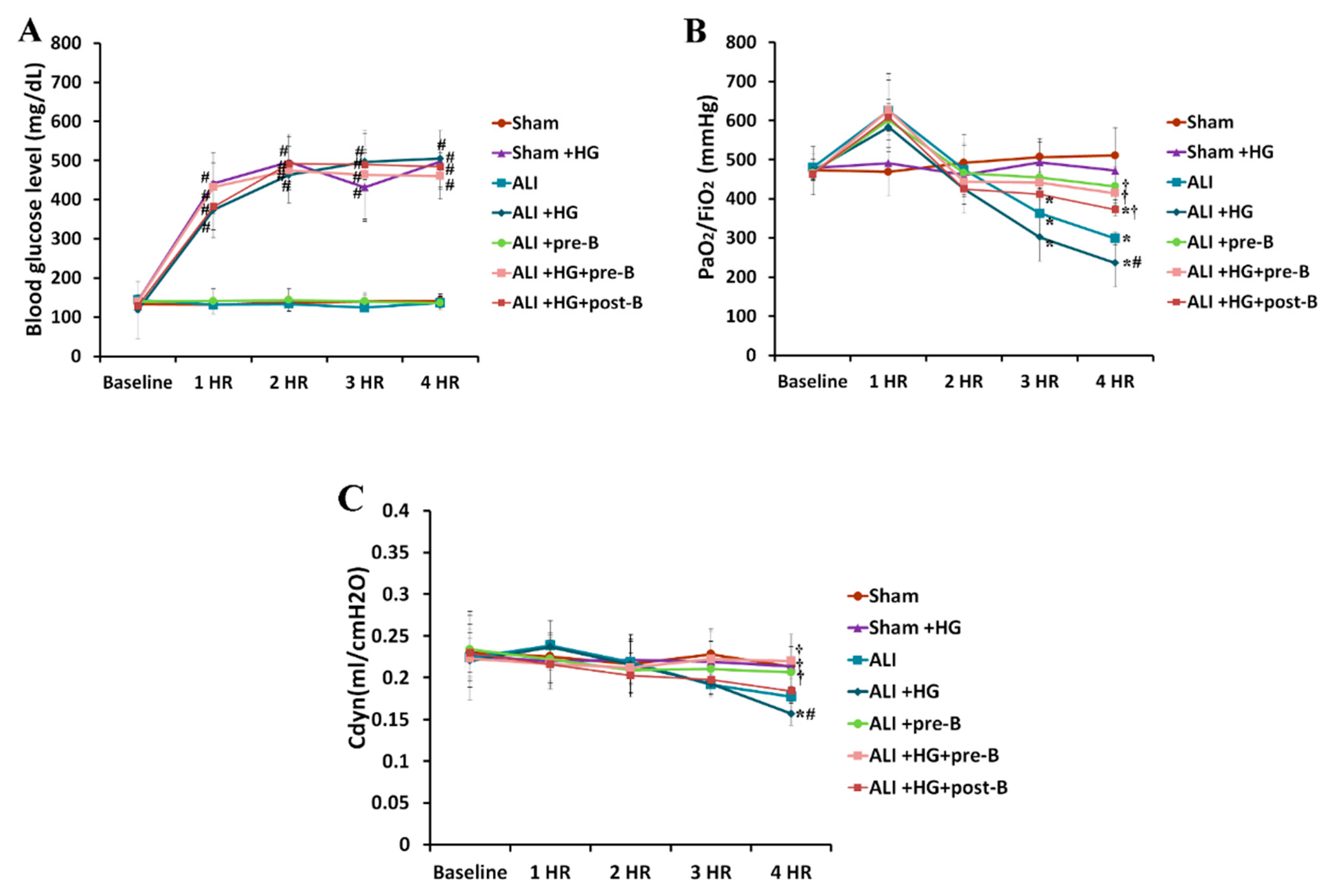
Figure 1.
Blood glucose levels, oxygenation, and dynamic compliance of lungs. The blood glucose levels (A) in the hyperglycemic rats were 400–500 mg/dL, which were significantly higher than that in non-hyperglycemic rats (p < 0.05). PaO2/FiO2 (B) was significantly reduced in rats with ALI or ALI + HG (p < 0.05). PaO2/FiO2 was lower in rats with ALI + HG than in rats with ALI (p < 0.05). The rats that received the NKCC1 inhibitor had higher PaO2/FiO2 than those without NKCC1 inhibitor (p < 0.05). The Cdyn (C) was significantly reduced in rats with ALI or ALI + HG than in those from the sham group (p < 0.05). The Cdyn was lower in rats with ALI + HG than in those with ALI (p < 0.05). The rats with the NKCC1 inhibitor had higher Cdyn than those without the NKCC1 inhibitor (p < 0.05). * Significant difference, as compared to the sham group (p < 0.05), # Significant difference, comparing the groups with HG to those without HG (I vs. II, III vs. IV, V vs. VI, and V vs. VII, p < 0.05), † Significant difference, comparing the ALI groups with B to those without B (III vs. V, IV vs. VI, and IV vs. VII, p < 0.05) Abbreviation: PaO2 = partial pressure of oxygen in arterial blood, FiO2 = fraction of inspired oxygen, Cdyn = dynamic compliance of lungs, ALI = acute lung injury, HG = hyperglycemia, pre-B = pretreatment with bumetanide, post-B = post-treatment with bumetanide.
