Remodeling Matrix Synthesis in a Rat Model of Aortocaval Fistula and the Cyclic Stretch: Impaction in Pulmonary Arterial Hypertension-Congenital Heart Disease


Abstract
:1. Introduction
2. Results
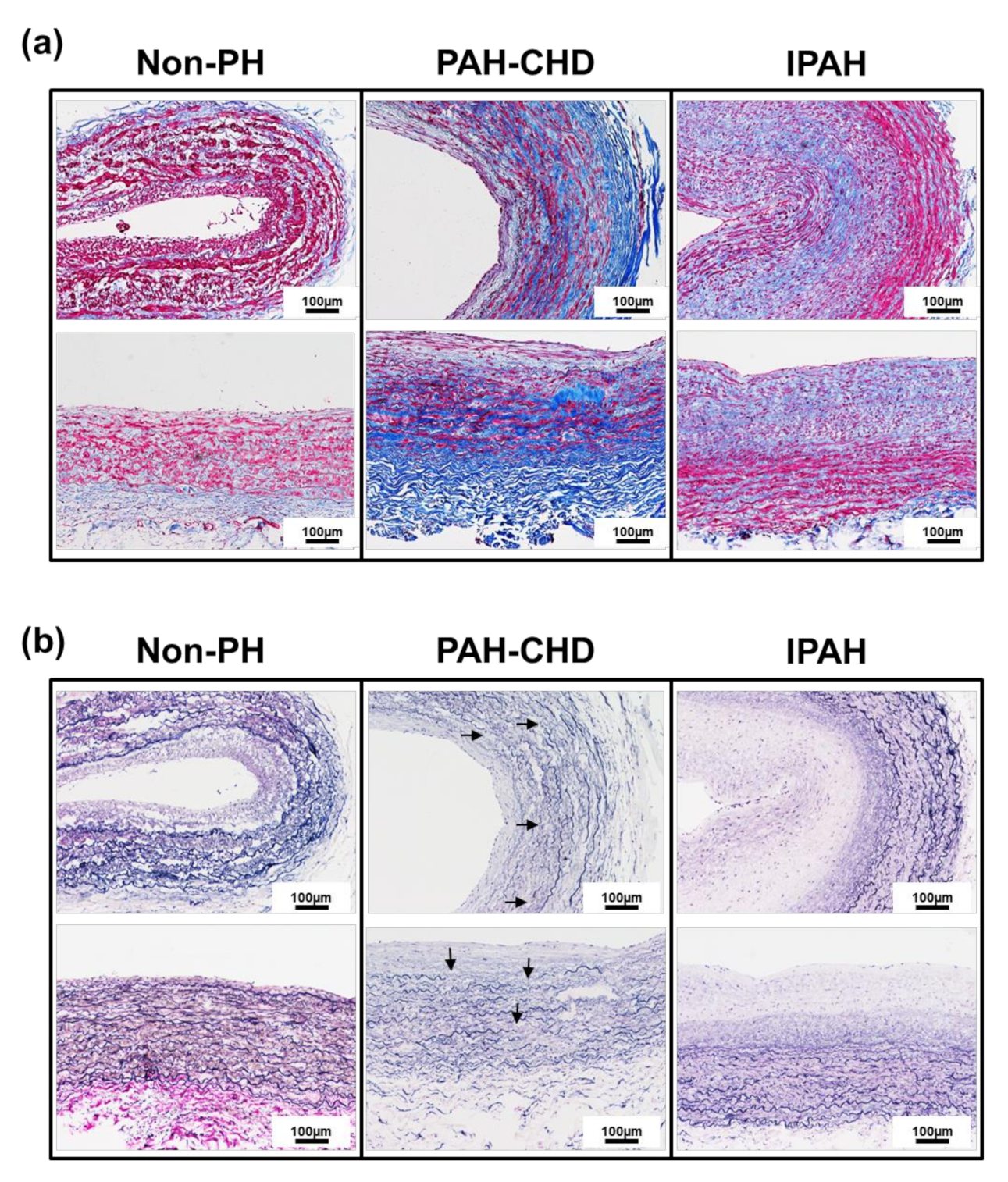
2.1. Pulmonary Arteries of Patients with PAH-CHD Are Characterized by Collagen Upregulation and Elastic Laminae Degradation
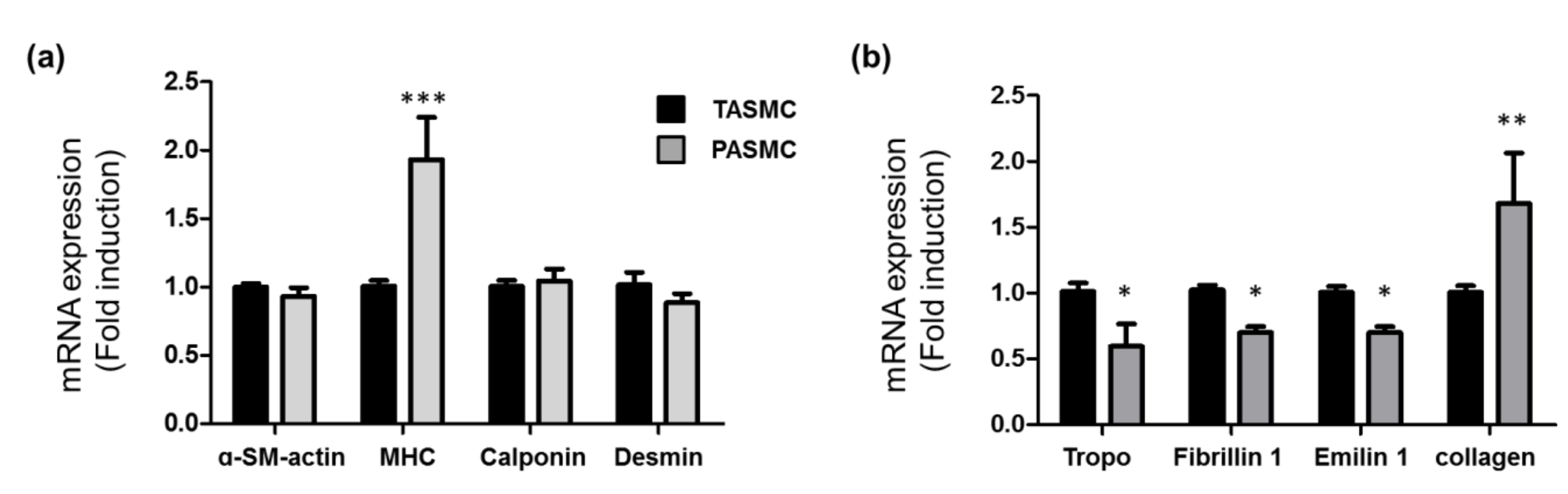
2.2. Gene Profile of SMC Markers, Extracellular Matrix and Collagen in SMCs from Pulmonary and Thorax Vessels
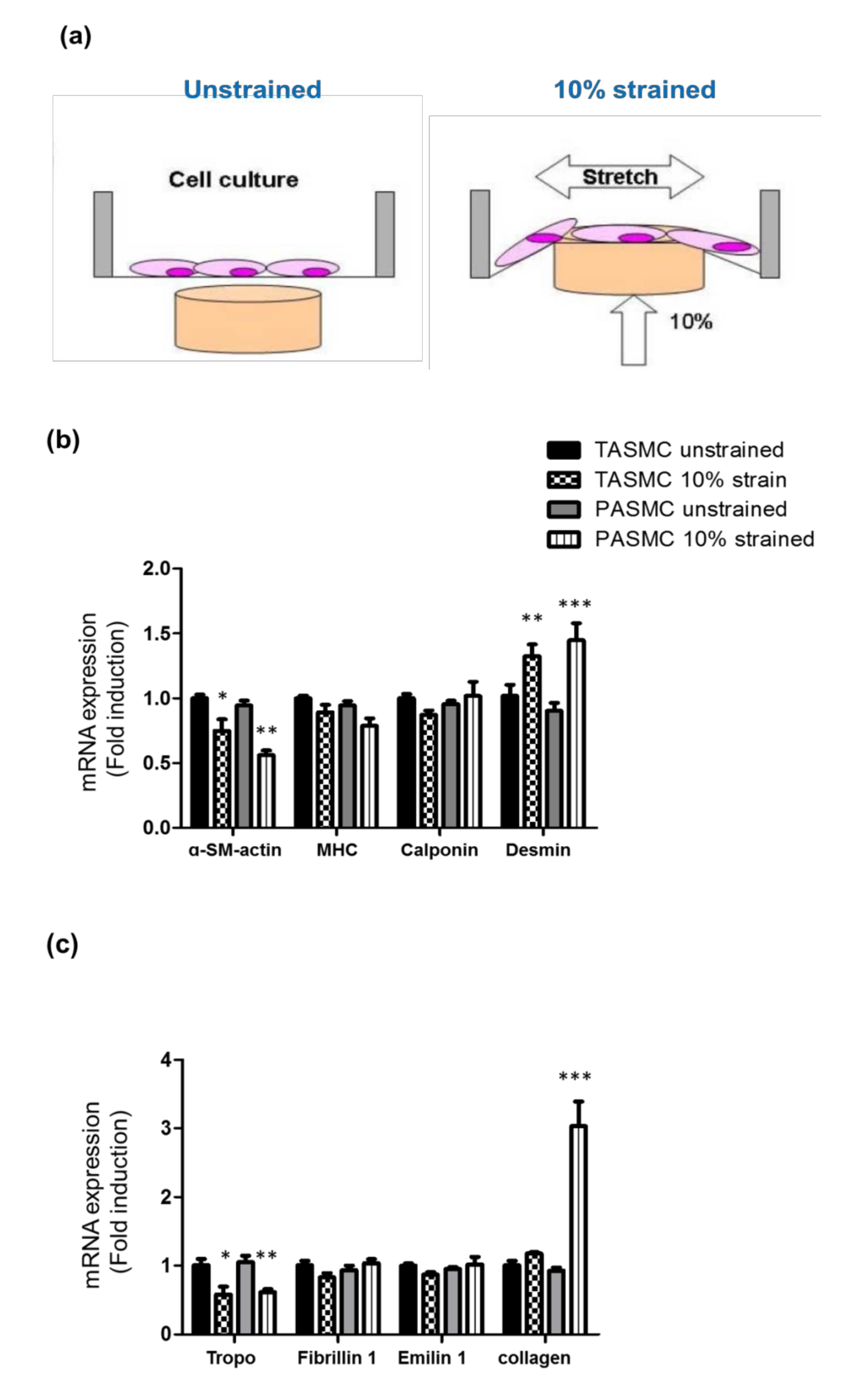
2.3. Cyclic Mechanical Stretch Affects Collagen Expression Specifically in PASMCs
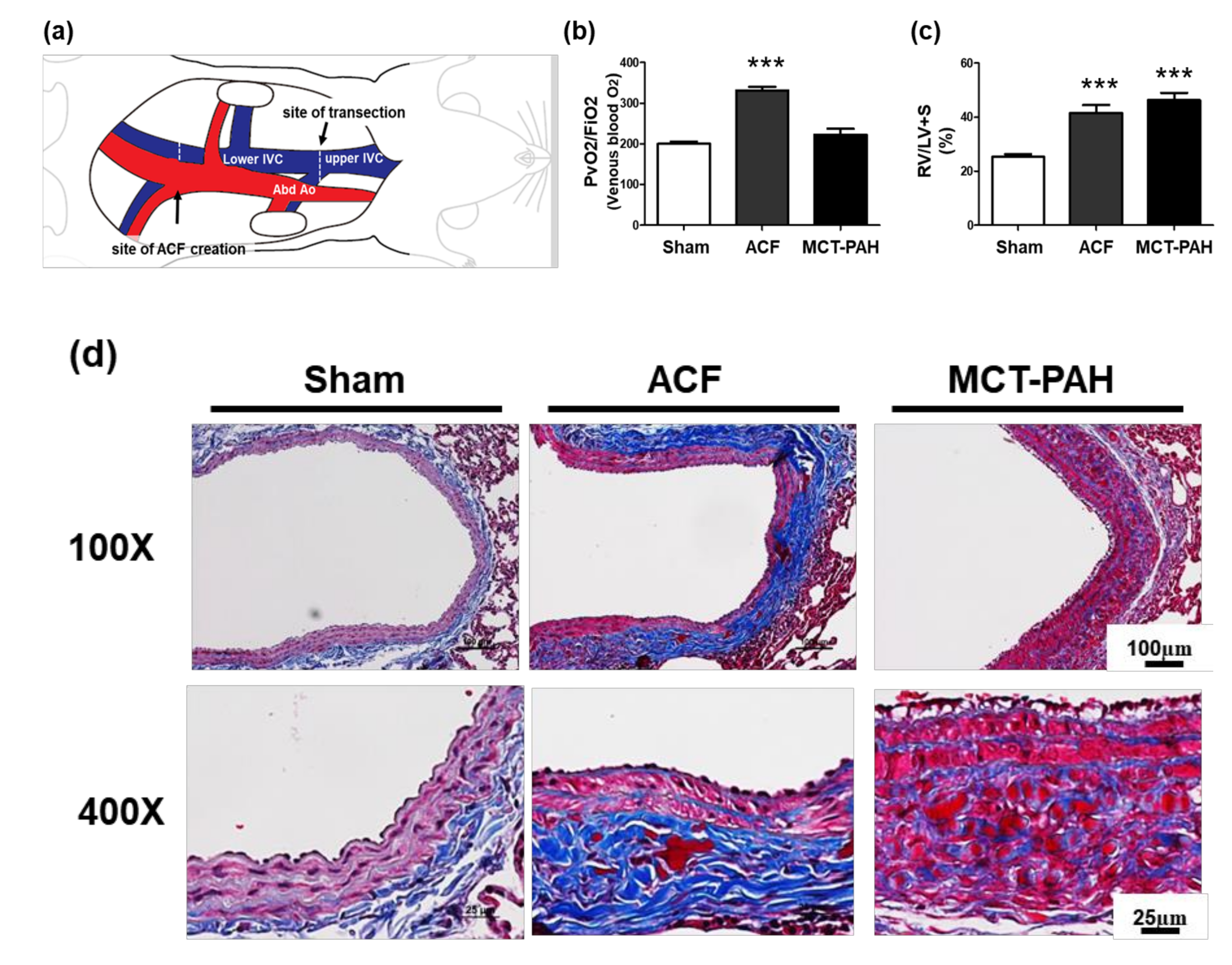
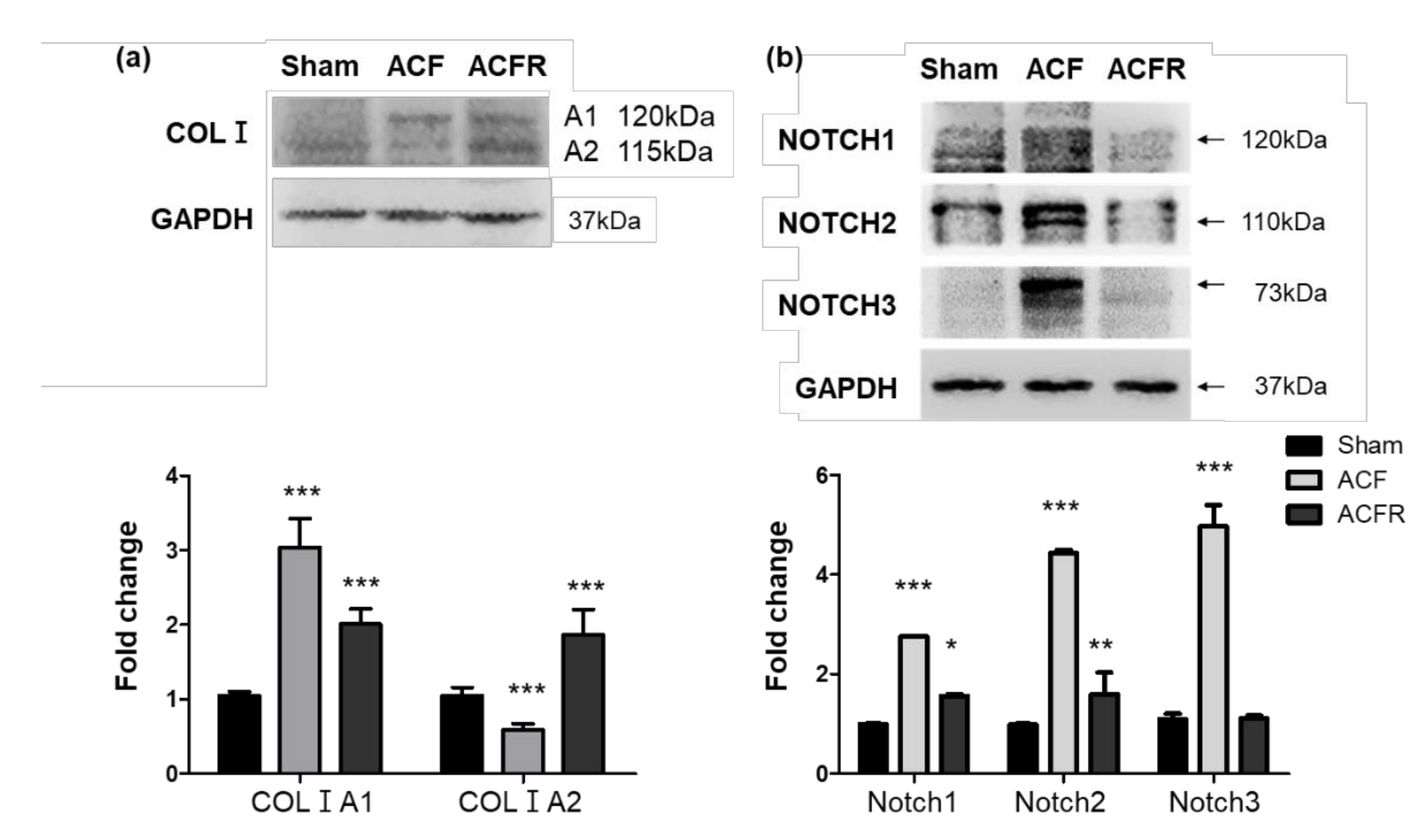
2.4. ACF Impacts Right Heart Hypertrophy and the Overexpression of Collagen, Leading to PA Stiffness
2.5. Effect of Flow Stress on Notch and Collagen Activation in the Pulmonary Circulation
3. Discussion
4. Materials and Methods
4.1. Patient Characteristics
4.2. Rat Model of ACF
4.3. Monocrotaline-Treated (MCT) Rats
4.4. Isolation and Culture of Animal Arterial SMCs
4.5. Mechanical Stretch Assay
4.6. Histological Examination and Morphometric Analysis
4.7. Total RNA Extraction and Quantitative Real-Time Reverse Transcription-Polymerase Chain Reaction
4.8. Immunoblotting
4.9. Statistical Analysis
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| ACF | Aortocaval fistula |
| ACFRs | ACF rats |
| CHD | Congenital heart disease |
| COLIA1 | Collagen I A1 |
| COLIA2 | Collagen I A2 |
| ECM | Extracellular matrix |
| GAPDH | Glyceraldehyde 3-phosphate dehydrogenase |
| IACUC | Institutional animal care and use committee |
| IPAH | Idiopathic pulmonary arterial hypertension |
| i.p. | Intraperitoneal |
| IVC | Inferior vena cava |
| MCT | Monocrotaline |
| MHC | Myosin heavy chain |
| PAH-CHD | Pulmonary arterial hypertension-congenital heart disease |
| PaO2 | Partial pressure of oxygen |
| PAs | pulmonary arteries |
| PASMCs | Pulmonary artery smooth muscle cells |
| PH | Pulmonary hypertension |
| PRN | pro re nata |
| PVR | Pulmonary vascular resistance |
| RV/LV+S | Right Ventricle weight to Left Ventricle plus septum weight |
| SE | Standard error |
| SD | Sprague-Dawley |
| SMC | Smooth muscle cell |
| TASMC | Thoracic aorta smooth muscle cell |
