On the Catalytic Activity of the Engineered Coiled-Coil Heptamer Mimicking the Hydrolase Enzymes: Insights from a Computational Study





Abstract
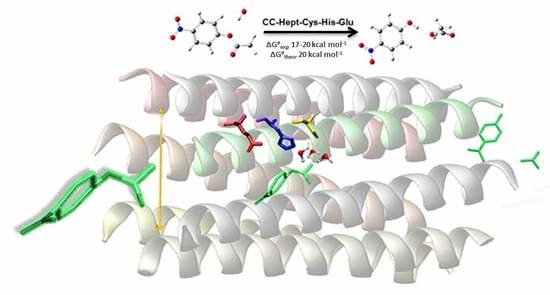
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
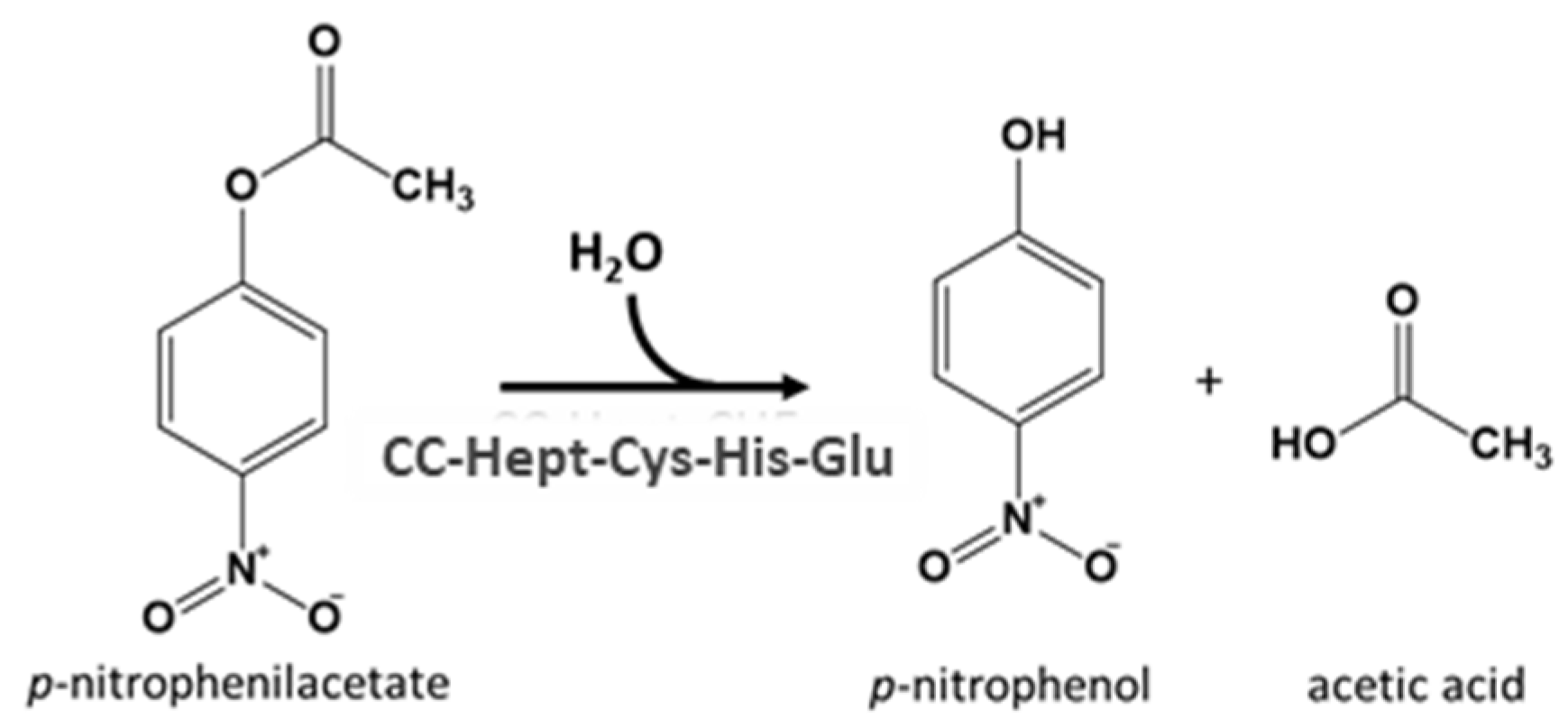
1. Introduction
2. Results
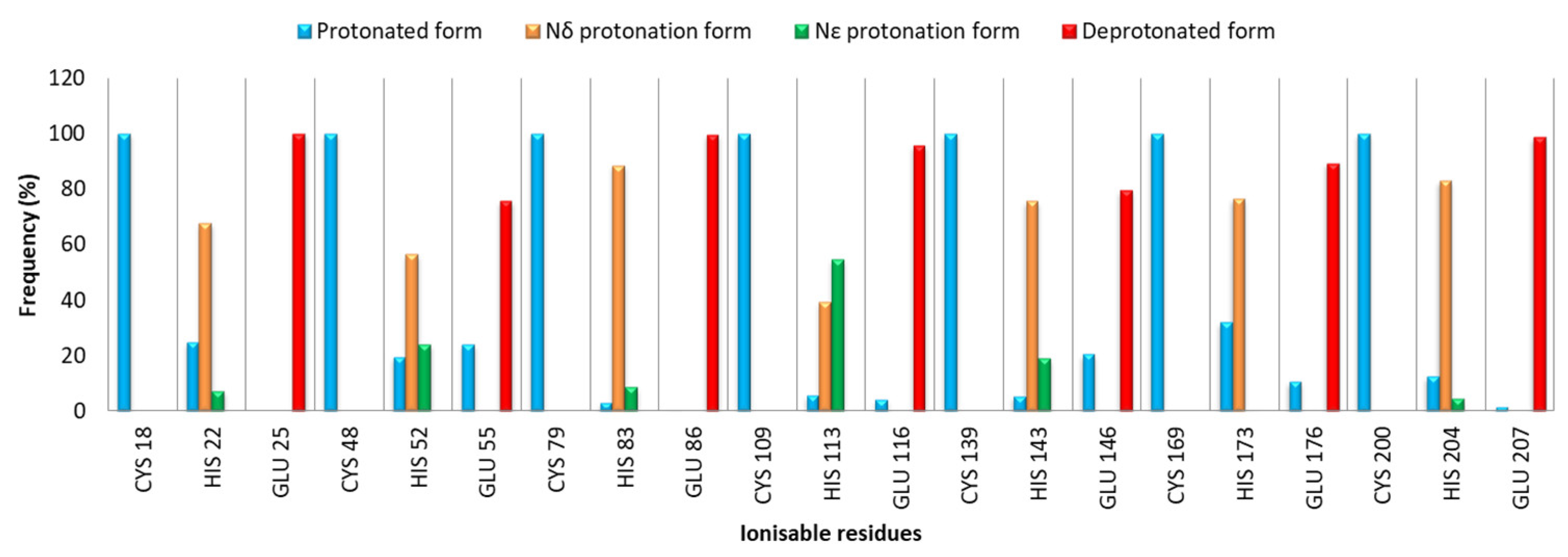
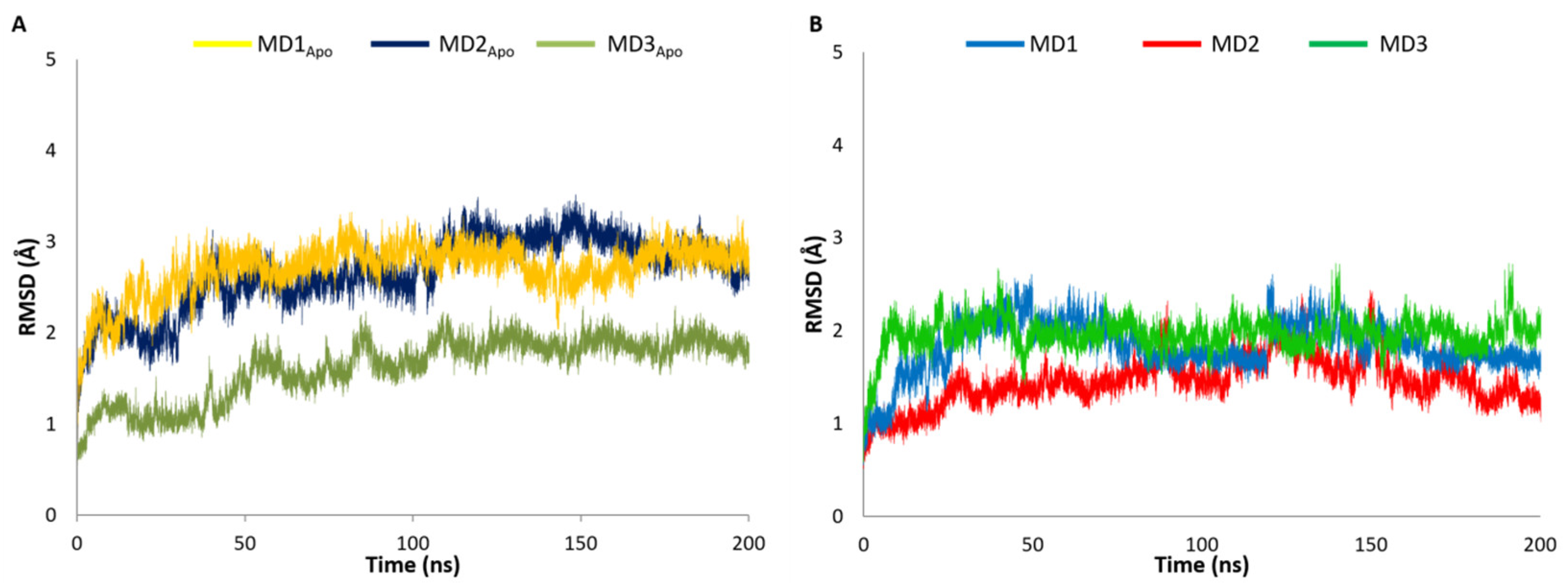
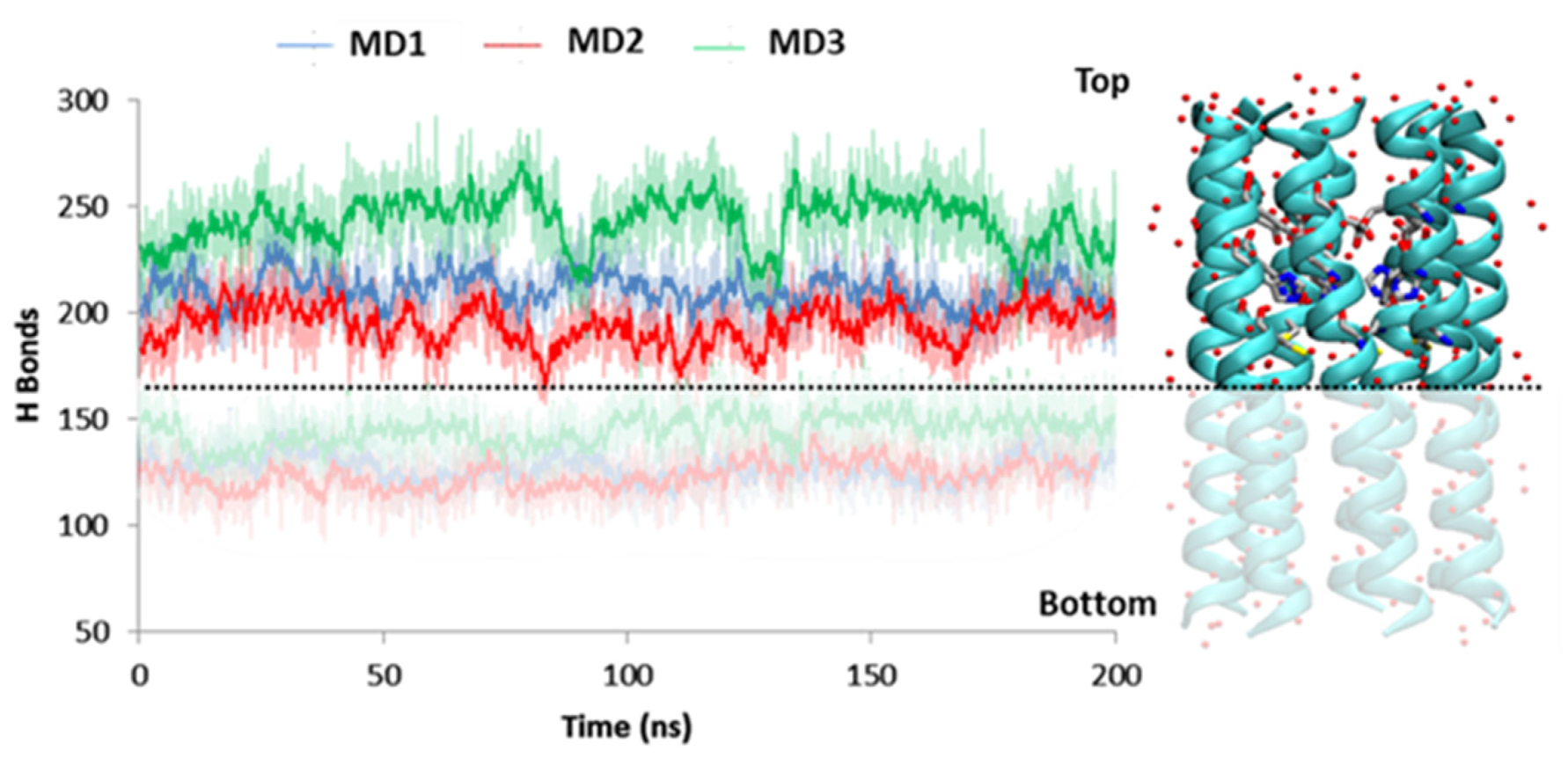
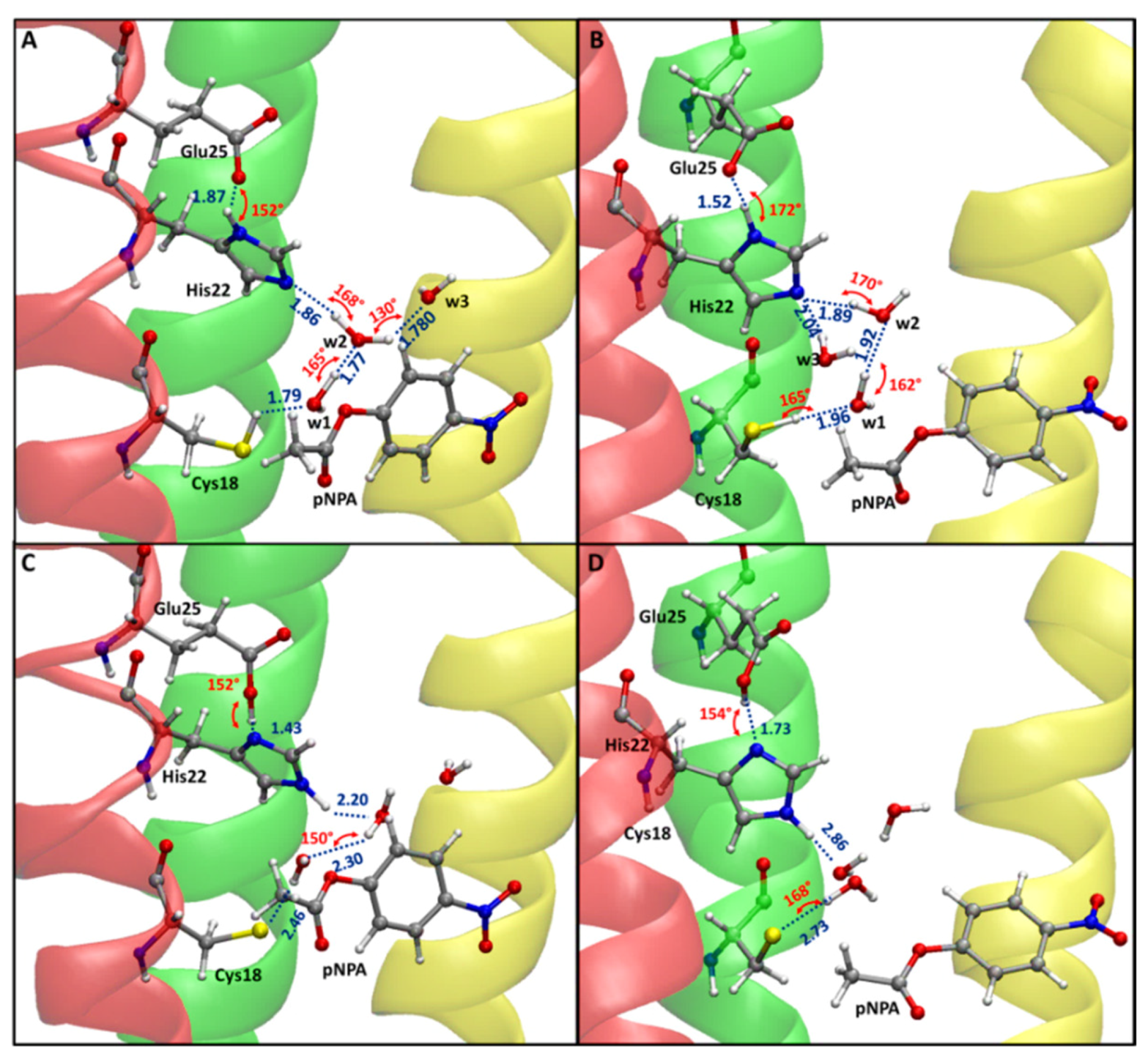
2.1. MD Investigation
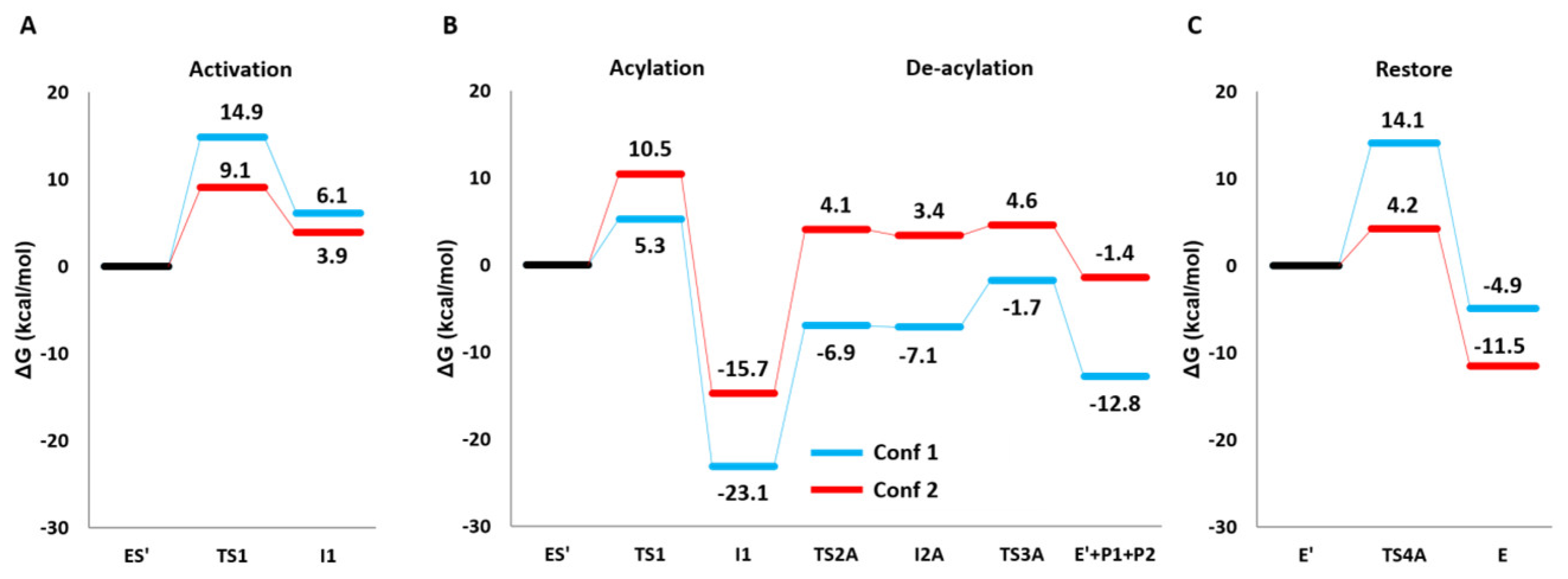
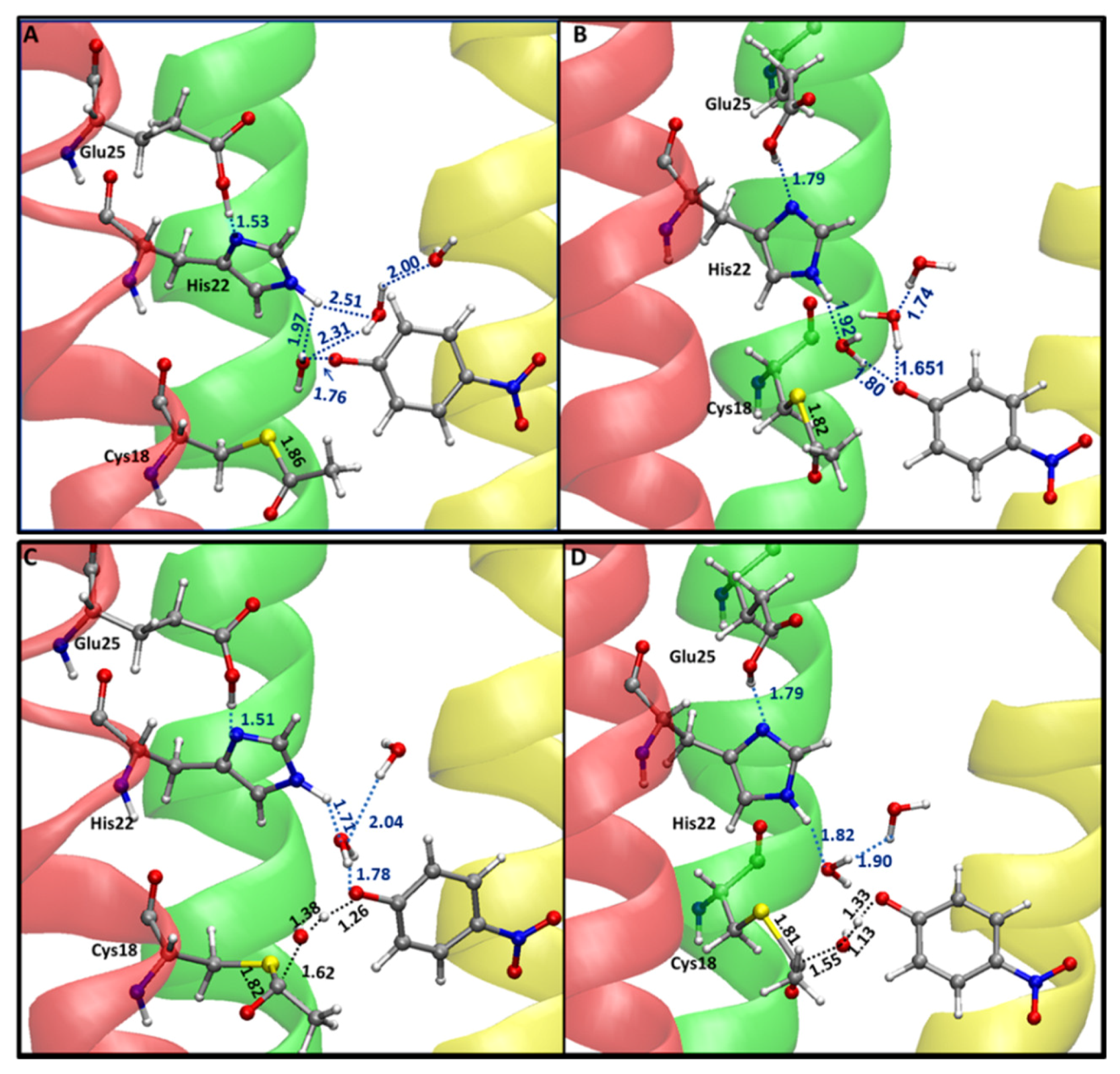
2.2. Reaction Mechanism
3. Discussion
4. Materials and Methods
4.1. MDs for CC-Hept-Cys-His-Glu and pNPA/CC-Hept-Cys-His-Glu
4.2. QM/MM
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| B3LYP | Becke-Lee-Yang-Parr |
| CC-Hept | Coiled Coil-Heptamer |
| CC-Hept-Cys-His-Glu | Coiled Coil-Heptamer-Cysteine-Histidine-Glutamate |
| CpHMD | Constant pH Molecular Dynamics |
| DFT | Density Functional Theory |
| FES | Free Energy Surfaces |
| GAFF | General Amber Force Field |
| MD | Molecular Dynamics |
| ONIOM | Our own N-layered Integrated MO and MM |
| pNPA | Para-NitroPhenylAcetate |
| QMMM | Quantum Mechanics/Molecular Mechanics |
| RESP | Restrained ElectroStatic Potential |
| RMSD | Root Mean Square Deviation |
| ZPE | Zero Point Energy |
References
- Silverman, R.B. Group Transfer Reactions: Hydrolysis, Amination, Phosphorylation. In The Organic Chemistry of Enzyme—Catalyzed Reactions, 2nd ed.; Silverman, R.B., Ed.; Elsevier. Academic Press: San Diego, CA, USA, 2002; pp. 39–94. [Google Scholar]