Figure 1.
Blood glucose levels, oxygenation, and dynamic compliance of lungs. The blood glucose levels (A) in the hyperglycemic rats were 400–500 mg/dL, which were significantly higher than that in non-hyperglycemic rats (p < 0.05). PaO2/FiO2 (B) was significantly reduced in rats with ALI or ALI + HG (p < 0.05). PaO2/FiO2 was lower in rats with ALI + HG than in rats with ALI (p < 0.05). The rats that received the NKCC1 inhibitor had higher PaO2/FiO2 than those without NKCC1 inhibitor (p < 0.05). The Cdyn (C) was significantly reduced in rats with ALI or ALI + HG than in those from the sham group (p < 0.05). The Cdyn was lower in rats with ALI + HG than in those with ALI (p < 0.05). The rats with the NKCC1 inhibitor had higher Cdyn than those without the NKCC1 inhibitor (p < 0.05). * Significant difference, as compared to the sham group (p < 0.05), # Significant difference, comparing the groups with HG to those without HG (I vs. II, III vs. IV, V vs. VI, and V vs. VII, p < 0.05), † Significant difference, comparing the ALI groups with B to those without B (III vs. V, IV vs. VI, and IV vs. VII, p < 0.05) Abbreviation: PaO2 = partial pressure of oxygen in arterial blood, FiO2 = fraction of inspired oxygen, Cdyn = dynamic compliance of lungs, ALI = acute lung injury, HG = hyperglycemia, pre-B = pretreatment with bumetanide, post-B = post-treatment with bumetanide.
![Ijms 21 04803 g001]()
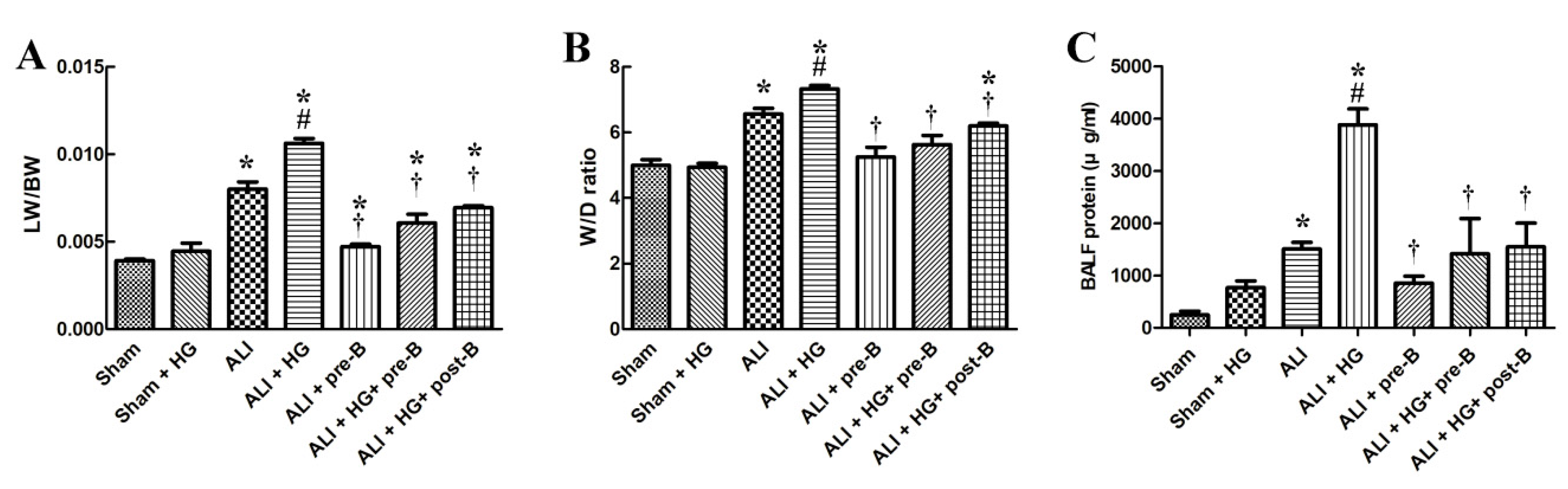
Figure 2.
Effects of hyperglycemia and NKCC1 inhibitor treatment on pulmonary edema and alveolar protein leakage. The LW/BW (A) and W/D ratios (B) were significantly increased in rats with ALI and further increased in rats with ALI + HG (p < 0.05). The NKCC1 inhibitor reduced pulmonary edema in both ALI and ALI + HG groups (p < 0.05). The TP concentration in BALF (C) was prominent in rats with ALI and further increased in rats with ALI + HG (p < 0.05). The NKCC1 inhibitor reduced alveolar protein leakage in both ALI and ALI + HG rats (p < 0.05). * Significant difference, as compared to the sham group (p < 0.05), # Significant difference, comparing the groups with HG to those without HG (I vs. II, III vs. IV, V vs. VI, and V vs. VII, p < 0.05), † Significant difference, comparing the ALI groups with B to those without B (III vs. V, IV vs. VI, and IV vs. VII, p < 0.05). Abbreviation: LW/BW = lung weight/body weight, W/D = lung wet/dry weight ratio. ALI = acute lung injury, HG = hyperglycemia, pre-B = pretreatment with bumetanide, post-B = post-treatment with bumetanide, BALF = bronchoalveolar lavaged fluid, TP = total protein.