| VSD | Ventricular septal defect |
| VSMCs | Vascular smooth muscle cells |
References
- Gan, H.L.; Zhang, J.Q.; Zhou, Q.W.; Feng, L.; Chen, F.; Yang, Y. Patients with congenital systemic-to-pulmonary shunts and increased pulmonary vascular resistance: What predicts postoperative survival? PLoS ONE 2014, 9, e83976. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sawada, H.; Mitani, Y.; Nakayama, T.; Fukushima, H.; Kogaki, S.; Igarashi, T.; Ichida, F.; Ono, Y.; Nakanishi, T.; Doi, S.; et al. Detection of Pediatric Pulmonary Arterial Hypertension by School Electrocardiography Mass Screening. Am. J. Respir. Crit. Care Med. 2019, 199, 1397–1406. [Google Scholar] [CrossRef] [PubMed]
- Galie, N.; Humbert, M.; Vachiery, J.L.; Gibbs, S.; Lang, I.; Torbicki, A.; Simonneau, G.; Peacock, A.; Noordegraaf, A.V.; Beghetti, M.; et al. 2015 ESC/ERS Guidelines for the diagnosis and treatment of pulmonary hypertension: The Joint Task Force for the Diagnosis and Treatment of Pulmonary Hypertension of the European Society of Cardiology (ESC) and the European Respiratory Society (ERS): Endorsed by: Association for European Paediatric and Congenital Cardiology (AEPC), International Society for Heart and Lung Transplantation (ISHLT). Eur. Heart J. 2016, 37, 67–119. [Google Scholar] [PubMed]
- Huang, W.-C.; Hsu, C.-H.; Sung, S.-H.; Ho, W.-J.; Chu, C.-Y.; Chang, C.-P.; Chiu, Y.-W.; Wu, C.-H.; Chang, W.-T.; Lin, L.; et al. 2018 TSOC guideline focused update on diagnosis and treatment of pulmonary arterial hypertension. J. Formos. Med Assoc. 2019, 118, 1584–1609. [Google Scholar] [CrossRef] [PubMed]
- Galiè, N.; Torbicki, A.; Barst, R.; Dartevelle, P.; Haworth, S.; Higenbottam, T.; Olschewski, H.; Peacock, A.; Pietra, G.; Rubin, L.J.; et al. Guidelines on diagnosis and treatment of pulmonary arterial hypertension. The Task Force on Diagnosis and Treatment of Pulmonary Arterial Hypertension of the European Society of Cardiology. Eur. Heart J. 2004, 25, 2243–2278. [Google Scholar]
- An, H.S.; Kim, G.B.; Song, M.K.; Bang, J.S.; Lee, S.Y.; Bae, E.J.; Noh, C.I. Eisenmenger Syndrome in Adults: Treatment Pattern and Prognostic Factors in the Advanced Pulmonary Vasodilator Era. Pediatr. Cardiol. 2018, 40, 23–28. [Google Scholar] [CrossRef]