- Burton, A.J.; Thomson, A.R.; Dawson, W.M.; Brady, R.L.; Woolfson, D.N. Installing hydrolytic activity into a completely de novo protein framework. Nat. Chem. 2016, 8, 837–844. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Smith, A.J.; Müller, R.; Toscano, M.D.; Kast, P.; Hellinga, H.W.; Hilvert, D.; Houk, K. Structural reorganization and preorganization in enzyme active sites: Comparisons of experimental and theoretically ideal active site geometries in the multistep serine esterase reaction cycle. J. Am. Chem. Soc. 2008, 130, 15361–15373. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Makhlynets, O.V.; Korendovych, I.V. Enzyme design: Functional Frankensteins. Nat. Chem. 2016, 8, 823–824. [Google Scholar] [CrossRef] [PubMed]
- Richter, F.; Blomberg, R.; Khare, S.D.; Kiss, G.; Kuzin, A.P.; Smith, A.J.; Gallaher, J.; Pianowski, Z.; Helgeson, R.C.; Grjasnow, A. Computational design of catalytic dyads and oxyanion holes for ester hydrolysis. J. Am. Chem. Soc. 2012, 134, 16197–16206. [Google Scholar] [CrossRef] [Green Version]
- Der, B.S.; Edwards, D.R.; Kuhlman, B. Catalysis by a de novo zinc-mediated protein interface: Implications for natural enzyme evolution and rational enzyme engineering. Biochemistry 2012, 51, 3933–3940. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ma, S.; Devi-Kesavan, L.S.; Gao, J. Molecular dynamics simulations of the catalytic pathway of a cysteine protease: A combined QM/MM study of human cathepsin K. J. Am. Chem. Soc. 2007, 129, 13633–13645. [Google Scholar] [CrossRef] [Green Version]
- Amaro, R.E.; Baron, R.; McCammon, J.A. An improved relaxed complex scheme for receptor flexibility in computer-aided drug design. J. Comput. Aid. Mol. Des. 2008, 22, 693–705. [Google Scholar] [CrossRef] [Green Version]
- Thomas, F.; Dawson, W.M.; Lang, E.J.; Burton, A.J.; Bartlett, G.J.; Rhys, G.G.; Mulholland, A.J.; Woolfson, D.N. De Novo-Designed α-Helical Barrels as Receptors for Small Molecules. ACS Synth. Biol. 2018, 7, 1808–1816. [Google Scholar] [CrossRef] [Green Version]
