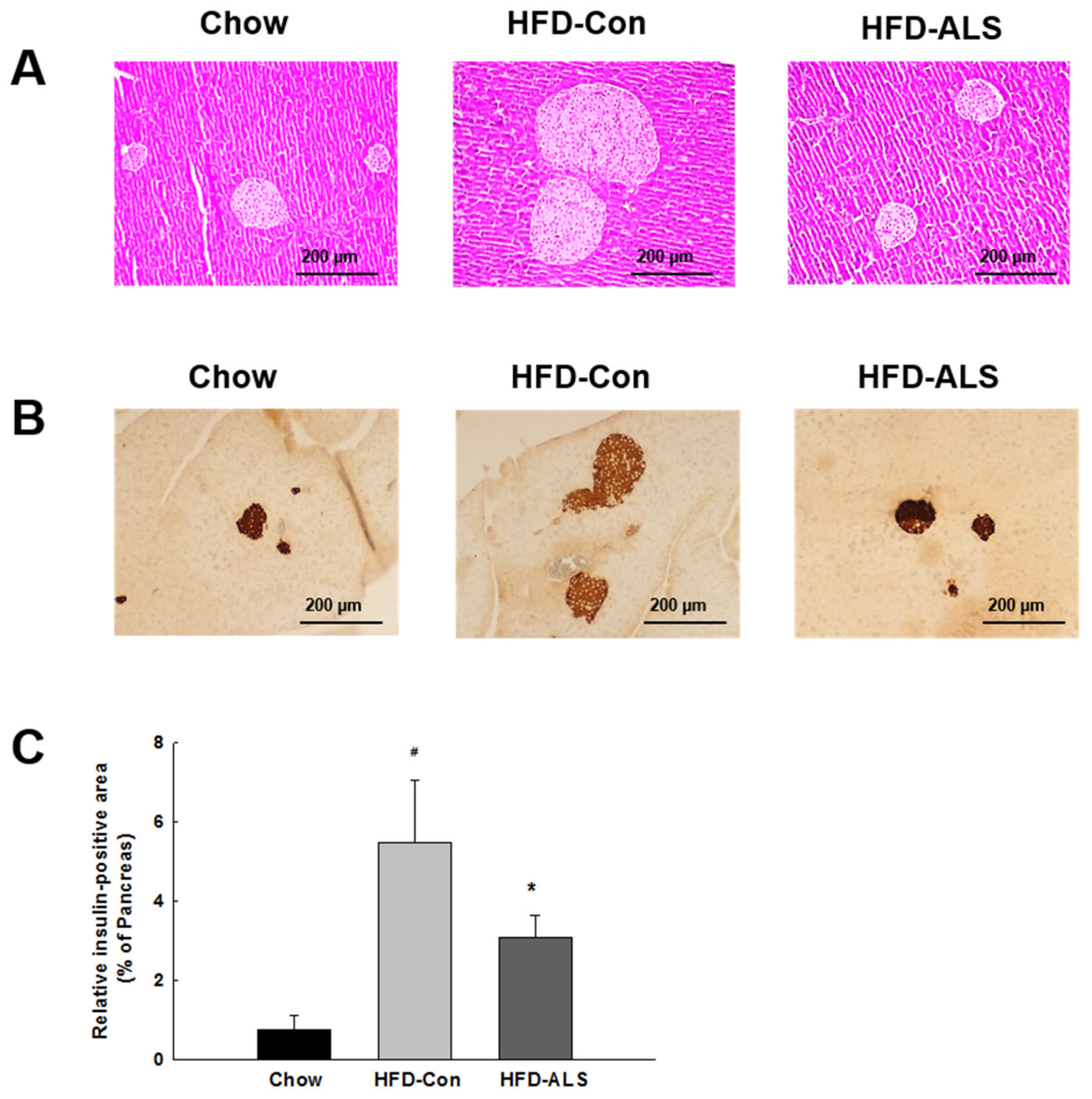
2.3. ALS-L1023 Normalizes Insulin-Positive β-Cell Mass in HFD-Fed Obese Mice
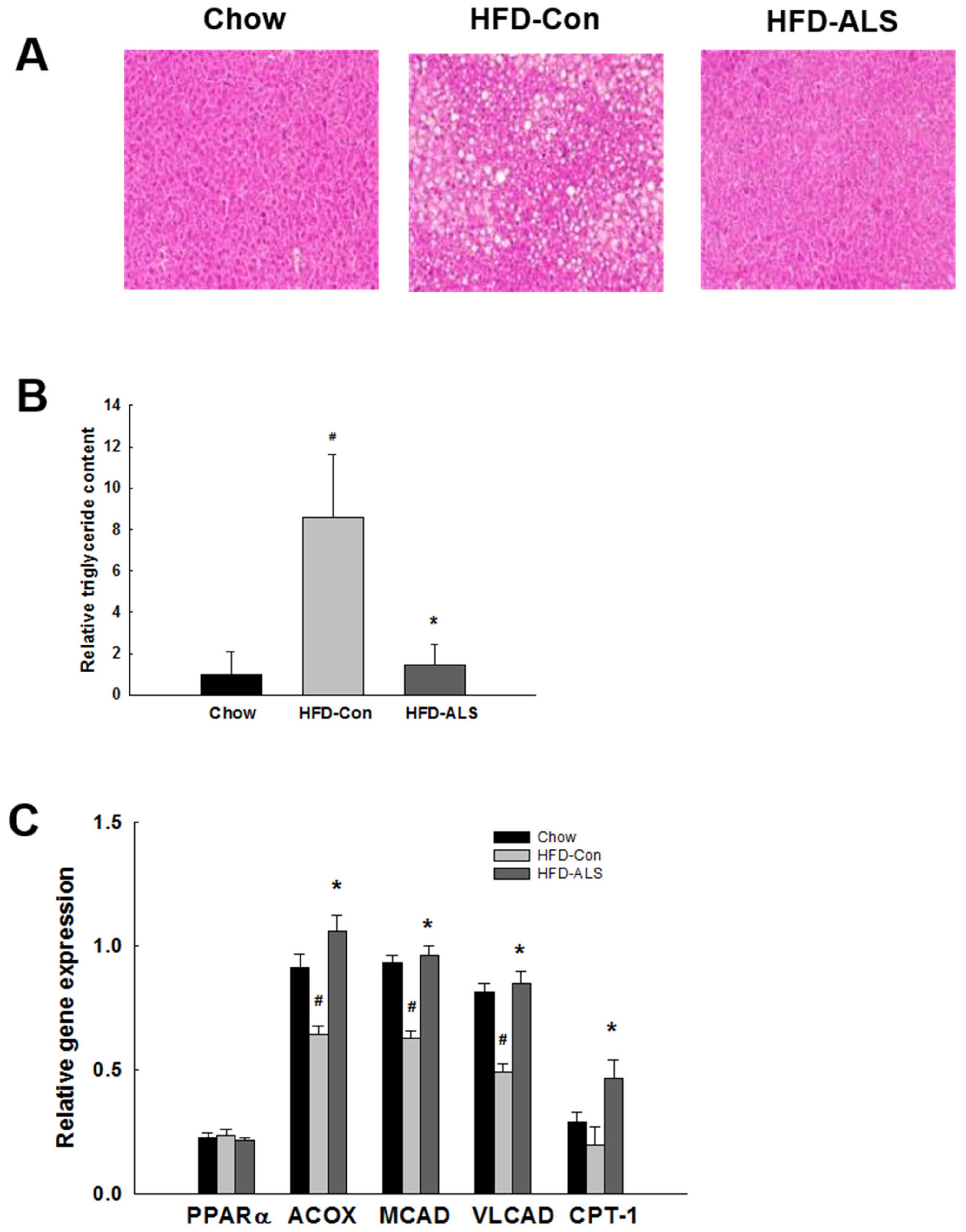
2.4. ALS-L1023 Inhibits Hepatic Steatosis and Increases Hepatic PPARα Target Gene Expression in HFD-Fed Obese Mice
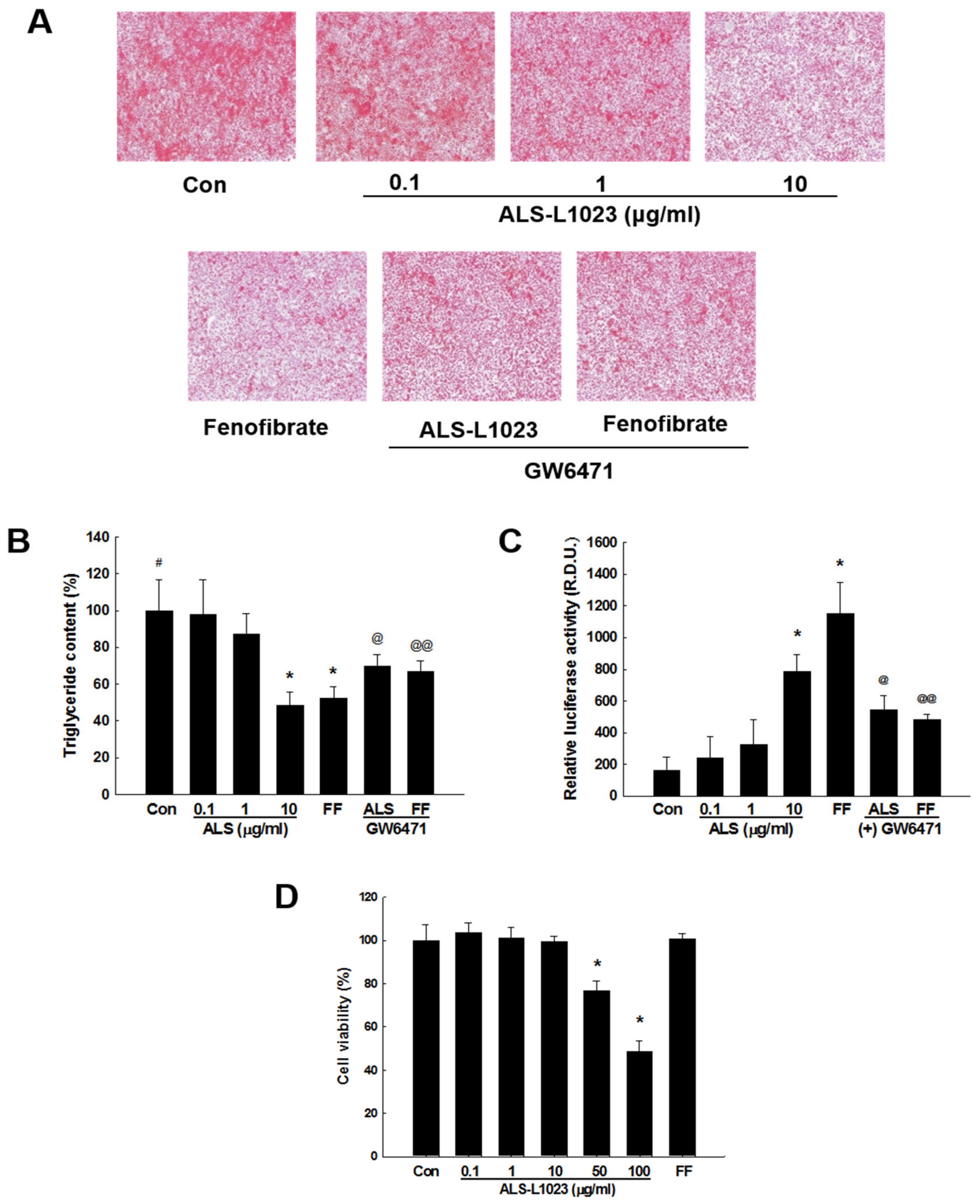
2.5. ALS-L1023 Decreases Lipid Accumulation and Increases PPARα Activity in HepG2 Cells
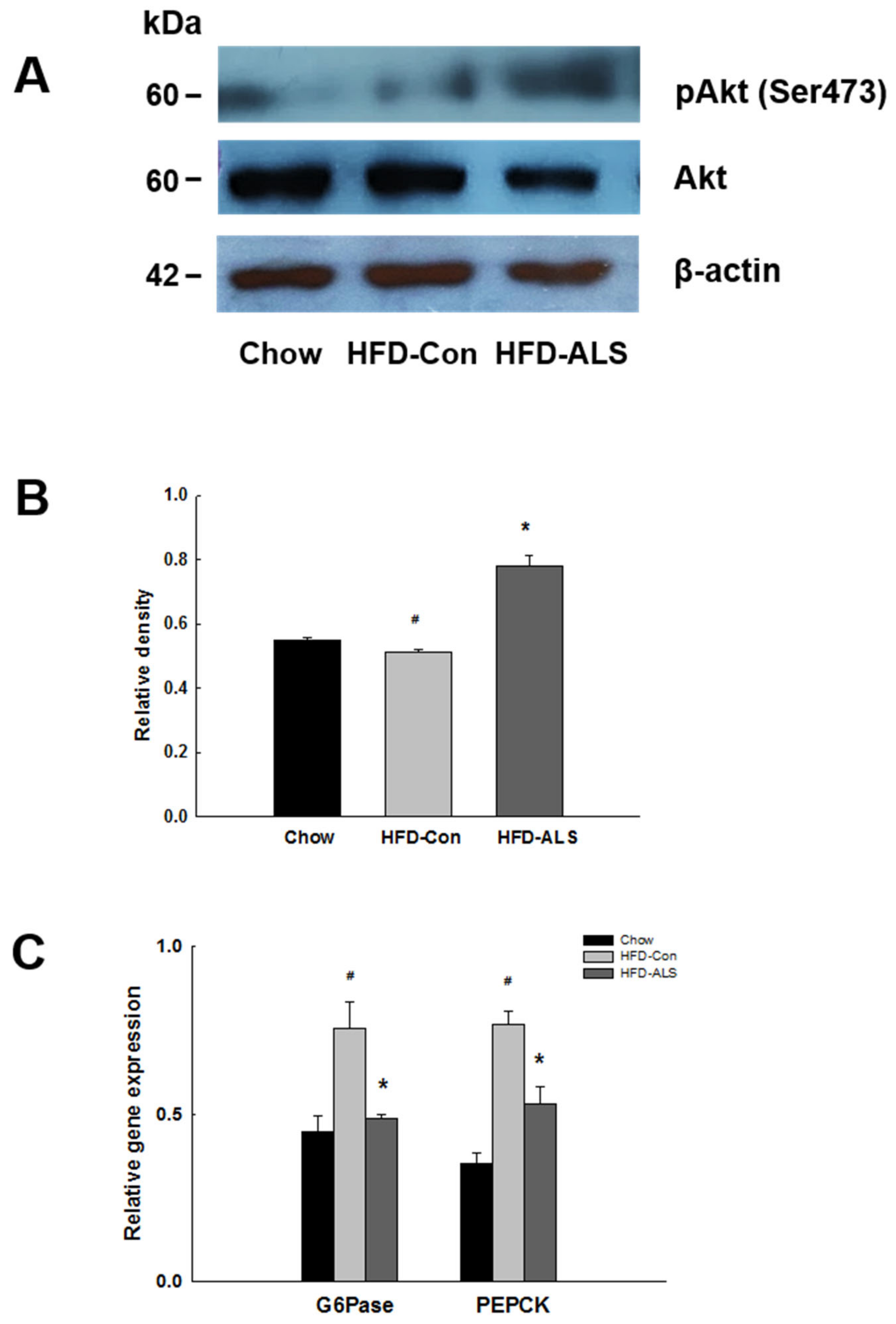
2.6. ALS-L1023 Activates Akt Phosphorylation and Decreases Expression of Gluconeogenesis Genes in HFD-Fed Obese Mice
3. Discussion
4. Materials and Methods
4.1. Preparation of ALS-L1023
4.2. Animals and Treatments
4.3. Cell Culture, Treatment of Free Fatty Acids, and Analysis of Triglyceride Content
4.4. In Vitro Cytotoxicity Test
4.5. Blood Analysis
4.6. Histological Analysis
4.7. Western Blot Analysis
4.8. Quantitative Reverse Transcription Polymerase Chain Reaction (qRT-PCR)
4.9. Transient Transfection Assay
4.10. Statistical Analysis
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Ahren, B.; Pacini, G. Insufficient islet compensation to insulin resistance vs. reduced glucose effectiveness in glucose-intolerant mice. Am. J. Physiol. Metab. 2002, 283, E738–E744. [Google Scholar] [CrossRef] [PubMed]
- Kahn, S.E. The relative contributions of insulin resistance and beta-cell dysfunction to the pathophysiology of Type 2 diabetes. Diabetology 2003, 46, 3–19. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Prentki, M.; Nolan, C.J. Islet β cell failure in type 2 diabetes. J. Clin. Investig. 2006, 116, 1802–1812. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lara-Castro, C.; Garvey, W.T. Intracellular Lipid Accumulation in Liver and Muscle and the Insulin Resistance Syndrome. Endocrinol. Metab. Clin. North Am. 2008, 37, 841–856. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Krššák, M.; Petersen, K.F.; Dresner, A.; di Pietro, L.; Vogel, S.M.; Rothman, D.L.; Shulman, G.I.; Roden, M. Intramyocellular lipid concentrations are correlated with insulin sensitivity in humans: A 1 H NMR spectroscopy study. Diabetology 1999, 42, 113–116. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kelley, D.E.; McKolanis, T.M.; Hegazi, R.A.F.; Kuller, L.H.; Kalhan, S.C. Fatty liver in type 2 diabetes mellitus: Relation to regional adiposity, fatty acids, and insulin resistance. Am. J. Physiol. Metab. 2003, 285, E906–E916. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ryysy, L.; Hakkinen, A.M.; Goto, T.; Vehkavaara, S.; Westerbacka, J.; Halavaara, J.; Yki-Jarvinen, H. Hepatic fat content and insulin action on free fatty acids and glucose metabolism rather than insulin absorption are associated with insulin requirements during insulin therapy in type 2 diabetic patients. Diabetes 2000, 49, 749–758. [Google Scholar] [CrossRef] [Green Version]
- Tolman, K.G.; Fonseca, V.; Dalpiaz, A.; Tan, M.H. Spectrum of Liver Disease in Type 2 Diabetes and Management of Patients with Diabetes and Liver Disease. Diabetes Care 2007, 30, 734–743. [Google Scholar] [CrossRef] [Green Version]
- Petersen, K.F.; Dufour, S.; Feng, J.; Befroy, U.; Dziura, J.; Man, C.D.; Cobelli, C.; Shulman, G.I. Increased prevalence of insulin resistance and nonalcoholic fatty liver disease in Asian-Indian men. Proc. Natl. Acad. Sci. USA 2006, 103, 18273–18277. [Google Scholar] [CrossRef] [Green Version]
- Fabbrini, E.; Sullivan, S.; Klein, S. Obesity, and nonalcoholic fatty liver disease: Biochemical, metabolic, and clinical implications. Hepatology 2010, 51, 679–689. [Google Scholar] [CrossRef]
- Perry, R.J.; Samuel, V.T.; Petersen, K.F.; Shulman, G.I. The role of hepatic lipids in hepatic insulin resistance and type 2 diabetes. Nature 2014, 510, 84–91. [Google Scholar] [CrossRef] [PubMed]