Figure 2.
Effects of hyperglycemia and NKCC1 inhibitor treatment on pulmonary edema and alveolar protein leakage. The LW/BW (A) and W/D ratios (B) were significantly increased in rats with ALI and further increased in rats with ALI + HG (p < 0.05). The NKCC1 inhibitor reduced pulmonary edema in both ALI and ALI + HG groups (p < 0.05). The TP concentration in BALF (C) was prominent in rats with ALI and further increased in rats with ALI + HG (p < 0.05). The NKCC1 inhibitor reduced alveolar protein leakage in both ALI and ALI + HG rats (p < 0.05). * Significant difference, as compared to the sham group (p < 0.05), # Significant difference, comparing the groups with HG to those without HG (I vs. II, III vs. IV, V vs. VI, and V vs. VII, p < 0.05), † Significant difference, comparing the ALI groups with B to those without B (III vs. V, IV vs. VI, and IV vs. VII, p < 0.05). Abbreviation: LW/BW = lung weight/body weight, W/D = lung wet/dry weight ratio. ALI = acute lung injury, HG = hyperglycemia, pre-B = pretreatment with bumetanide, post-B = post-treatment with bumetanide, BALF = bronchoalveolar lavaged fluid, TP = total protein.
![Ijms 21 04803 g002]()
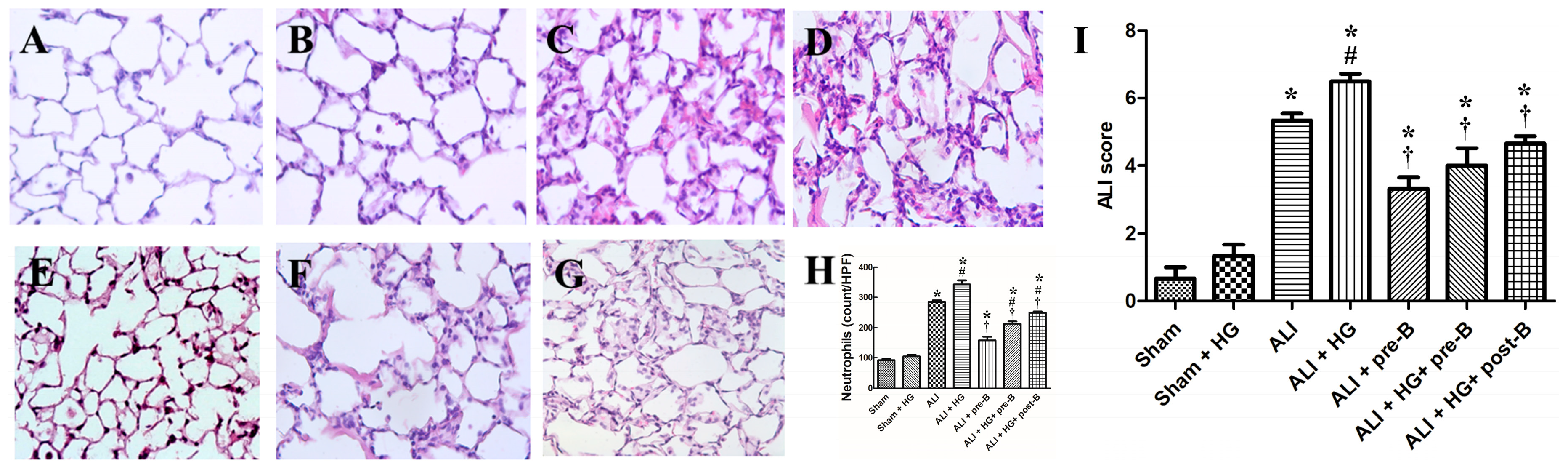
Figure 3.
Effects of hyperglycemia and NKCC1 inhibitor treatment on the histopathological features in ALI and neutrophil sequestration. The sham (A) and sham + HG (B) groups exhibited normal histology. Sections from rats of the ALI (C) and ALI + HG (D) groups revealed severe lung injury with prominent alveolar exudation, alveolar septum thickening, and neutrophil sequestration. Pretreatment or post-ALI treatment with the NKCC1 inhibitor reduced lung injury, which was observable in (E) ALI + pre-B, (F) ALI + HG + pre-B, and (G) ALI + HG + post-B groups. The neutrophil counts (H) were high in rats with ALI and further increased in the ALI + HG group (p < 0.05). The NKCC1 inhibitor reduced neutrophil counts in both ALI and ALI + HG groups (p < 0.05). The ALI scores (I) were high in rats with ALI and further increased in the ALI + HG group (p < 0.05). The NKCC1 inhibitor reduced ALI scores in both ALI and ALI + HG groups (p < 0.05). * Significant difference, as compared to the sham group (p < 0.05), # Significant difference, comparing the groups with HG to those without HG (I vs. II, III vs. IV, V vs. VI, and V vs. VII, p < 0.05), † Significant difference, comparing the ALI groups with B to those without B (III vs. V, IV vs. VI, and IV vs. VII, p < 0.05). Abbreviation: ALI = acute lung injury, HG = hyperglycemia, pre-B = pretreatment with bumetanide, post-B = post-treatment with bumetanide.
Figure 3.
