Investigating REPAIRv2 as a Tool to Edit CFTR mRNA with Premature Stop Codons

 , , , ,
, , , , 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
2.1. ‘Spacer’ Design and Cloning into the Vector pC0043-PspCas13b
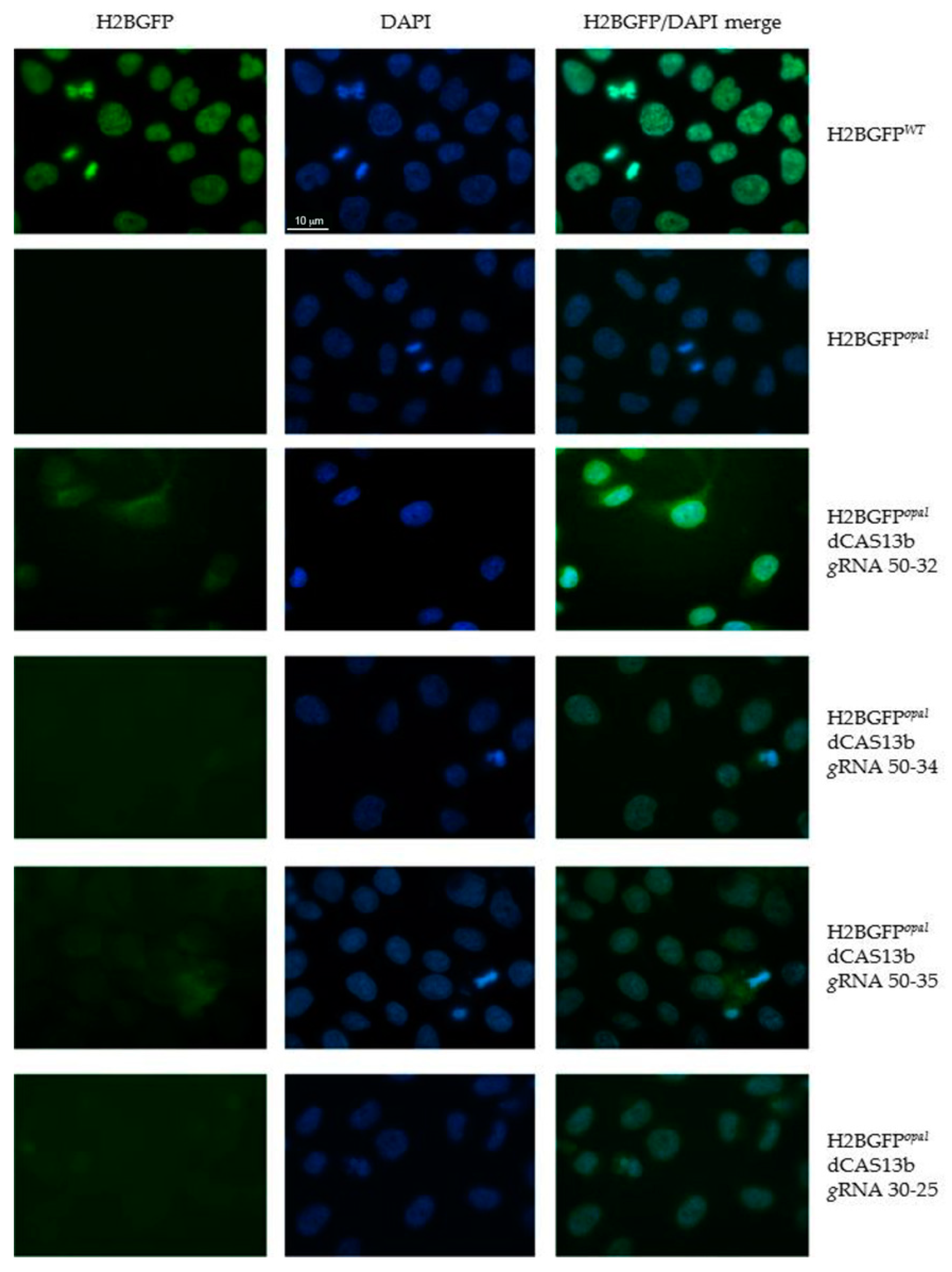
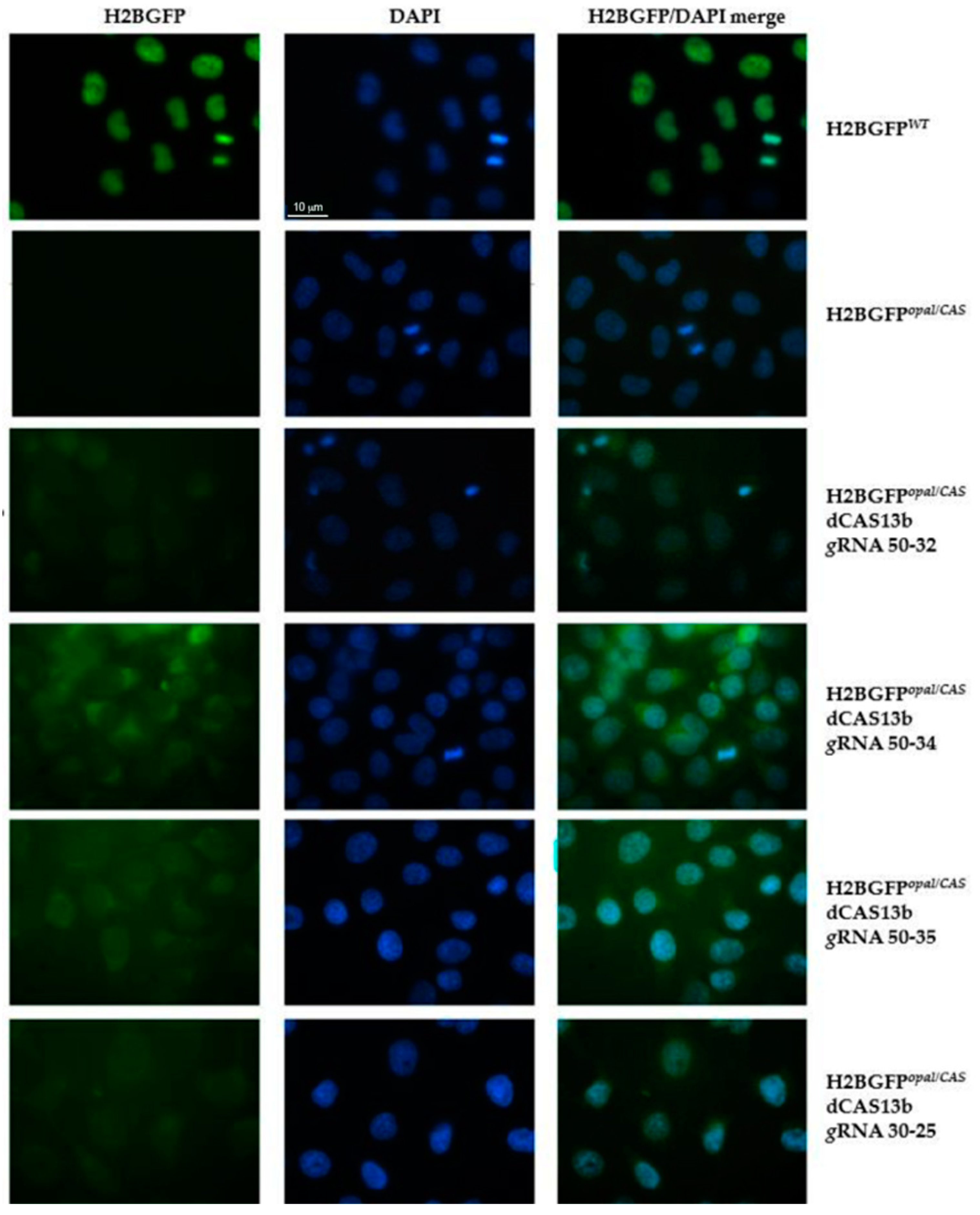
2.2. Evaluation by Fluorescence Microscopy of RNA Editing of the H2BGFPopal Stop Mutation
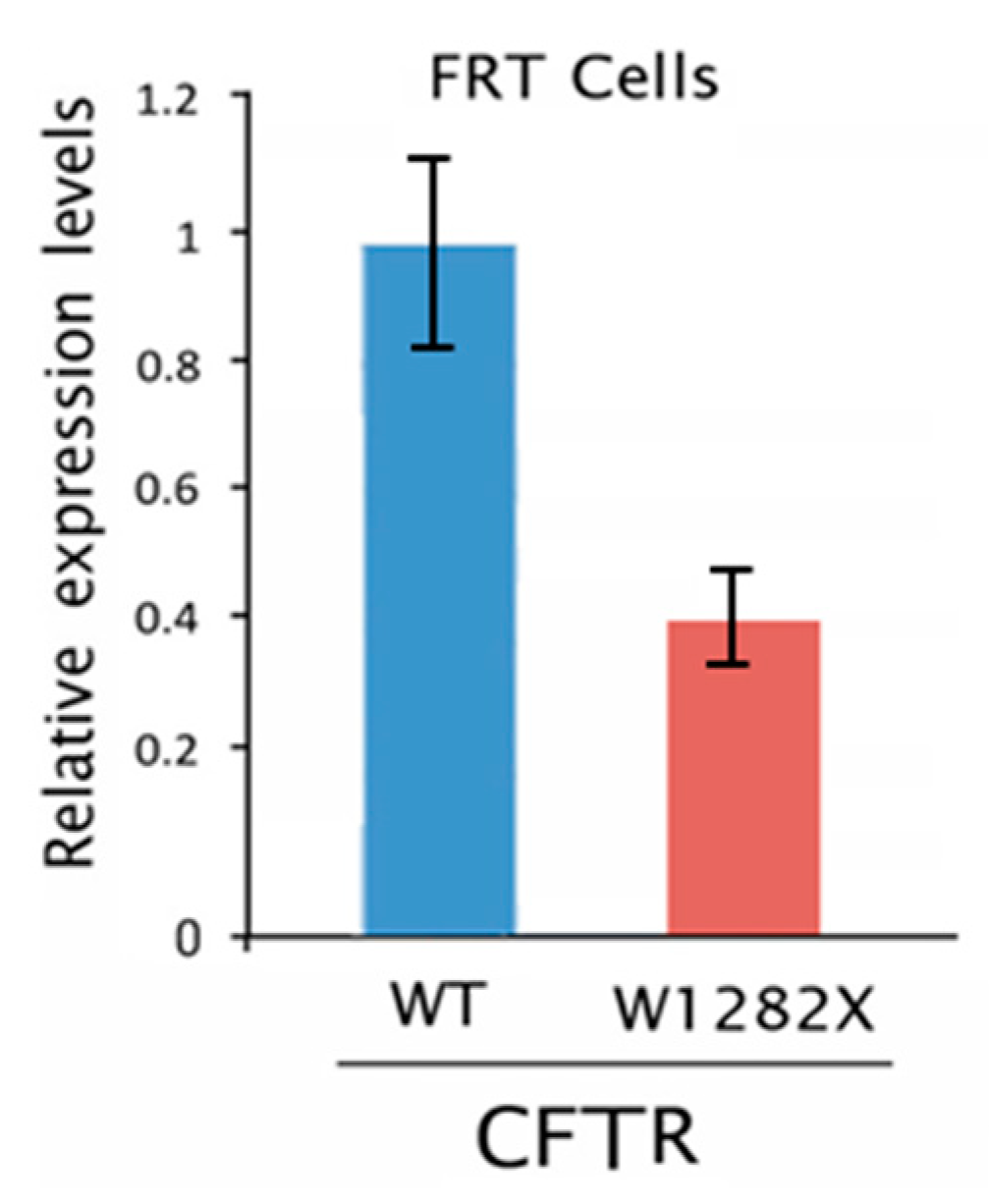
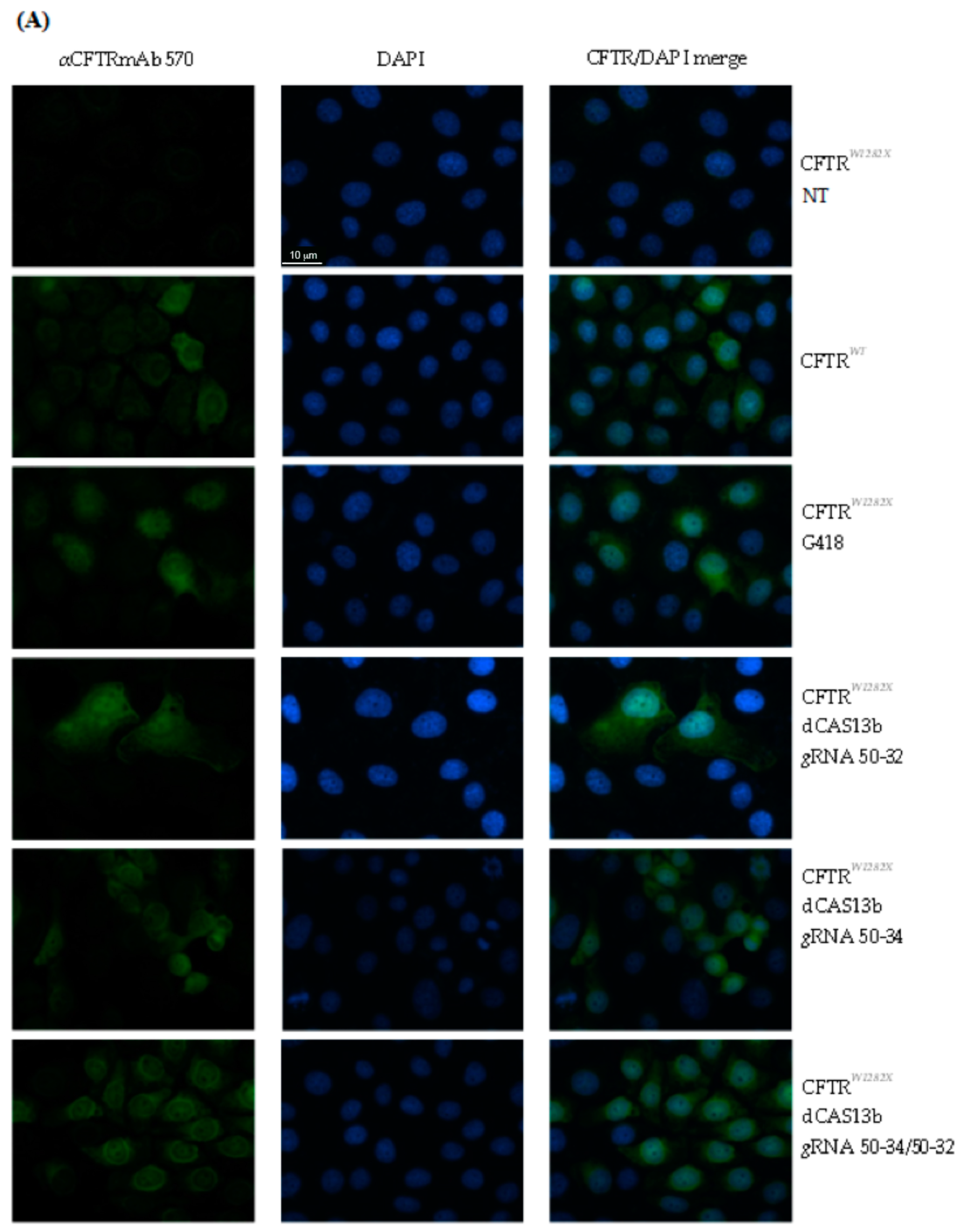
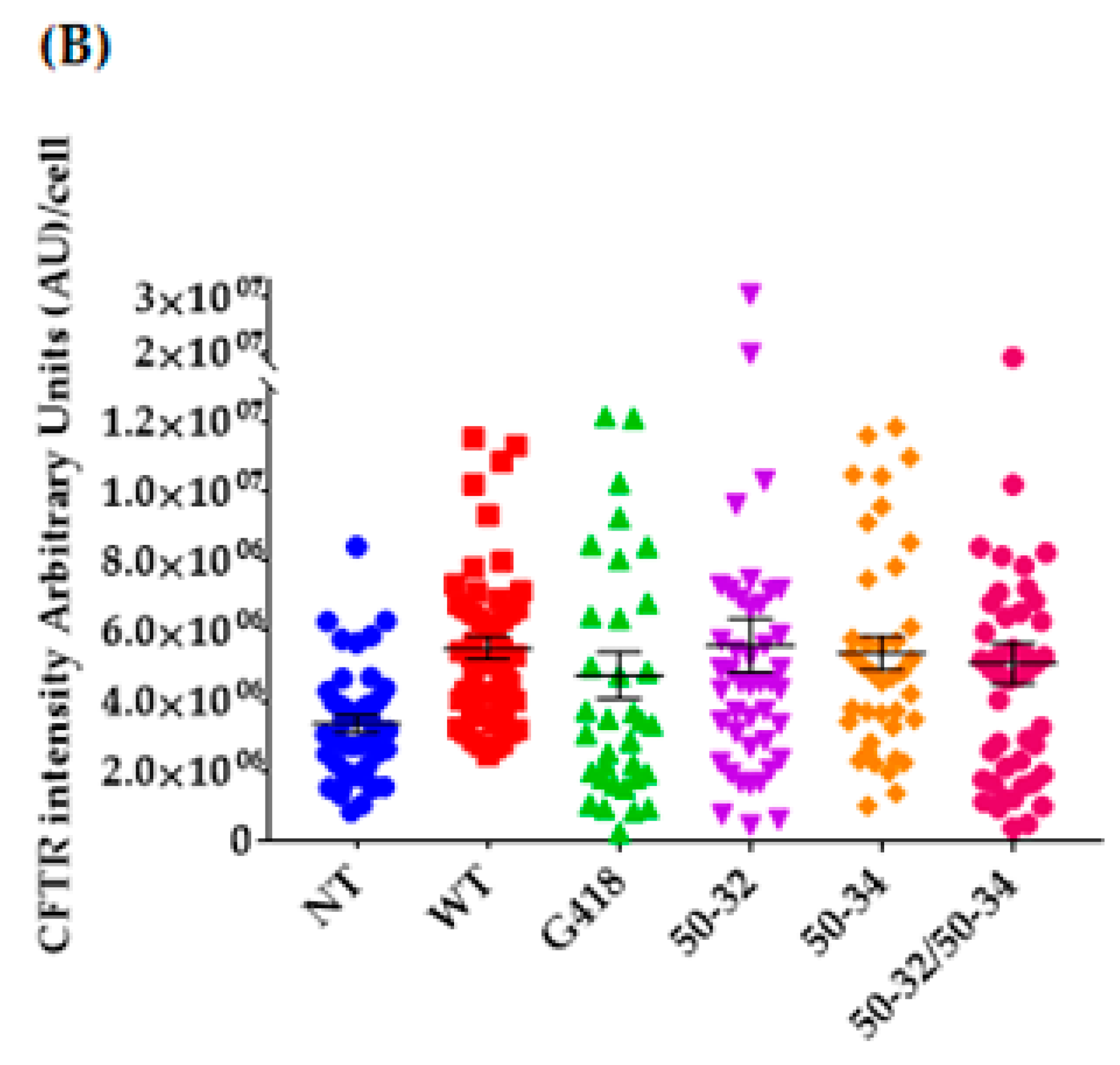
2.3. Editing of the CFTR Transcripts with UGA Nonsense Mutation in FRT-CFTRW1282X Cells
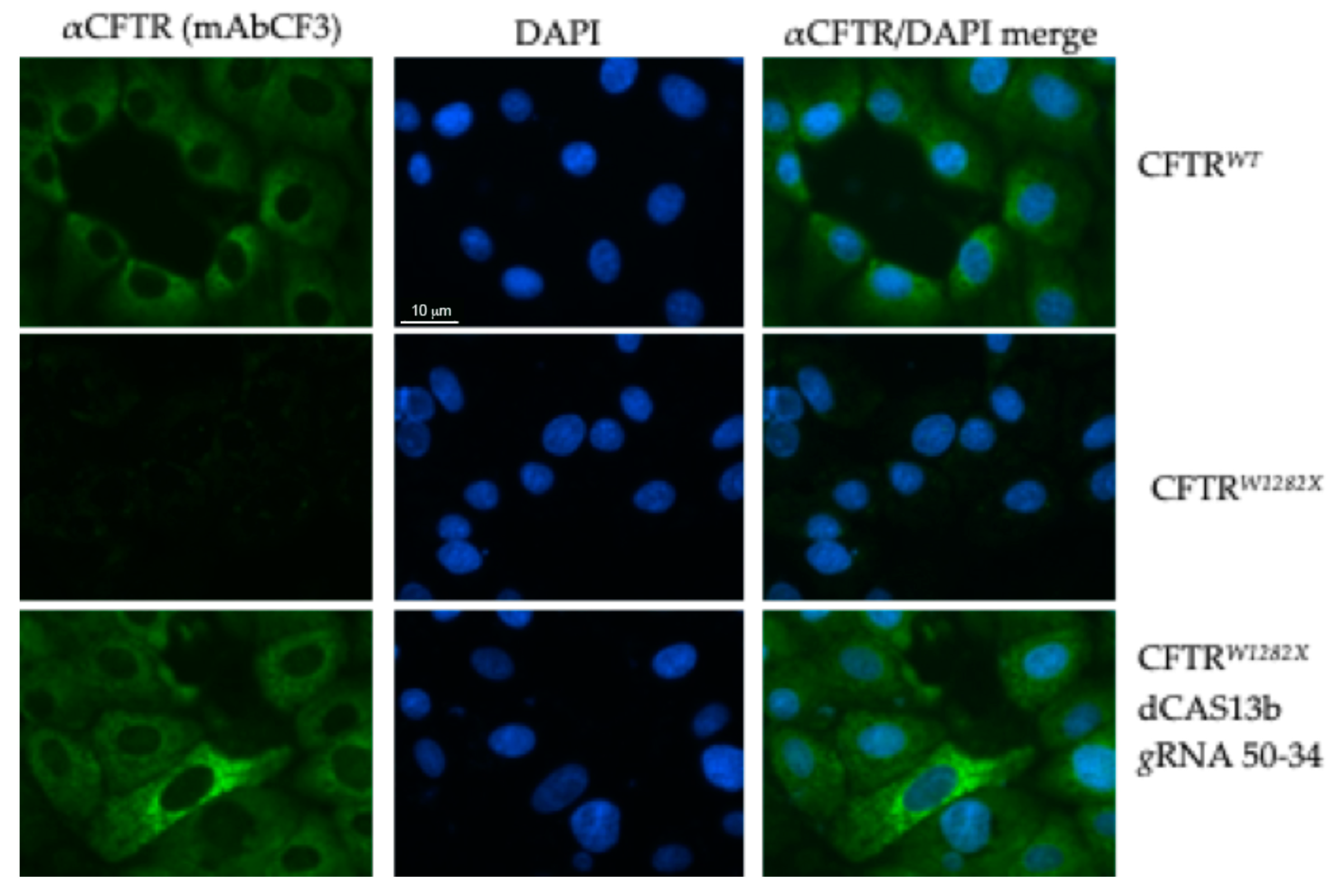
2.4. Evaluation of CFTR Rescue Following Editing CFTRW1282X in IB3-1 Human Cells
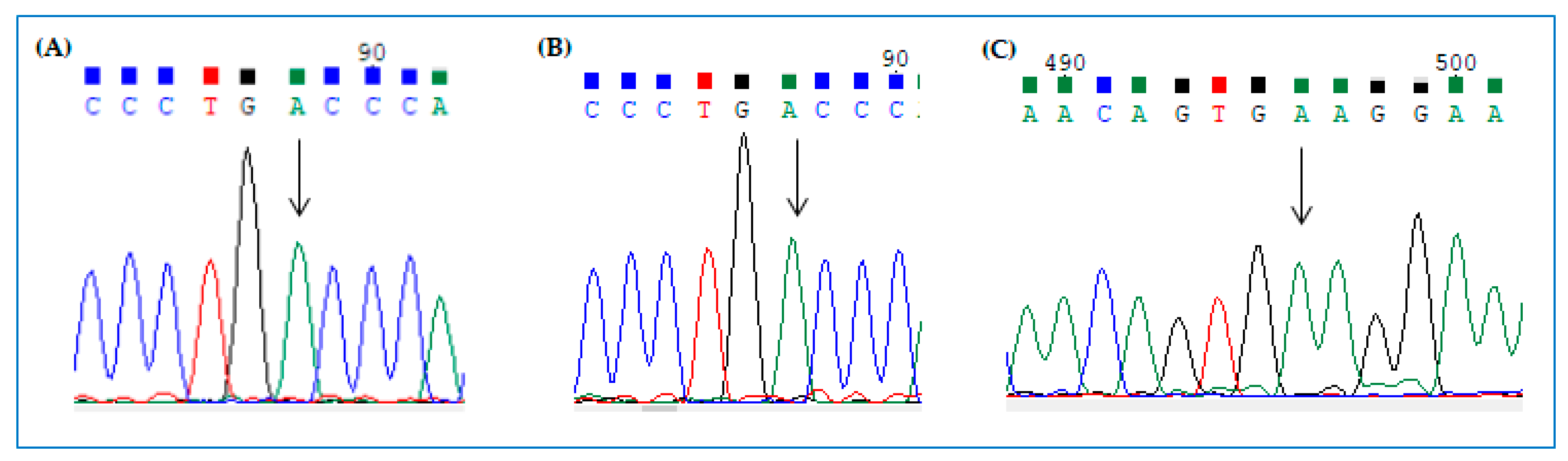
2.5. Visualization of Editing by cDNA Sequencing
3. Discussion
4. Materials and Methods
4.1. Site-Directed Mutagenesis and Bacterial Clone Selection
4.2. Vectors and Clones Used in This Study
4.3. Spacer Cloning
4.4. Cell Culture Conditions
4.5. Cell Transfection
4.6. Immunofluorescence
4.7. RT-qPCR
4.8. The cDNA Synthesis and RT-PCR
4.9. Western Blotting and Immunoprecipitation
4.10. Quantifications of the Immunofluorescence
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Riordan, J.R.; Rommens, J.M.; Kerem, B.; Alon, N.; Rozmahel, R.; Grzelczak, Z.; Zielenski, J.; Lok, S.; Plavsic, N.; Chou, J.L.; et al. Identification of the cystic fibrosis gene: Cloning and characterization of complementary DNA. Science 1989, 245, 1066–1073. [Google Scholar] [CrossRef]