- Marino, T.; Russo, N.; Prejanò, M. QM cluster or QM/MM in computational enzymology: The test case of LigW-decarboxylase. Front. Chem. 2018, 6, 249. [Google Scholar]
- Piazzetta, P.; Marino, T.; Russo, N. Promiscuous ability of human carbonic anhydrase: QM and QM/MM investigation of carbon dioxide and carbodiimide hydration. Inorg. Chem. 2014, 53, 3488–3493. [Google Scholar] [CrossRef]
- Sousa, S.F.; Ribeiro, A.J.; Neves, R.P.; Brás, N.F.; Cerqueira, N.M.; Fernandes, P.A.; Ramos, M.J. Application of quantum mechanics/molecular mechanics methods in the study of enzymatic reaction mechanisms. Wiley Interdiscip. Rev. Comput. Mol. Sci. 2017, 7, e1281. [Google Scholar] [CrossRef]
- Piazzetta, P.; Marino, T.; Russo, N. The working mechanism of the β-carbonic anhydrase degrading carbonyl sulphide (COSase): A theoretical study. Phys. Chem. Chem. Phys. 2015, 17, 14843–14848. [Google Scholar] [CrossRef]
- Medina, F.E.; Neves, R.P.; Ramos, M.J.; Fernandes, P.A. QM/MM Study of the Reaction Mechanism of the Dehydratase Domain from Mammalian Fatty Acid Synthase. ACS Catal. 2018, 8, 10267–10278. [Google Scholar] [CrossRef]
- Prejanò, M.; Marino, T.; Russo, N. On the Inhibition Mechanism of Glutathione Transferase P1 by Piperlongumine. Insight from Theory. Front. Chem. 2018, 6, 606. [Google Scholar] [CrossRef] [PubMed]
- Prejanò, M.; Medina, F.E.; Ramos, M.J.; Russo, N.; Fernandes, P.A.; Marino, T. How the Destabilization of a Reaction Intermediate Affects Enzymatic Efficiency: The Case of Human Transketolase. ACS Catal. 2020, 10, 2872–2881. [Google Scholar] [CrossRef]
- Bender, M.L.; Kezdy, F.J.; Wedler, F.C. α-Chymotrypsin enzyme concentration and kinetics. J. Chem. Educ. 1967, 44, 84–87. [Google Scholar] [CrossRef] [PubMed]