- Kelley, D.E.; He, J.; Menshikova, E.V.; Ritov, V.B. Dysfunction of mitochondria in human skeletal muscle in type 2 diabetes. Diabetes 2002, 51, 2944–2950. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Petersen, K.F.; Dufour, S.; Befroy, U.; Garcia, R.; Shulman, G.I. Impaired mitochondrial activity in the insulin-resistant offspring of patients with type 2 diabetes. New Engl. J. Med. 2004, 350, 664–671. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Petersen, K.F.; Befroy, U.; Dufour, S.; Dziura, J.; Ariyan, C.; Rothman, U.L.; DiPietro, L.; Cline, G.W.; Shulman, G.I. Mitochondrial Dysfunction in the Elderly: Possible Role in Insulin Resistance. Science 2003, 300, 1140–1142. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Neschen, S.; Morino, K.; Dong, J.; Wang-Fischer, Y.; Cline, G.W.; Romanelli, A.J.; Rossbacher, J.C.; Moore, I.K.; Regittnig, W.; Munoz, D.S.; et al. n-3 Fatty Acids Preserve Insulin Sensitivity In Vivo in a Peroxisome Proliferator-Activated Receptor—Dependent Manner. Diabetes 2007, 56, 1034–1041. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, N.; Liu, Z.-X.; Choi, C.S.; Tian, L.; Kibbey, R.; Dong, J.; Cline, G.W.; Wood, P.A.; Shulman, G.I. Mitochondrial dysfunction due to long-chain Acyl-CoA dehydrogenase deficiency causes hepatic steatosis and hepatic insulin resistance. Proc. Natl. Acad. Sci. USA 2007, 104, 17075–17080. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Broskey, N.T.; Obanda, D.N.; Burton, J.; Cefalu, W.T.; Ravussin, E. Skeletal muscle ceramides and daily fat oxidation in obesity and diabetes. Metabolism 2018, 82, 118–123. [Google Scholar] [CrossRef]
- McGarry, J.D. Banting lecture 2001: Dysregulation of fatty acid metabolism in the etiology of type 2 diabetes. Diabetes 2002, 51, 7–18. [Google Scholar] [CrossRef] [Green Version]
- Yoon, M. The role of PPARα in lipid metabolism and obesity: Focusing on the effects of estrogen on PPARα actions. Pharmacol. Res. 2009, 60, 151–159. [Google Scholar] [CrossRef]
- Yoon, M. PPARα in Obesity: Sex Difference and Estrogen Involvement. PPAR Res. 2010, 2010, 1–16. [Google Scholar] [CrossRef] [Green Version]