- De Boeck, K.; Zolin, A.; Cuppens, H.; Olesen, H.V.; Viviani, L. The relative frequency of CFTR mutation classes in European patients with cystic fibrosis. J. Cys. Fibros. 2014, 13, 403–409. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Manuvakhova, M.; Keeling, K.; Bedwell, D.M. Aminoglycoside antibiotics mediate context-dependent suppression of termination codons in a mammalian translation system. Rna 2000, 6, 1044–1055. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Prayle, A.; Smyth, A.R. Aminoglycoside use in cystic fibrosis: Therapeutic strategies and toxicity. Curr. Op. Pulm. Med. 2010, 16, 604–610. [Google Scholar] [CrossRef] [PubMed]
- Roy, B.; Friesen, W.J.; Tomizawa, Y.; Leszyk, J.D.; Zhuo, J.; Johnson, B.; Dakka, J.; Trotta, C.R.; Xue, X.; Mutyam, V.; et al. Ataluren stimulates ribosomal selection of near-cognate tRNAs to promote nonsense suppression. Proc. Natl. Acad.Sci. USA 2016, 113, 12508–12513. [Google Scholar] [CrossRef] [Green Version]
- Xue, X.; Mutyam, V.; Thakerar, A.; Mobley, J.; Bridges, R.J.; Rowe, S.M.; Keeling, K.M.; Bedwell, D.M. Identification of the amino acids inserted during suppression of CFTR nonsense mutations and determination of their functional consequences. Hum. Mol. Genet. 2017, 26, 3116–3129. [Google Scholar] [CrossRef] [Green Version]
- Zomer-van Ommen, D.D.; Vijftigschild, L.A.; Kruisselbrink, E.; Vonk, A.M.; Dekkers, J.F.; Janssens, H.M.; de Winter-de Groot, K.M.; van der Ent, C.K.; Beekman, J.M. Limited premature termination codon suppression by read-through agents in cystic fibrosis intestinal organoids. J. Cys. Fibros. 2016, 15, 158–162. [Google Scholar] [CrossRef]