- Kezdy, F.J.; Bender, M.L. The kinetics of the α-chymotrypsin-catalyzed hydrolysis of p-nitrophenyl acetate. Biochemistry 1962, 1, 1097–1106. [Google Scholar] [CrossRef]
- Roos, G.; Foloppe, N.; Messens, J. Understanding the PKa of Redox Cysteines: The Key Role of Hydrogen Bonding. Antioxid. Redox Sign. 2013, 18, 94–127. [Google Scholar] [CrossRef]
- Kuntz, I.D.; Chen, K.; Sharp, K.A.; Kollman, P.A. The Maximal Affinity of Ligands. Proc. Natl. Acad. Sci. USA 1999, 96, 9997–10002. [Google Scholar] [CrossRef] [Green Version]
- Klebe, G. Applying Thermodynamic Profiling in Lead Finding and Optimization. Nat. Rev. Drug Discov. 2015, 14, 95–110. [Google Scholar] [CrossRef]
- Silva, J.R.A.; Cianni, L.; Araujo, D.; Batista, P.H.J.; de Vita, D.; Rosini, F.; Litao, A.; Lameira, J.; Montanari, C.A. Assessment of the Cruzain Cysteine Protease Reversible and Irreversible Covalent Inhibition Mechanism. J. Chem. Inf. Model. 2020, 60, 1666–1677. [Google Scholar] [CrossRef] [PubMed]
- Keillor, J.W.; Brown, R.S. Attack of Zwitterionic Ammonium Thiolates on a Distorted Anilide as a Model for the Acylation of Papain by Amides. A Simple Demonstration of a Bell-Shaped PH/ Rate Profile. J. Am. Chem. Soc. 1992, 114, 7983–7989. [Google Scholar] [CrossRef]
- Creighton, D.J.; Gessouroun, M.S.; Heapes, J.M. Is the Thiolate-Imidazolium Ion Pair the Catalytically Important Form of Papain? FEBS Lett. 1980, 110, 319–322. [Google Scholar] [CrossRef] [Green Version]
- Wei, D.; Huang, X.; Liu, J.; Tang, M.; Zhan, C.G. Reaction Pathway and Free Energy Profile for Papain-Catalysed Hydrolysis of N-Acetyl-Phe-Gly 4-Nitroanilide. Biochemistry 2013, 52, 5145–5154. [Google Scholar] [CrossRef] [Green Version]