- Jeong, S.; Kim, M.; Han, M.; Lee, H.; Ahn, J.; Kim, M.; Song, Y.-H.; Shin, C.; Nam, K.-H.; Kim, T.W.; et al. Fenofibrate prevents obesity and hypertriglyceridemia in low-density lipoprotein receptor-null mice. Metabolism 2004, 53, 607–613. [Google Scholar] [CrossRef] [PubMed]
- Jeong, S.; Yoon, M. Inhibition of the Actions of Peroxisome Proliferator-activated Receptor α on Obesity by Estrogen. Obesity 2007, 15, 1430–1440. [Google Scholar] [CrossRef] [PubMed]
- Blumenthal, M.; Goldberg, A.; Brinckmann, J. Herbal Medicine-Expanded Commission E. Mongraphs; Integrative Medicine Communications: Newton, MA, USA, 2000; pp. 230–232. [Google Scholar]
- Akhondzadeh, S.; Noroozian, M.; Mohammadi, M.; Ohadinia, S.; Jamshidi, A.H.; Khani, M. Melissa officinalis extract in the treatment of patients with mild to moderate Alzheimer’s disease: A double blind, randomized, placebo-controlled trial. J. Neuro. Neurosurg. Psychiatr. 2003, 6, 625–632. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Park, B.Y.; Lee, H.; Woo, S.; Yoon, M.; Kim, J.; Hong, Y.; Lee, H.S.; Park, E.K.; Hahm, J.C.; Kim, J.W.; et al. Reduction of Adipose Tissue Mass by the Angiogenesis Inhibitor ALS-L1023 from Melissa officinalis. PLoS ONE 2015, 10, e0141612. [Google Scholar] [CrossRef]
- Kim, J.; Lee, H.; Lim, J.; Oh, J.; Shin, S.S.; Yoon, M. The Angiogenesis Inhibitor ALS-L1023 from Lemon-Balm Leaves Attenuates High-Fat Diet-Induced Nonalcoholic Fatty Liver Disease through Regulating the Visceral Adipose-Tissue Function. Int. J. Mol. Sci. 2017, 18, 846. [Google Scholar] [CrossRef]
- Woo, S.; Yoon, M.; Kim, J.; Hong, Y.; Kim, M.-Y.; Shin, S.S.; Yoon, M. The anti-angiogenic herbal extract from Melissa officinalis inhibits adipogenesis in 3T3-L1 adipocytes and suppresses adipocyte hypertrophy in high fat diet-induced obese C57BL/6J mice. J. Ethnopharmacol. 2016, 178, 238–250. [Google Scholar] [CrossRef]