- Goldmann, T.; Overlack, N.; Moller, F.; Belakhov, V.; van Wyk, M.; Baasov, T.; Wolfrum, U.; Nagel-Wolfrum, K. A comparative evaluation of NB30, NB54 and PTC124 in translational read-through efficacy for treatment of an USH1C nonsense mutation. EMBO Mol. Med. 2012, 4, 1186–1199. [Google Scholar] [CrossRef]
- Welch, E.M.; Barton, E.R.; Zhuo, J.; Tomizawa, Y.; Friesen, W.J.; Trifillis, P.; Paushkin, S.; Patel, M.; Trotta, C.R.; Hwang, S.; et al. PTC124 targets genetic disorders caused by nonsense mutations. Nature 2007, 447, 87–91. [Google Scholar] [CrossRef]
- Lentini, L.; Melfi, R.; Di Leonardo, A.; Spinello, A.; Barone, G.; Pace, A.; Palumbo Piccionello, A.; Pibiri, I. Toward a rationale for the PTC124 (Ataluren) promoted readthrough of premature stop codons: A computational approach and GFP-reporter cell-based assay. Mol. Pharm. 2014, 11, 653–664. [Google Scholar] [CrossRef] [PubMed]
- Pibiri, I.; Lentini, L.; Melfi, R.; Gallucci, G.; Pace, A.; Spinello, A.; Barone, G.; Di Leonardo, A. Enhancement of premature stop codon readthrough in the CFTR gene by Ataluren (PTC124) derivatives. Eur. J. Med. Chem. 2015, 101, 236–244. [Google Scholar] [CrossRef] [PubMed]
- Pibiri, I.; Lentini, L.; Tutone, M.; Melfi, R.; Pace, A.; Di Leonardo, A. Exploring the readthrough of nonsense mutations by non-acidic Ataluren analogues selected by ligand-based virtual screening. Eur. J. Med. Chem. 2016, 122, 429–435. [Google Scholar] [CrossRef] [PubMed]