- Radkiewicz, J.L.; Brooks, C.L. Protein dynamics in enzymatic catalysis: Exploration of dihydrofolate reductase. J. Am. Chem. Soc. 2000, 122, 225–231. [Google Scholar] [CrossRef]
- Elsasser, B.; Zauner, F.B.; Massner, J.; Soh, W.T.; Dall, E.; Brandstetter, H. Distinct Roles of Catalytic Cysteine and Histidine in the Protease and Ligase Mechanisms of Human Legumain As Revealed by DFT-Based QM/MM Simulations. ACS Catal. 2017, 7, 5585–5593. [Google Scholar] [CrossRef]
- Fuhrmann, C.N.; Daugherty, M.D.; Agard, D.A. Subangstrom crystallography reveals that short ionic hydrogen bonds, and not a His-Asp low-barrier hydrogen bond, stabilize the transition state in serine protease catalysis. J. Am. Chem. Soc. 2006, 128, 9086–9102. [Google Scholar] [CrossRef]
- Nothling, M.D.; Xiao, Z.; Bhaskaran, A.; Blyth, M.T.; Bennett, C.; Coote, M.L.; Connal, L.A. Synthetic Catalysts Inspired by Hydrolytic Enzymes. ACS Catal. 2019, 9, 168–187. [Google Scholar] [CrossRef]
- Case, D.A.; Cheatham, T.E.; Darden, T.; Gohlke, H.; Luo, R.; Merz, K.M.; Onufriev, A.; Simmerling, C.; Wang, B.; Woods, R.J. The Amber biomolecular simulation programs. J. Comput. Chem. 2005, 26, 1668–1688. [Google Scholar] [CrossRef] [Green Version]
- Schafmeister, C.; Ross, W.; Romanovski, V. LEaP; University of California: San Francisco, CA, USA, 1995. [Google Scholar]
- Maier, J.A.; Martinez, C.; Kasavajhala, K.; Wickstrom, L.; Hauser, K.E.; Simmerling, C. ff14SB: Improving the accuracy of protein side chain and backbone parameters from ff14SB. J. Chem. Theory Comput. 2015, 11, 3696–3713. [Google Scholar] [CrossRef] [Green Version]