- Rabinovitch, M. Molecular pathogenesis of pulmonary arterial hypertension. J. Clin. Investig. 2008, 118, 2372–2379. [Google Scholar] [CrossRef] [Green Version]
- Schermuly, R.T.; Ghofrani, H.A.; Wilkins, M.R.; Grimminger, F. Mechanisms of disease: pulmonary arterial hypertension. Nat. Rev. Cardiol. 2011, 8, 443–455. [Google Scholar] [CrossRef]
- Van Der Feen, D.E.; Bartelds, B.; De Boer, R.A.; Berger, R.M. Pulmonary arterial hypertension in congenital heart disease: translational opportunities to study the reversibility of pulmonary vascular disease. Eur. Heart J. 2017, 38, 2034–2041. [Google Scholar] [CrossRef] [Green Version]
- Simonneau, G.; Montani, D.; Celermajer, D.S.; Denton, C.P.; Gatzoulis, M.A.; Krowka, M.; Williams, P.G.; Souza, R. Haemodynamic definitions and updated clinical classification of pulmonary hypertension. Eur. Respir. J. 2019, 53, 1801913. [Google Scholar] [CrossRef]
- Tojais, N.F.; Cao, A.; Lai, Y.-J.; Wang, L.; Chen, P.-I.; Alcazar, M.A.A.; Perez, V.A.D.J.; Hopper, R.K.; Rhodes, C.J.; Bill, M.A.; et al. Codependence of Bone Morphogenetic Protein Receptor 2 and Transforming Growth Factor-β in Elastic Fiber Assembly and Its Perturbation in Pulmonary Arterial Hypertension. Arter. Thromb. Vasc. Boil. 2017, 37, 1559–1569. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Humbert, M.; Guignabert, C.; Bonnet, S.; Dorfmüller, P.; Klinger, J.R.; Nicolls, M.R.; Olschewski, A.J.; Pullamsetti, S.S.; Schermuly, R.T.; Stenmark, K.R.; et al. Pathology and pathobiology of pulmonary hypertension: state of the art and research perspectives. Eur. Respir. J. 2019, 53, 1801887. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tuder, R.M.; Archer, S.L.; Dorfmüller, P.; Erzurum, S.C.; Guignabert, C.; Michelakis, E.; Rabinovitch, M.; Schermuly, R.T.; Stenmark, K.R.; Morrell, N.W. Relevant issues in the pathology and pathobiology of pulmonary hypertension. J. Am. Coll. Cardiol. 2013, 62, D4–D12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rensen, S.S.; Doevendans, P.A.F.M.; Van Eys, G.J.J.M. Regulation and characteristics of vascular smooth muscle cell phenotypic diversity. Neth. Heart J. 2007, 15, 100–108. [Google Scholar] [CrossRef] [Green Version]