- Pibiri, I.; Lentini, L.; Melfi, R.; Tutone, M.; Baldassano, S.; Ricco Galluzzo, P.; Di Leonardo, A.; Pace, A. Rescuing the CFTR protein function: Introducing 1,3,4-oxadiazoles as translational readthrough inducing drugs. Eur. J. Med. Chem. 2018, 159, 126–142. [Google Scholar] [CrossRef] [PubMed]
- Crooke, S.T.; Witztum, J.L.; Bennett, C.F.; Baker, B.F. RNA-Targeted Therapeutics. Cell Metab. 2018, 27, 714–739. [Google Scholar] [CrossRef] [PubMed]
- Montiel-Gonzalez, M.F.; Diaz Quiroz, J.F.; Rosenthal, J.J.C. Current strategies for Site-Directed RNA Editing using ADARs. Methods 2019, 156, 16–24. [Google Scholar] [CrossRef]
- Aquino-Jarquin, G. Novel Engineered Programmable Systems for ADAR-Mediated RNA Editing. Molecular Ther. Nucleic Acids 2020, 19, 1065–1072. [Google Scholar] [CrossRef]
- Chen, G.; Katrekar, D.; Mali, P. RNA-Guided Adenosine Deaminases: Advances and Challenges for Therapeutic RNA Editing. Biochemistry 2019, 58, 1947–1957. [Google Scholar] [CrossRef]
- Merkle, T.; Merz, S.; Reautschnig, P.; Blaha, A.; Li, Q.; Vogel, P.; Wettengel, J.; Li, J.B.; Stafforst, T. Precise RNA editing by recruiting endogenous ADARs with antisense oligonucleotides. Nat. Biotechnol. 2019, 37, 133–138. [Google Scholar] [CrossRef]
- Qu, L.; Yi, Z.; Zhu, S.; Wang, C.; Cao, Z.; Zhou, Z.; Yuan, P.; Yu, Y.; Tian, F.; Liu, Z.; et al. Programmable RNA editing by recruiting endogenous ADAR using engineered RNAs. Nat. Biotechnol. 2019, 37, 1059–1069. [Google Scholar] [CrossRef]