- Anandakrishnan, R.; Aguilar, B.; Onufriev, A.V. H++ 3.0: Automating pK prediction and the preparation of biomolecular structures for atomistic molecular modeling and simulation. Nucleic Acids Res. 2012, 40, W537–W541. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wails, J.M.; York, D.M.; Roitberg, A.E. Constant pH replica exchange molecular dynamics in explicit solvent using discrete protonation states: Implementation, testing, and validation. J. Chem. Theory Comput. 2014, 10, 1341–1352. [Google Scholar]
- Laurie, A.T.; Jackson, R.M. Q-SiteFinder: An energy-based method for the prediction of protein–ligand binding sites. Bioinformatics 2005, 21, 1908–1916. [Google Scholar] [CrossRef] [PubMed]
- Berendsen, H.J.C.; Postma, J.P.M.; van Gunsteren, W.F.; Di Nola, A.; Haak, J.R. Molecular dynamics with coupling to an external bath. J. Chem. Phys. 1984, 81, 3684–3690. [Google Scholar] [CrossRef] [Green Version]
- Darden, T.; York, D.; Pedersen, L. Particle mesh Ewald: An N⋅log (N) method for Ewald sums in large systems. J. Chem. Phys. 1993, 98, 10089–10092. [Google Scholar] [CrossRef] [Green Version]
- Romeo, I.; Marascio, N.; Pavia, G.; Talarico, C.; Costa, G.; Alcaro, S.; Artese, A.; Torti, C.; Liberto, M.C.; Focà, A. Structural Modeling of New Polymorphism Clusters of HCV Polymerase Isolated from Direct-Acting Antiviral Naïve Patients: Focus on Dasabuvir and Setrobuvir Binding Affinity. ChemistrySelect 2018, 3, 6009–6017. [Google Scholar] [CrossRef]
- Baron, R.; McCammon, J.A. Dynamics, Hydration, and Motional Averaging of a Loop-Gated Artificial Protein Cavity: The W191G Mutant of Cytochrome c Peroxidase in Water as Revealed by Molecular Dynamics Simulations. Biochemistry 2007, 46, 10629–10642. [Google Scholar] [CrossRef]
- Trott, O.; Olson, A.J. AutoDock Vina: Improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J. Comput. Chem. 2010, 31, 455–461. [Google Scholar] [CrossRef] [Green Version]
- Wang, J.; Wolf, R.M.; Caldwell, J.W.; Kollman, P.A.; Case, D.A. Development and testing of a general amber force field. J. Comput. Chem. 2004, 25, 1157–1174. [Google Scholar] [CrossRef]