Effects of hyperglycemia and NKCC1 inhibitor treatment on the histopathological features in ALI and neutrophil sequestration. The sham (A) and sham + HG (B) groups exhibited normal histology. Sections from rats of the ALI (C) and ALI + HG (D) groups revealed severe lung injury with prominent alveolar exudation, alveolar septum thickening, and neutrophil sequestration. Pretreatment or post-ALI treatment with the NKCC1 inhibitor reduced lung injury, which was observable in (E) ALI + pre-B, (F) ALI + HG + pre-B, and (G) ALI + HG + post-B groups. The neutrophil counts (H) were high in rats with ALI and further increased in the ALI + HG group (p < 0.05). The NKCC1 inhibitor reduced neutrophil counts in both ALI and ALI + HG groups (p < 0.05). The ALI scores (I) were high in rats with ALI and further increased in the ALI + HG group (p < 0.05). The NKCC1 inhibitor reduced ALI scores in both ALI and ALI + HG groups (p < 0.05). * Significant difference, as compared to the sham group (p < 0.05), # Significant difference, comparing the groups with HG to those without HG (I vs. II, III vs. IV, V vs. VI, and V vs. VII, p < 0.05), † Significant difference, comparing the ALI groups with B to those without B (III vs. V, IV vs. VI, and IV vs. VII, p < 0.05). Abbreviation: ALI = acute lung injury, HG = hyperglycemia, pre-B = pretreatment with bumetanide, post-B = post-treatment with bumetanide.
![Ijms 21 04803 g003]()
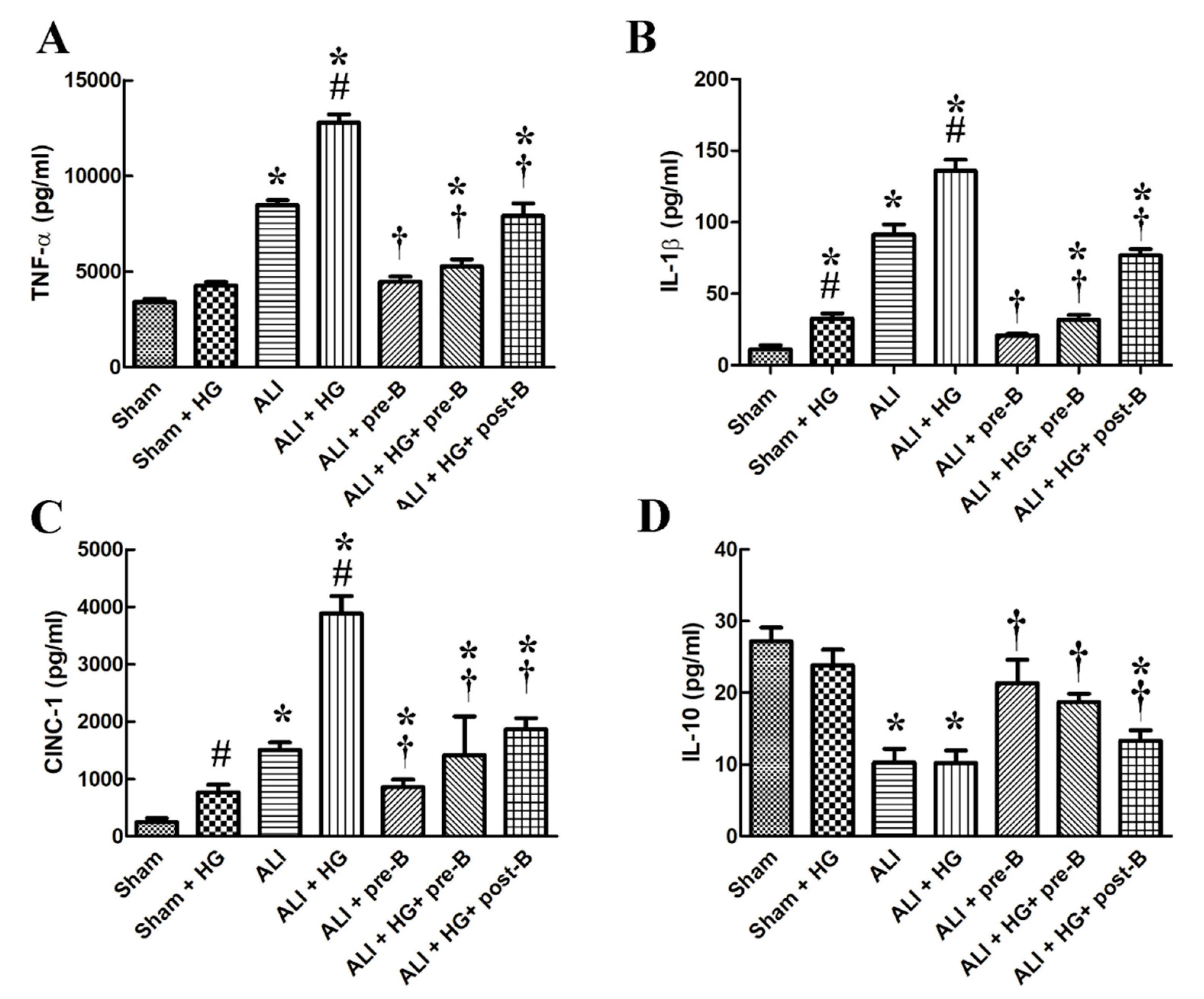
Figure 4.