- Katrekar, D.; Chen, G.; Meluzzi, D.; Ganesh, A.; Worlikar, A.; Shih, Y.R.; Varghese, S.; Mali, P. In vivo RNA editing of point mutations via RNA-guided adenosine deaminases. Nat. Methods 2019, 16, 239–242. [Google Scholar] [CrossRef]
- Wettengel, J.; Reautschnig, P.; Geisler, S.; Kahle, P.J.; Stafforst, T. Harnessing human ADAR2 for RNA repair - Recoding a PINK1 mutation rescues mitophagy. Nucleic Acids Res. 2017, 45, 2797–2808. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vogel, P.; Moschref, M.; Li, Q.; Merkle, T.; Selvasaravanan, K.D.; Li, J.B.; Stafforst, T. Efficient and precise editing of endogenous transcripts with SNAP-tagged ADARs. Nat. Methods 2018, 15, 535–538. [Google Scholar] [CrossRef] [PubMed]
- Montiel-Gonzalez, M.F.; Vallecillo-Viejo, I.; Yudowski, G.A.; Rosenthal, J.J. Correction of mutations within the cystic fibrosis transmembrane conductance regulator by site-directed RNA editing. Proc. Natl. Acad.Sci. USA 2013, 110, 18285–18290. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cong, L.; Ran, F.A.; Cox, D.; Lin, S.; Barretto, R.; Habib, N.; Hsu, P.D.; Wu, X.; Jiang, W.; Marraffini, L.A.; et al. Multiplex genome engineering using CRISPR/Cas systems. Science 2013, 339, 819–823. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cox, D.B.T.; Gootenberg, J.S.; Abudayyeh, O.O.; Franklin, B.; Kellner, M.J.; Joung, J.; Zhang, F. RNA editing with CRISPR-Cas13. Science 2017, 358, 1019–1027. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vallecillo-Viejo, I.C.; Liscovitch-Brauer, N.; Montiel-Gonzalez, M.F.; Eisenberg, E.; Rosenthal, J.J.C. Abundant off-target edits from site-directed RNA editing can be reduced by nuclear localization of the editing enzyme. RNA Biol. 2018, 15, 104–114. [Google Scholar] [CrossRef] [Green Version]