- Bayly, C.I.; Cieplak, P.; Cornell, W.; Kollman, P.A. A well-behaved electrostatic potential based method using charge restraints for deriving atomic charges: The RESP model. J. Phys. Chem. 1993, 97, 10269–10280. [Google Scholar] [CrossRef]
- Svensson, M.; Humbel, S.; Froese, R.D.; Matsubara, T.; Sieber, S.; Morokuma, K. ONIOM: A multilayered integrated MO+ MM method for geometry optimizations and single point energy predictions. A test for Diels—Alder reactions and Pt (P (t-Bu)3)2+ H2 oxidative addition. J. Phys. Chem. 1996, 100, 19357–19363. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Petersson, G.A.; Nakatsuji, H. Gaussian 09, Revision D.01; Gaussian Inc.: Wallingford, CT, USA, 2009. [Google Scholar]
- Vreven, T.; Byun, K.S.; Komáromi, I.; Dapprich, S.; Montgomery, J.A., Jr.; Morokuma, K.; Frisch, M.J. Combining quantum mechanics methods with molecular mechanics methods in ONIOM. J. Chem. Theory Comput. 2006, 2, 815–826. [Google Scholar] [CrossRef] [PubMed]
- Becke, A.D. Density-functional thermochemistry. III. The role of exact exchange. J. Phys. Chem. 1993, 98, 5648–5652. [Google Scholar] [CrossRef] [Green Version]
- Lee, C.; Yang, W.; Parr, R.G. Development of the Colle-Salvetti correlation-energy formula into a functional of the electron density. Phys. Rev. B 1988, 37, 785–789. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Blomberg, M.R.; Borowski, T.; Himo, F.; Liao, R.Z.; Siegbahn, P.E. Quantum chemical studies of mechanisms for metalloenzymes. Chem. Rev. 2014, 114, 3601–3658. [Google Scholar] [CrossRef] [PubMed]