- Tanaka, Y.; Schuster, D.P.; Davis, E.C.; A Patterson, G.; Botney, M.D. The role of vascular injury and hemodynamics in rat pulmonary artery remodeling. J. Clin. Investig. 1996, 98, 434–442. [Google Scholar] [CrossRef] [Green Version]
- Yanagisawa, H.; Davis, E.C. Unraveling the mechanism of elastic fiber assembly: The roles of short fibulins. Int. J. Biochem. Cell Boil. 2010, 42, 1084–1093. [Google Scholar] [CrossRef] [Green Version]
- Tamez, Z.S.E.; Safdar, Z.; Frost, A.; Guffey, D.; Minard, C.G.; Entman, M.L. Collagen Metabolism Biomarkers and Health Related Quality of Life in Pulmonary Arterial Hypertension. Int. J. Cardiovasc. Res. 2015, 4. [Google Scholar] [CrossRef]
- Wang, X.; Shi, K.; Li, J.; Chen, T.; Guo, Y.; Liu, Y.; Yang, Y.; Yang, S. Effects of angiotensin II intervention on MMP-2, MMP-9, TIMP-1, and collagen expression in rats with pulmonary hypertension. Genet. Mol. Res. 2015, 14, 1707–1717. [Google Scholar] [CrossRef]
- Wagenseil, J.E.; Mecham, R.P. Vascular extracellular matrix and arterial mechanics. Physiol. Rev. 2009, 89, 957–989. [Google Scholar] [CrossRef] [Green Version]
- Raaz, U.; Schellinger, I.N.; Chernogubova, E.; Warnecke, C.; Kayama, Y.; Penov, K.; Hennigs, J.K.; Salomons, F.; Eken, S.M.; Emrich, F.C.; et al. Transcription Factor Runx2 Promotes Aortic Fibrosis and Stiffness in Type 2 Diabetes Mellitus. Circ. Res. 2015, 117, 513–524. [Google Scholar] [CrossRef] [Green Version]
- Komarova, E.A.; Diatchenko, L.; Rokhlin, O.W.; E Hill, J.; Wang, Z.J.; I Krivokrysenko, V.; Feinstein, E.; Gudkov, A.V. Stress-induced secretion of growth inhibitors: a novel tumor suppressor function of p53. Oncogene 1998, 17, 1089–1096. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Niwa, K.; Perloff, J.K.; Bhuta, S.M.; Laks, H.; Drinkwater, D.C.; Child, J.S.; Miner, P.D. Structural abnormalities of great arterial walls in congenital heart disease: light and electron microscopic analyses. Circulation 2001, 103, 393–400. [Google Scholar] [CrossRef] [PubMed]
- Maruyama, K.; Ye, C.L.; Woo, M.; Venkatacharya, H.; Lines, L.D.; Silver, M.M.; Rabinovitch, M. Chronic hypoxic pulmonary hypertension in rats and increased elastolytic activity. Am. J. Physiol. Circ. Physiol. 1991, 261, H1716–H1726. [Google Scholar] [CrossRef] [PubMed]
- Wagenseil, J.E.; Mecham, R.P. New insights into elastic fiber assembly. Birth Defects Res. Part C Embryo Today Rev. 2007, 81, 229–240. [Google Scholar] [CrossRef] [PubMed]