Effects of hyperglycemia and NKCC1 inhibitor treatment on the levels of cytokines. Higher levels of serum TNF-α (A), IL-1β (B), CINC-1, (C) and lower level of IL-10 (D) were observed in rats from the ALI and ALI + HG groups (p < 0.05). However, the higher levels of pro-inflammatory cytokines and lower level of anti-inflammatory cytokine was noted in the ALI + HG group than in the ALI group (p < 0.05). The NKCC1 inhibitor reduced the levels of these pro-inflammatory cytokines and increased the levels of anti-inflammatory cytokine in rats from both ALI and ALI + HG groups (p < 0.05). * Significant difference, as compared to the sham group (p < 0.05), # Significant difference, comparing the groups with HG to those without HG (I vs. II, III vs. IV, V vs. VI, and V vs. VII, p < 0.05), † Significant difference, comparing the ALI groups with B to those without B (III vs. V, IV vs. VI, and IV vs. VII, p < 0.05). Abbreviation: TNF-α = tumor necrosis factor alpha, IL-1β = interleukin 1 beta, CINC-1 = cytokine-induced neutrophil chemoattractant 1, IL-10 = interleukin 10, ALI = acute lung injury, HG = hyperglycemia, pre-B = pretreatment with bumetanide, post-B = post-treatment with bumetanide.
Figure 4.
Effects of hyperglycemia and NKCC1 inhibitor treatment on the levels of cytokines. Higher levels of serum TNF-α (A), IL-1β (B), CINC-1, (C) and lower level of IL-10 (D) were observed in rats from the ALI and ALI + HG groups (p < 0.05). However, the higher levels of pro-inflammatory cytokines and lower level of anti-inflammatory cytokine was noted in the ALI + HG group than in the ALI group (p < 0.05). The NKCC1 inhibitor reduced the levels of these pro-inflammatory cytokines and increased the levels of anti-inflammatory cytokine in rats from both ALI and ALI + HG groups (p < 0.05). * Significant difference, as compared to the sham group (p < 0.05), # Significant difference, comparing the groups with HG to those without HG (I vs. II, III vs. IV, V vs. VI, and V vs. VII, p < 0.05), † Significant difference, comparing the ALI groups with B to those without B (III vs. V, IV vs. VI, and IV vs. VII, p < 0.05). Abbreviation: TNF-α = tumor necrosis factor alpha, IL-1β = interleukin 1 beta, CINC-1 = cytokine-induced neutrophil chemoattractant 1, IL-10 = interleukin 10, ALI = acute lung injury, HG = hyperglycemia, pre-B = pretreatment with bumetanide, post-B = post-treatment with bumetanide.
![Ijms 21 04803 g004]()
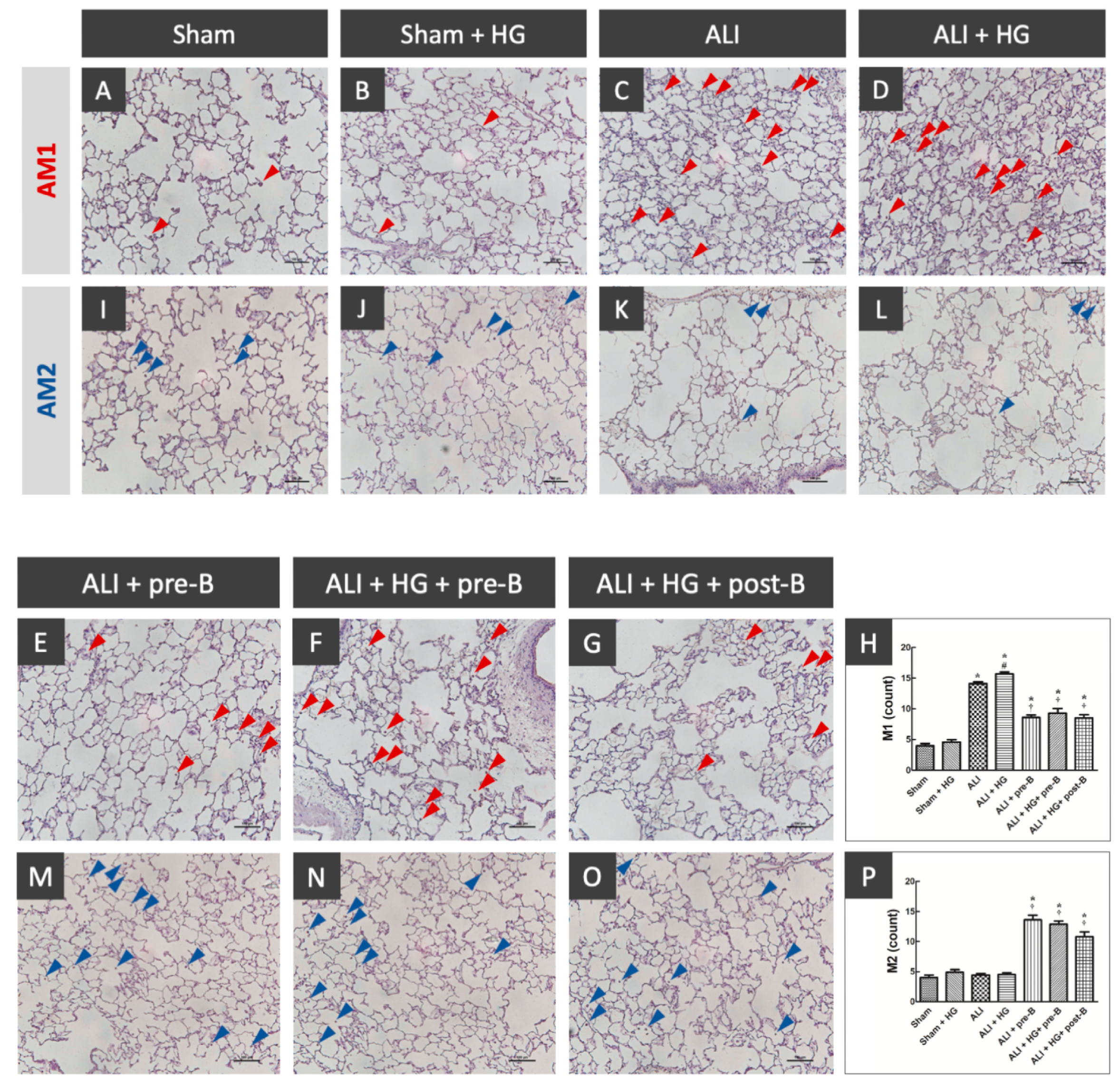
Figure 5.