- Samuel, V.T.; Shulman, G.I. The pathogenesis of insulin resistance: Integrating signaling pathways and substrate flux. J. Clin. Investig. 2016, 126, 12–22. [Google Scholar] [CrossRef] [Green Version]
- Reuter, T.Y. Diet-induced models for obesity and type 2 diabetes. Drug Discov. Today Dis. Model. 2007, 4, 3–8. [Google Scholar] [CrossRef]
- Winzell, M.S.; Ahren, B. The high-fat diet-fed mouse: A model for studying mechanisms and treatment of impaired glucose tolerance and type 2 diabetes. Diabetes 2004, 53, S215–S219. [Google Scholar] [CrossRef] [Green Version]
- Winzell, M.S.; Holm, C.; Ahren, B. Downregulation of islet hormone-sensitive lipase during long-term high-fat feeding. Biochem. Biophys. Res. Commun. 2003, 304, 273–278. [Google Scholar] [CrossRef]
- Hotamisligil, G.; Shargill, N.; Spiegelman, B. Adipose expression of tumor necrosis factor-alpha: Direct role in obesity-linked insulin resistance. Science 1993, 259, 87–91. [Google Scholar] [CrossRef] [PubMed]
- Taylor, S.I.; Barr, V.; Reitman, M.L. Does Leptin Contribute to Diabetes Caused by Obesity? Science 1996, 274, 1151. [Google Scholar] [CrossRef] [PubMed]
- Boden, G. Role of fatty acids in the pathogenesis of insulin resistance and NIDDM. Diabetes 1997, 46, 3–10. [Google Scholar] [CrossRef] [PubMed]
- Okuno, A.; Tamemoto, H.; Tobe, K.; Ueki, K.; Mori, Y.; Iwamoto, K.; Umesono, K.; Akanuma, Y.; Fujiwara, T.; Horikoshi, H.; et al. Troglitazone increases the number of small adipocytes without the change of white adipose tissue mass in obese Zucker rats. J. Clin. Investig. 1998, 101, 1354–1361. [Google Scholar] [CrossRef]
- de Souza, C.J.; Eckhardt, M.; Gagen, K.; Dong, M.; Chen, W.; Laurent, D.; Burkey, B.F. Effects of pioglitazone on adipose tissue remodeling within the setting of obesity and insulin resistance. Diabetes 2001, 50, 1863–1871. [Google Scholar] [CrossRef] [Green Version]
- Ahrén, J.; Ahren, B.; Wierup, N. Increased? Cell volume in mice fed a high-fat diet: A dynamic study over 12 months. Islets 2010, 2, 353–356. [Google Scholar] [CrossRef] [Green Version]