- Nave, A.H.; Ikova, I.M.; Niess, G.; Steenbock, H.; Reichenberger, F.; Talavera, M.L.; Veit, F.; Herold, S.; Mayer, K.; Weissmann, N.; et al. Lysyl Oxidases Play a Causal Role in Vascular Remodeling in Clinical and Experimental Pulmonary Arterial Hypertension. Arter. Thromb. Vasc. Boil. 2014, 34, 1446–1458. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rabinovitch, M.; Bothwell, T.; Hayakawa, B.N.; Williams, W.G.; A Trusler, G.; Rowe, R.D.; Olley, P.M.; Cutz, E. Pulmonary artery endothelial abnormalities in patients with congenital heart defects and pulmonary hypertension. A correlation of light with scanning electron microscopy and transmission electron microscopy. Lab. Investig. 1986, 55, 632–653. [Google Scholar] [PubMed]
- Spiekerkoetter, E.; Alvira, C.M.; Kim, Y.M.; Bruneau, A.; Pricola, K.L.; Wang, L.; Ambartsumian, N.; Rabinovitch, M. Reactivation of gammaHV68 induces neointimal lesions in pulmonary arteries of S100A4/Mts1-overexpressing mice in association with degradation of elastin. Am. J. Physiol. Lung Cell Mol. Physiol. 2008, 294, L276–L289. [Google Scholar] [CrossRef]
- Shifren, J.L.; Rifai, N.; Desindes, S.; McIlwain, M.; Doros, G.; Mazer, N.A. A comparison of the short-term effects of oral conjugated equine estrogens versus transdermal estradiol on C-reactive protein, other serum markers of inflammation, and other hepatic proteins in naturally menopausal women. J. Clin. Endocrinol. Metab. 2008, 93, 1702–1710. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chang, C.-J.; Hsu, H.-C.; Ho, W.-J.; Chang, G.-J.; Pang, J.-H.S.; Chen, W.-J.; Huang, C.-C.; Lai, Y.-J. Cathepsin S promotes the development of pulmonary arterial hypertension. Am. J. Physiol. Cell. Mol. Physiol. 2019, 317, L1–L13. [Google Scholar] [CrossRef] [PubMed]
- Ye, C.L.; Rabinovitch, M. Inhibition of elastolysis by SC-37698 reduces development and progression of monocrotaline pulmonary hypertension. Am. J. Physiol. Circ. Physiol. 1991, 261, H1255–H1267. [Google Scholar] [CrossRef]
- Provencher, S.; Archer, S.L.; Ramirez, F.D.; Hibbert, B.; Paulin, R.; Boucherat, O.; Lacasse, Y.; Bonnet, S. Standards and Methodological Rigor in Pulmonary Arterial Hypertension Preclinical and Translational Research. Circ. Res. 2018, 122, 1021–1032. [Google Scholar] [CrossRef] [PubMed]