- Kim, Y.B.; Komor, A.C.; Levy, J.M.; Packer, M.S.; Zhao, K.T.; Liu, D.R. Increasing the genome-targeting scope and precision of base editing with engineered Cas9-cytidine deaminase fusions. Nat. Biotechnol. 2017, 35, 371–376. [Google Scholar] [CrossRef]
- Kanda, T.; Sullivan, K.F.; Wahl, G.M. Histone-GFP fusion protein enables sensitive analysis of chromosome dynamics in living mammalian cells. Curr. Biol. 1998, 8, 377–385. [Google Scholar] [CrossRef] [Green Version]
- Costa, G.; Barra, V.; Lentini, L.; Cilluffo, D.; Di Leonardo, A. DNA demethylation caused by 5-Aza-2’-deoxycytidine induces mitotic alterations and aneuploidy. Oncotarget 2016, 7, 3726–3739. [Google Scholar] [CrossRef] [Green Version]
- Zeitlin, P.L.; Lu, L.; Rhim, J.; Cutting, G.; Stetten, G.; Kieffer, K.A.; Craig, R.; Guggino, W.B. A cystic fibrosis bronchial epithelial cell line: Immortalization by adeno-12-SV40 infection. Am. J. Resp. Cell Mol. Biol. 1991, 4, 313–319. [Google Scholar] [CrossRef]
- Maquat, L.E. Nonsense-mediated mRNA decay in mammals. J. Cell Sci. 2005, 118, 1773–1776. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Doma, M.K.; Parker, R. Endonucleolytic cleavage of eukaryotic mRNAs with stalls in translation elongation. Nature 2006, 440, 561–564. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Harigaya, Y.; Parker, R. No-go decay: A quality control mechanism for RNA in translation. Wiley Interdiscip. Rev. RNA 2010, 1, 132–141. [Google Scholar] [CrossRef] [PubMed]