- Jeong, S.; Yoon, M. Fenofibrate inhibits adipocyte hypertrophy and insulin resistance by activating adipose PPARalpha in high fat diet-induced obese mice. Exp. Mol. Med. 2009, 41, 397–405. [Google Scholar] [CrossRef] [Green Version]
- Lake, B.G. Mechanisms of hepatocarcinogenicity of peroxisome-proliferating drugs and chemicals. Annu. Rev. Pharmacol. Toxicol. 1995, 35, 483–507. [Google Scholar] [CrossRef]
- Minnich, A.; Tian, N.; Byan, L.; Bilder, G. A potent PPARalpha agonist stimulates mitochondrial fatty acid beta-oxidation in liver and skeletal muscle. Am. J. Physiol. Endocrinol. Metab. 2001, 280, E270–E279. [Google Scholar] [CrossRef] [Green Version]
- Staels, B.; Dallongeville, J.; Auwerx, J.; Schoonjans, K.; Leitersdorf, E.; Fruchart, J.C. Mechanism of action of fibrates on lipid and lipoprotein metabolism. Circulation 1998, 98, 2088–2093. [Google Scholar] [CrossRef] [Green Version]
- Jeong, S.; Han, M.; Lee, H.; Kim, M.; Kim, J.; Nicol, C.J.; Kim, B.H.; Choi, J.H.; Nam, K.-H.; Oh, G.T.; et al. Effects of fenofibrate on high-fat diet-induced body weight gain and adiposity in female 57BL/6J mice. Metabolism 2004, 53, 1284–1289. [Google Scholar] [CrossRef] [PubMed]
- Oh, K.S.; Kim, M.; Lee, J.; Kim, M.J.; Nam, Y.S.; Ham, J.E.; Kim, B.H.; Choi, J.H.; Nam, K.H.; Oh, G.T.; et al. Liver PPARalpha and UCP2 are involved in the regulation of obesity and lipid metabolism by swim training in genetically obese db/db mice. Biochem. Biophys. Res. Commun. 2006, 34, 1232–1239. [Google Scholar] [CrossRef] [PubMed]
- Samuel, V.T.; Liu, Z.X.; Wang, A.; Beddow, S.A.; Geisler, J.G.; Kahn, M.; Zhang, X.M.; Monia, B.P.; Bhanot, S.; Shulman, G.I. Inhibition of protein kinase Cepsilon prevents hepatic insulin resistance in nonalcoholic fatty liver disease. J. Clin. Invest. 2007, 117, 739–745. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kumashiro, N.; Erion, D.M.; Zhang, N.; Kahn, M.; Beddow, S.A.; Chu, X.; Still, C.D.; Gerhard, G.S.; Han, X.; Dziura, J.; et al. Cellular mechanism of insulin resistance in nonalcoholic fatty liver disease. Proc. Natl. Acad. Sci. USA 2011, 108, 16381–16385. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Taniguchi, C.M.; Emanuelli, B.; Kahn, C.R. Critical nodes in signalling pathways: Insights into insulin action. Nat. Rev. Mol. Cell Boil. 2006, 7, 85–96. [Google Scholar] [CrossRef] [PubMed]
- Guerre-Millo, M.; Gervois, P.; Raspé, E.; Madsen, L.; Poulain, P.; Derudas, B.; Herbert, J.-M.; Winegar, D.A.; Willson, T.M.; Fruchart, J.-C.; et al. Peroxisome Proliferator-activated Receptor α Activators Improve Insulin Sensitivity and Reduce Adiposity. J. Boil. Chem. 2000, 275, 16638–16642. [Google Scholar] [CrossRef] [Green Version]
- Srivastava, R.A. Fenofibrate ameliorates diabetic and dyslipidemic profiles in KKAy mice partly via down-regulation of 11beta-HSD1, PEPCK and DGAT2. Comparison of PPARalpha, PPARgamma, and liver x receptor agonists. Eur. J. Pharmacol. 2009, 607, 258–263. [Google Scholar] [CrossRef]
- Nakasatomi, M.; Kim, H.; Arai, T.; Hirako, S.; Shioda, S.; Iizuka, Y.; Sakurai, K.; Matsumoto, A. Fish oil and fenofibrate inhibit pancreatic islet hypertrophy, and improve glucose and lipid metabolic dysfuntions with different ways in diabetic KK mice. Obes. Res. Clin. Pr. 2018, 12, 29–38. [Google Scholar] [CrossRef] [Green Version]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Genes | Gene Bank No. | Primer Sequences | Size (bp) | Amplification Efficiency (%) |
|---|---|---|---|---|
| ACOX | NM_015729.3 | Forward: 5′-GCCCAACTGTGACTTCCATT-3′ | 113 | 94 |
| Reverse: 5′-GGCATGTAACCCGTAGCACT-3′ | ||||
| CPT-1 | NM_013495.2 | Forward: 5′-CAGCAGCAGGTGGAACTGT-3′ | 99 | 93 |
| Reverse: 5′-GGAAACACCATAGCCGTCAT-3′ | ||||
| G6Pase | NM_010493.2 | Forward: 5′-ACACCGACTACTACAGCAACAG-3′ | 151 | 99 |
| Reverse: 5′-CCTCGAAAGATAGCAAGAGTAG-3′ | ||||