Effects of hyperglycemia and NKCC1 inhibitor treatment on macrophages. The sections collected from rats of the sham (A) and sham + HG (B) groups showed rare distribution of AM1 (red arrow). Higher AM1 infiltration was noted in the rats of the ALI (C) and ALI + HG (D) groups. Decreased AM1 infiltration was noted in rats of the ALI-pre-B (E), ALI + HG + pre-B (F) and ALI + HG + post-B groups (G). The AM1 counts (H) were higher in rats with ALI and further increased in the ALI + HG group (p < 0.05). The NKCC1 inhibitor reduced AM1 counts in both ALI and ALI + HG groups (p < 0.05). The AM2 infiltration (blue arrow) was rare in the sham (I), sham + HG (J), ALI (K) and ALI + HG (L) groups. Increased AM2 infiltration was noted in rats of the ALI + pre-B (M), ALI + HG + pre-B (N) and ALI + HG + post-B groups (O). The AM2 counts (P) were low in rats of sham, sham +HG, ALI and ALI + HG. The NKCC1 inhibitor increased AM2 counts in both ALI and ALI + HG groups (p < 0.05). * Significant difference, as compared to the sham group (p < 0.05), # Significant difference, comparing the groups with HG to those without HG (I vs. II, III vs. IV, V vs. VI, and V vs. VII, p < 0.05), † Significant difference, comparing the ALI groups with B to those without B (III vs. V, IV vs. VI, and IV vs. VII, p < 0.05). Abbreviation: AM1 = alveolar macrophage 1, AM2 = alveolar macrophage 2, ALI = acute lung injury, HG = hyperglycemia, pre-B = pretreatment with bumetanide, post-B = post-treatment with bumetanide.
Figure 5.
Effects of hyperglycemia and NKCC1 inhibitor treatment on macrophages. The sections collected from rats of the sham (A) and sham + HG (B) groups showed rare distribution of AM1 (red arrow). Higher AM1 infiltration was noted in the rats of the ALI (C) and ALI + HG (D) groups. Decreased AM1 infiltration was noted in rats of the ALI-pre-B (E), ALI + HG + pre-B (F) and ALI + HG + post-B groups (G). The AM1 counts (H) were higher in rats with ALI and further increased in the ALI + HG group (p < 0.05). The NKCC1 inhibitor reduced AM1 counts in both ALI and ALI + HG groups (p < 0.05). The AM2 infiltration (blue arrow) was rare in the sham (I), sham + HG (J), ALI (K) and ALI + HG (L) groups. Increased AM2 infiltration was noted in rats of the ALI + pre-B (M), ALI + HG + pre-B (N) and ALI + HG + post-B groups (O). The AM2 counts (P) were low in rats of sham, sham +HG, ALI and ALI + HG. The NKCC1 inhibitor increased AM2 counts in both ALI and ALI + HG groups (p < 0.05). * Significant difference, as compared to the sham group (p < 0.05), # Significant difference, comparing the groups with HG to those without HG (I vs. II, III vs. IV, V vs. VI, and V vs. VII, p < 0.05), † Significant difference, comparing the ALI groups with B to those without B (III vs. V, IV vs. VI, and IV vs. VII, p < 0.05). Abbreviation: AM1 = alveolar macrophage 1, AM2 = alveolar macrophage 2, ALI = acute lung injury, HG = hyperglycemia, pre-B = pretreatment with bumetanide, post-B = post-treatment with bumetanide.