- Zegarra-Moran, O.; Romio, L.; Folli, C.; Caci, E.; Becq, F.; Vierfond, J.M.; Mettey, Y.; Cabrini, G.; Fanen, P.; Galietta, L.J. Correction of G551D-CFTR transport defect in epithelial monolayers by genistein but not by CPX or MPB-07. Br. J. Pharmacol. 2002, 137, 504–512. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Galietta, L.V.; Jayaraman, S.; Verkman, A.S. Cell-based assay for high-throughput quantitative screening of CFTR chloride transport agonists. Am. J. Physiol. Cell Physiol. 2001, 281, C1734–C1742. [Google Scholar] [CrossRef]
- Veneziano, L.; Barra, V.; Cilluffo, D.; Di Leonardo, A. Proliferation of aneuploid cells induced by CENP-E depletion is counteracted by the p14(ARF) tumor suppressor. Mol. Genet. Genomics 2019, 294, 149–158. [Google Scholar] [CrossRef]
- Veneziano, L.; Barra, V.; Lentini, L.; Spatafora, S.; Di Leonardo, A. p14(ARF) Prevents Proliferation of Aneuploid Cells by Inducing p53-Dependent Apoptosis. J. Cell. Physiol. 2016, 231, 336–344. [Google Scholar] [CrossRef] [Green Version]
- Coppola, A.; Tomasello, L.; Pizzolanti, G.; Pucci-Minafra, I.; Albanese, N.; Di Cara, G.; Cancemi, P.; Pitrone, M.; Bommarito, A.; Carissimi, E.; et al. In vitro phenotypic, genomic and proteomic characterization of a cytokine-resistant murine beta-TC3 cell line. PLoS ONE 2012, 7, e32109. [Google Scholar] [CrossRef] [Green Version]