- Frid, M.G.; Dempsey, E.C.; Durmowicz, A.G.; Stenmark, K.R. Smooth muscle cell heterogeneity in pulmonary and systemic vessels. Importance in vascular disease. Arter. Thromb. Vasc. Boil. 1997, 17, 1203–1209. [Google Scholar] [CrossRef] [PubMed]
- Chang, C.-J.; Chen, C.-C.; Hsu, L.-A.; Chang, G.-J.; Ko, Y.-H.; Chen, C.-F.; Chen, M.-Y.; Yang, S.-H.; Pang, J.-H.S. Degradation of the Internal Elastic Laminae in Vein Grafts of Rats with Aortocaval Fistulae. Am. J. Pathol. 2009, 174, 1837–1846. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, X.; Zhang, X.; Leathers, R.; Makino, A.; Huang, C.; Parsa, P.; Macias, J.; Yuan, J.X.-J.; Jamieson, S.W.; Thistlethwaite, P.A. Notch3 signaling promotes the development of pulmonary arterial hypertension. Nat. Med. 2009, 15, 1289–1297. [Google Scholar] [CrossRef] [Green Version]
- Lai, Y.-J.; Chang, G.-J.; Yeh, Y.-H.; Pang, J.-H.S.; Huang, C.-C.; Chen, W.-J. Propylthiouracil Attenuates Experimental Pulmonary Hypertension via Suppression of Pen-2, a Key Component of Gamma-Secretase. PLOS ONE 2015, 10, e0137426. [Google Scholar] [CrossRef]
- Wagenvoort, C. Open lung biopsies in congenital heart disease for evaluation of pulmonary vascular disease. Predictive value with regard to corrective operability. Histopathol. 1985, 9, 417–436. [Google Scholar] [CrossRef]
- Haworth, S.G. Pulmonary vascular disease in ventricular septal defect: Structural and functional correlations in lung biopsies from 85 patients, with outcome of intra-cardiac repair. J. Pathol. 1987, 152, 157–168. [Google Scholar] [CrossRef] [PubMed]
- Baeten, J.; Lilly, B. Notch Signaling in Vascular Smooth Muscle Cells. HIV-1 Mol. Boil. Pathog. 2016, 78, 351–382. [Google Scholar]
- Morrow, D.; Guha, S.; Sweeney, C.; Birney, Y.; Walshe, T.; O’Brien, C.; Walls, D.; Redmond, E.M.; Cahill, P.A. Notch and Vascular Smooth Muscle Cell Phenotype. Circ. Res. 2008, 103, 1370–1382. [Google Scholar] [CrossRef] [Green Version]
- Van Loon, R.L.E.; Roofthooft, M.T.; Hillege, H.L.; Harkel, A.D.T.; Van Osch-Gevers, M.; Delhaas, T.; Kapusta, L.; Strengers, J.L.M.; Rammeloo, L.; Clur, S.-A.B.; et al. Pediatric Pulmonary Hypertension in the Netherlands. Circulation 2011, 124, 1755–1764. [Google Scholar] [CrossRef] [Green Version]