![Ijms 21 04803 g005]()
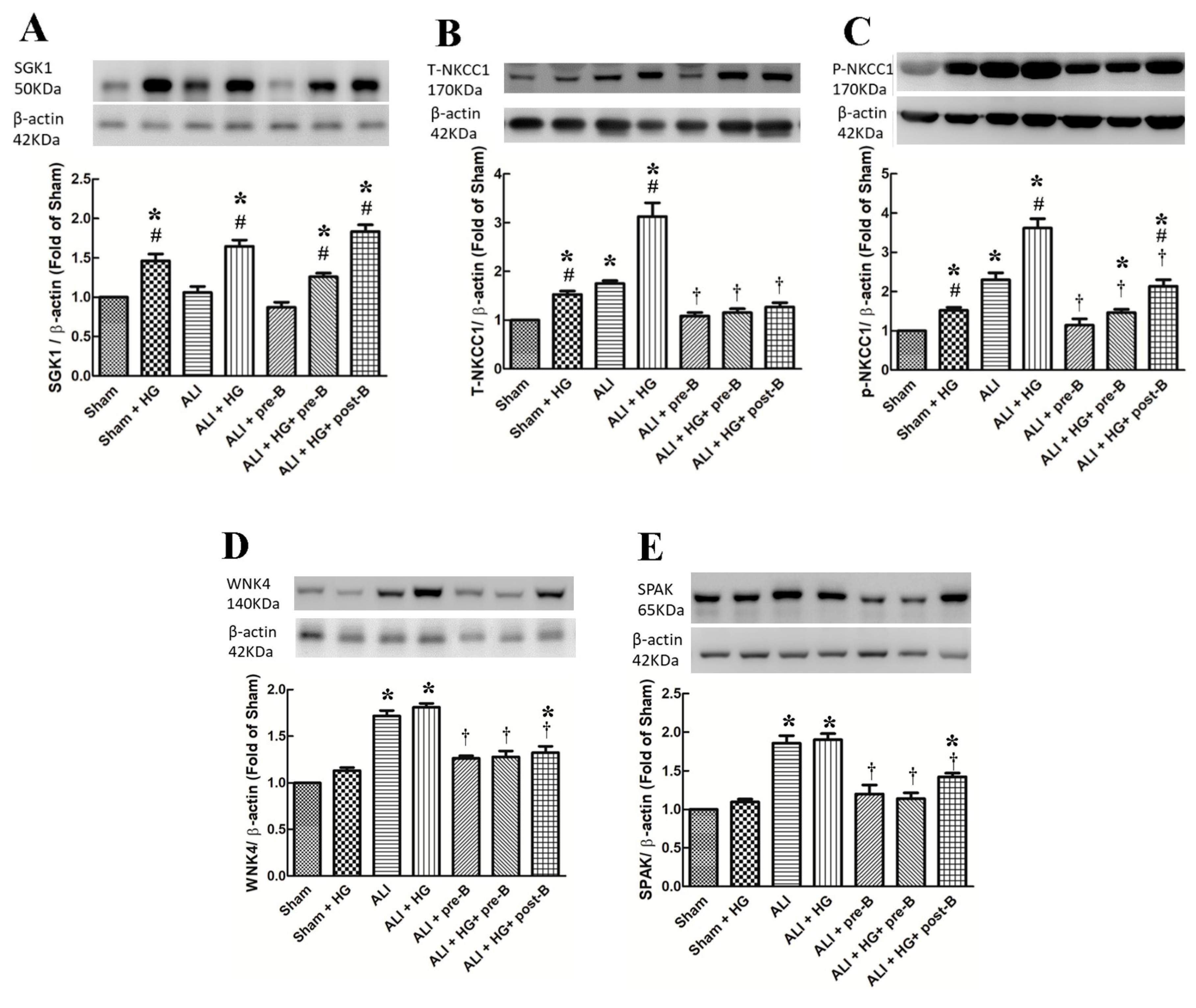
Figure 6.
Effects of hyperglycemia and NKCC1 inhibitor treatment on the levels of SGK1, NKCC1, WNK4 and SPAK. The levels of SGK1 (A) were significantly higher in hyperglycemic rats than in non-hyperglycemic rats (p < 0.05). High levels of T-NKCC1 (B) and p-NKCC1 (C) were observed in the ALI group, which increased further in the ALI + HG group (p < 0.05). The NKCC1 expression was lower in the rats of ALI + pre-B, ALI + HG + pre-B and ALI + HG + post B (p < 0.05). The levels of WNK4 (D), and SPAK (E) were higher in rats of ALI and ALI + HG (p < 0.05) and decreased in the rats of ALI + pre-B, ALI + HG + pre-B and ALI + HG + post B (p < 0.05). * Significant difference, as compared to the sham group (p < 0.05), # Significant difference, comparing the groups with HG to those without HG (I vs. II, III vs. IV, V vs. VI, and V vs. VII, p < 0.05), † Significant difference, comparing the ALI groups with B to those without B (III vs. V, IV vs. VI, and IV vs. VII, p < 0.05). Abbreviation: ALI = acute lung injury, SGK1 = serum-glucocorticoid kinase 1, T-NKCC1 = total sodium-potassium-chloride co-transporter one, p-NKCC1 = phosphorylated sodium-potassium-chloride co-transporter one, WNK4 = with-no-lysine kinases 4, SPAK = STE20/SPS1-related proline/alanine-rich kinase, HG = hyperglycemia, pre-B = pretreatment with bumetanide, post-B = post-treatment with bumetanide.
Figure 6.