| MCAD | NM_007382.5 | Forward: 5′-TGATCAACGCGCACATTC-3′ | 53 | 98 |
| Reverse: 5′-GAACGTTCCCAGGCCAAG-3′ | ||||
| PEPCK | NM_011044.2 | Forward: 5′-CATATGCTGATCCTGGGCATAAC-3′ | 163 | 90 |
| Reverse: 5′-CAAACTTCATCCAGGCAATGTC-3′ | ||||
| PPARα | NM_011144.6 | Forward: 5′-GCAGCTCGTACAGGTCATCA-3′ | 202 | 91 |
| Reverse: 5′-CTCTTCATCCCCAAGCGTAG-3′ | ||||
| VLCAD | NM_017366.3 | Forward: 5′-GCCCAGACACACAACCTTTG-3′ | 94 | 94 |
| Reverse: 5′-CCGAGCCGACTGCATCTC-3′ | ||||
| β-actin | NM_007393.5 | Forward: 5′-TACCACAGGCATTGTGATGG-3′ | 199 | 93 |
| Reverse: 5′-TTTGATGTCACGCACGATTT-3′ |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lee, D.; Shin, Y.; Roh, J.S.; Ahn, J.; Jeoong, S.; Shin, S.S.; Yoon, M. Lemon Balm Extract ALS-L1023 Regulates Obesity and Improves Insulin Sensitivity via Activation of Hepatic PPARα in High-Fat Diet-Fed Obese C57BL/6J Mice. Int. J. Mol. Sci. 2020, 21, 4256. https://doi.org/10.3390/ijms21124256
Lee D, Shin Y, Roh JS, Ahn J, Jeoong S, Shin SS, Yoon M. Lemon Balm Extract ALS-L1023 Regulates Obesity and Improves Insulin Sensitivity via Activation of Hepatic PPARα in High-Fat Diet-Fed Obese C57BL/6J Mice. International Journal of Molecular Sciences. 2020; 21(12):4256. https://doi.org/10.3390/ijms21124256
Chicago/Turabian StyleLee, Dongju, Yujin Shin, Jong Seong Roh, Jiwon Ahn, Sunhyo Jeoong, Soon Shik Shin, and Michung Yoon. 2020. "Lemon Balm Extract ALS-L1023 Regulates Obesity and Improves Insulin Sensitivity via Activation of Hepatic PPARα in High-Fat Diet-Fed Obese C57BL/6J Mice" International Journal of Molecular Sciences 21, no. 12: 4256. https://doi.org/10.3390/ijms21124256
APA StyleLee, D., Shin, Y., Roh, J. S., Ahn, J., Jeoong, S., Shin, S. S., & Yoon, M. (2020). Lemon Balm Extract ALS-L1023 Regulates Obesity and Improves Insulin Sensitivity via Activation of Hepatic PPARα in High-Fat Diet-Fed Obese C57BL/6J Mice. International Journal of Molecular Sciences, 21(12), 4256. https://doi.org/10.3390/ijms21124256