- Van Riel, A.C.; Schuuring, M.J.; Van Hessen, I.D.; Zwinderman, A.H.; Cozijnsen, L.; Reichert, C.L.; Hoorntje, J.C.; Wagenaar, L.J.; Post, M.C.; Van Dijk, A.; et al. Contemporary prevalence of pulmonary arterial hypertension in adult congenital heart disease following the updated clinical classification. Int. J. Cardiol. 2014, 174, 299–305. [Google Scholar] [CrossRef] [PubMed]
- Engelfriet, P.M.; Duffels, M.G.J.; Möller, T.; Boersma, E.; Tijssen, J.G.P.; Thaulow, E.; A Gatzoulis, M.; Mulder, B. Pulmonary arterial hypertension in adults born with a heart septal defect: the Euro Heart Survey on adult congenital heart disease. Heart 2006, 93, 682–687. [Google Scholar] [CrossRef] [Green Version]
- Owens, G.K.; Kumar, M.S.; Wamhoff, B.R. Molecular Regulation of Vascular Smooth Muscle Cell Differentiation in Development and Disease. Physiol. Rev. 2004, 84, 767–801. [Google Scholar] [CrossRef]
- Owens, G.K. Regulation of differentiation of vascular smooth muscle cells. Physiol. Rev. 1995, 75, 487–517. [Google Scholar] [CrossRef]
- Mantella, L.-E.; Quan, A.; Verma, S. Variability in vascular smooth muscle cell stretch-induced responses in 2D culture. Vasc. Cell 2015, 7, 7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lai, Y.-J.; Chen, I.-C.; Li, H.-H.; Huang, C.-C. EP4 Agonist L-902,688 Suppresses EndMT and Attenuates Right Ventricular Cardiac Fibrosis in Experimental Pulmonary Arterial Hypertension. Int. J. Mol. Sci. 2018, 19, 727. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lai, Y.-J.; Pullamsetti, S.S.; Dony, E.; Weissmann, N.; Butrous, G.; Banat, G.-A.; Ghofrani, H.A.; Seeger, W.; Grimminger, F.; Schermuly, R.T. Role of the Prostanoid EP4 Receptor in Iloprost-mediated Vasodilatation in Pulmonary Hypertension. Am. J. Respir. Crit. Care Med. 2008, 178, 188–196. [Google Scholar] [CrossRef] [Green Version]
- Chen, W.-J.; Lin, K.-H.; Lai, Y.-J.; Yang, S.-H.; Pang, J.-H.S. Protective Effect of Propylthiouracil Independent of Its Hypothyroid Effect on Atherogenesis in Cholesterol-Fed Rabbits. Circulation 2004, 110, 1313–1319. [Google Scholar] [CrossRef] [Green Version]