- Neves, R.P.P.; Fernandes, P.A.; Ramos, M.J. Mechanistic insights on the reduction of glutathione disulfide by protein disulfide isomerase. Proc. Natl. Acad. Sci. USA 2017, 114, 4724–4733. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grimme, S. Supramolecular binding thermodynamics by dispersion-corrected density functional theory. Chem. Eur. J. 2012, 18, 9955–9964. [Google Scholar] [CrossRef]
- Grimme, S.; Ehrlich, S.; Goerigk, L. Effect of the damping function in dispersion corrected density functional theory. J. Comput. Chem. 2011, 32, 1456–1465. [Google Scholar] [CrossRef]
- Glendening, E.; Reed, A.; Carpenter, J.; Weinhold, F. NBO Version 3.1; Gaussian Inc.: Pittsburgh, PA, USA, 2003. [Google Scholar]









© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Prejanò, M.; Romeo, I.; Russo, N.; Marino, T. On the Catalytic Activity of the Engineered Coiled-Coil Heptamer Mimicking the Hydrolase Enzymes: Insights from a Computational Study. Int. J. Mol. Sci. 2020, 21, 4551. https://doi.org/10.3390/ijms21124551
Prejanò M, Romeo I, Russo N, Marino T. On the Catalytic Activity of the Engineered Coiled-Coil Heptamer Mimicking the Hydrolase Enzymes: Insights from a Computational Study. International Journal of Molecular Sciences. 2020; 21(12):4551. https://doi.org/10.3390/ijms21124551
Chicago/Turabian StylePrejanò, Mario, Isabella Romeo, Nino Russo, and Tiziana Marino. 2020. "On the Catalytic Activity of the Engineered Coiled-Coil Heptamer Mimicking the Hydrolase Enzymes: Insights from a Computational Study" International Journal of Molecular Sciences 21, no. 12: 4551. https://doi.org/10.3390/ijms21124551
APA StylePrejanò, M., Romeo, I., Russo, N., & Marino, T. (2020). On the Catalytic Activity of the Engineered Coiled-Coil Heptamer Mimicking the Hydrolase Enzymes: Insights from a Computational Study. International Journal of Molecular Sciences, 21(12), 4551. https://doi.org/10.3390/ijms21124551