- Cancemi, P.; Albanese, N.N.; DiCara, G.; Marabeti, M.R.; Costantini, F.; Minafra, S.; Pucci-Minafra, I. Multiple changes induced by fibroblasts on breast cancer cells. Connect. Tissue Res. 2010, 51, 88–104. [Google Scholar] [CrossRef] [Green Version]
- Musso, R.; Di Cara, G.; Albanese, N.N.; Marabeti, M.R.; Cancemi, P.; Martini, D.; Orsini, E.; Giordano, C.; Pucci-Minafra, I. Differential proteomic and phenotypic behaviour of papillary and anaplastic thyroid cell lines. J. Proteomics 2013, 90, 115–125. [Google Scholar] [CrossRef]











© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Melfi, R.; Cancemi, P.; Chiavetta, R.; Barra, V.; Lentini, L.; Di Leonardo, A. Investigating REPAIRv2 as a Tool to Edit CFTR mRNA with Premature Stop Codons. Int. J. Mol. Sci. 2020, 21, 4781. https://doi.org/10.3390/ijms21134781
Melfi R, Cancemi P, Chiavetta R, Barra V, Lentini L, Di Leonardo A. Investigating REPAIRv2 as a Tool to Edit CFTR mRNA with Premature Stop Codons. International Journal of Molecular Sciences. 2020; 21(13):4781. https://doi.org/10.3390/ijms21134781
Chicago/Turabian StyleMelfi, Raffaella, Patrizia Cancemi, Roberta Chiavetta, Viviana Barra, Laura Lentini, and Aldo Di Leonardo. 2020. "Investigating REPAIRv2 as a Tool to Edit CFTR mRNA with Premature Stop Codons" International Journal of Molecular Sciences 21, no. 13: 4781. https://doi.org/10.3390/ijms21134781
APA StyleMelfi, R., Cancemi, P., Chiavetta, R., Barra, V., Lentini, L., & Di Leonardo, A. (2020). Investigating REPAIRv2 as a Tool to Edit CFTR mRNA with Premature Stop Codons. International Journal of Molecular Sciences, 21(13), 4781. https://doi.org/10.3390/ijms21134781