- Wipff, P.-J.; Majd, H.; Acharya, C.; Buscemi, L.; Meister, J.-J.; Hinz, B. The covalent attachment of adhesion molecules to silicone membranes for cell stretching applications. Biomaterials 2009, 30, 1781–1789. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Primer Name | Sequence | Amplicon Size (bp) |
|---|---|---|
| α-SM-actin | 5′-CTGCT GTCTC TGTCC TTCTA C-3′ | 151 |
| 5′-CCGAAGGACA GACCA GAAAC-3′ | ||
| MHC | 5′-CGACT TGACA ACCAA CCTA-3′ | 108 |
| 5′-TTCTT CAGCC TCACC TCTA-3′ | ||
| Calponin | 5′-CAAAGTTGCTTCCCAGAAAGG A-3′ | 161 |
| 5′-TTCTG CCTGG TAATCGCTAT CA-3′ | ||
| Desmin | 5′-GCAGG TCCAG GTAGA GAT-3′ | 97 |
| 5′-GATGT TCTTA GCCGC AATG-3′ | ||
| GAPDH | 5′ AGTCT ACTGG CGTCT TCA-3′ | 136 |
| 5′- TTGTC ATATT TCTCG TGGT-3′ | ||
| Fibrillin1 | 5′-CGTAT CTCCA TTGTC TCCC-3′ | 112 |
| 5′-ATGCG ACTGT CCACC TGA-3′ | ||
| tropoelastin | 5′-TGTTG GAGTT GGCGG AGTT-3′ | 147 |
| 5′-GGAAG ACCGA CACCA GGAA-3′ | ||
| Emilin-1 | 5′-AGGAC CGCTT CAACT CTACC-3′ | 185 |
| 5′-TGCCA CCTGC TCTTC CAATA-3′ | ||
| Collage I | 5′-TGACC GATGG ATTCC AGTTC-3′ | 132 |
| 5′-GCTGT TCTTG CAGTG ATAGG-3′ |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chang, C.-J.; Huang, C.-C.; Chen, P.-R.; Lai, Y.-J. Remodeling Matrix Synthesis in a Rat Model of Aortocaval Fistula and the Cyclic Stretch: Impaction in Pulmonary Arterial Hypertension-Congenital Heart Disease. Int. J. Mol. Sci. 2020, 21, 4676. https://doi.org/10.3390/ijms21134676
Chang C-J, Huang C-C, Chen P-R, Lai Y-J. Remodeling Matrix Synthesis in a Rat Model of Aortocaval Fistula and the Cyclic Stretch: Impaction in Pulmonary Arterial Hypertension-Congenital Heart Disease. International Journal of Molecular Sciences. 2020; 21(13):4676. https://doi.org/10.3390/ijms21134676
Chicago/Turabian StyleChang, Chi-Jen, Chung-Chi Huang, Po-Ru Chen, and Ying-Ju Lai. 2020. "Remodeling Matrix Synthesis in a Rat Model of Aortocaval Fistula and the Cyclic Stretch: Impaction in Pulmonary Arterial Hypertension-Congenital Heart Disease" International Journal of Molecular Sciences 21, no. 13: 4676. https://doi.org/10.3390/ijms21134676
APA StyleChang, C. -J., Huang, C. -C., Chen, P. -R., & Lai, Y. -J. (2020). Remodeling Matrix Synthesis in a Rat Model of Aortocaval Fistula and the Cyclic Stretch: Impaction in Pulmonary Arterial Hypertension-Congenital Heart Disease. International Journal of Molecular Sciences, 21(13), 4676. https://doi.org/10.3390/ijms21134676