Effects of hyperglycemia and NKCC1 inhibitor treatment on the levels of SGK1, NKCC1, WNK4 and SPAK. The levels of SGK1 (A) were significantly higher in hyperglycemic rats than in non-hyperglycemic rats (p < 0.05). High levels of T-NKCC1 (B) and p-NKCC1 (C) were observed in the ALI group, which increased further in the ALI + HG group (p < 0.05). The NKCC1 expression was lower in the rats of ALI + pre-B, ALI + HG + pre-B and ALI + HG + post B (p < 0.05). The levels of WNK4 (D), and SPAK (E) were higher in rats of ALI and ALI + HG (p < 0.05) and decreased in the rats of ALI + pre-B, ALI + HG + pre-B and ALI + HG + post B (p < 0.05). * Significant difference, as compared to the sham group (p < 0.05), # Significant difference, comparing the groups with HG to those without HG (I vs. II, III vs. IV, V vs. VI, and V vs. VII, p < 0.05), † Significant difference, comparing the ALI groups with B to those without B (III vs. V, IV vs. VI, and IV vs. VII, p < 0.05). Abbreviation: ALI = acute lung injury, SGK1 = serum-glucocorticoid kinase 1, T-NKCC1 = total sodium-potassium-chloride co-transporter one, p-NKCC1 = phosphorylated sodium-potassium-chloride co-transporter one, WNK4 = with-no-lysine kinases 4, SPAK = STE20/SPS1-related proline/alanine-rich kinase, HG = hyperglycemia, pre-B = pretreatment with bumetanide, post-B = post-treatment with bumetanide.
![Ijms 21 04803 g006]()
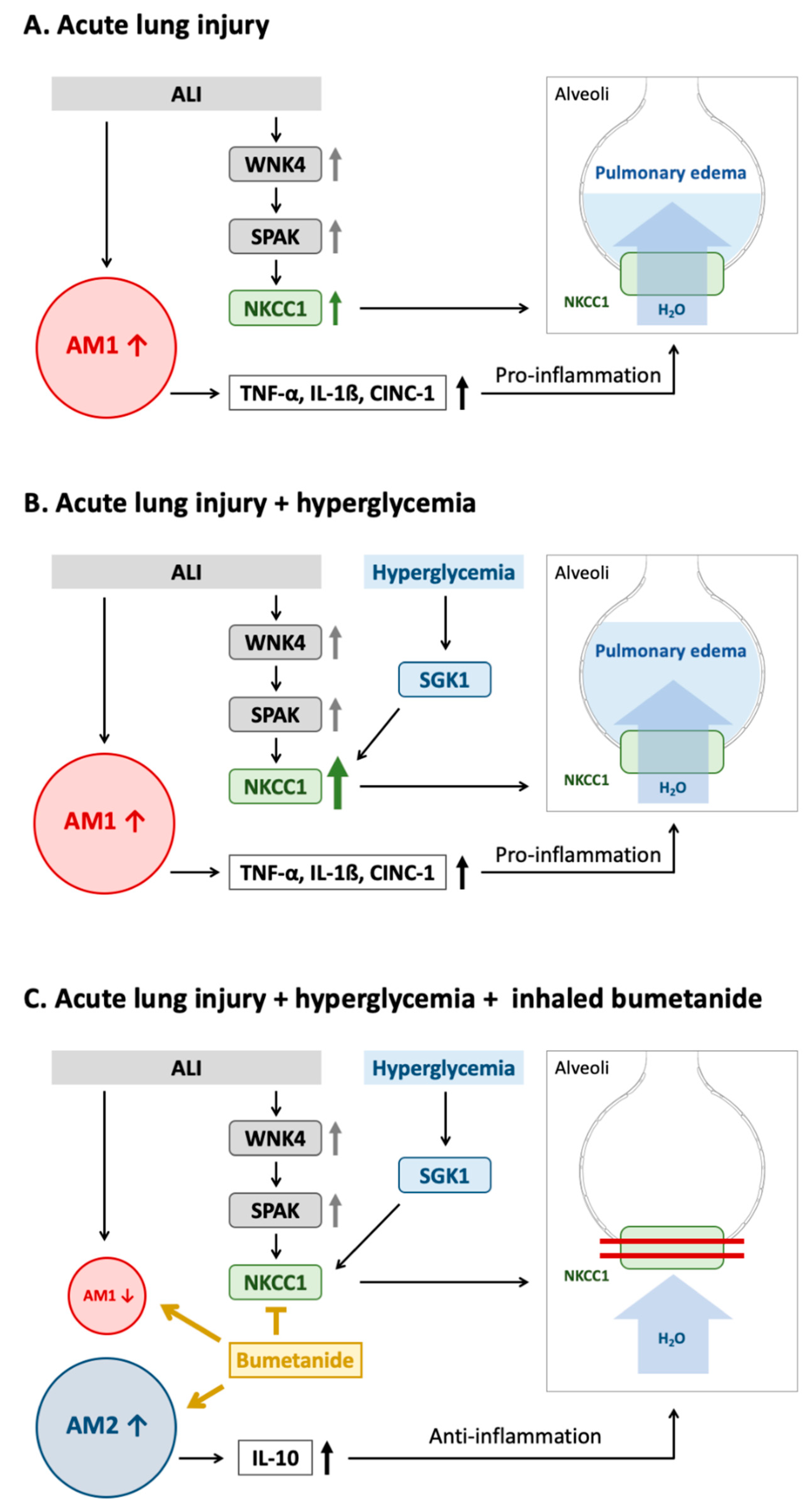
Figure 7.
Mechanism underlying the effect of acute hyperglycemia on SGK1–NKCC1 pathway in acute lung injury. (A) The WNK4–SPAK–NKCC1 pathway is activated in ALI. Higher expression of NKCC1 results in the activation of AM1, increased expression of pro-inflammatory cytokines and impaired alveolar fluid clearance, which result in pulmonary edema and inflammation. (B) Acute hyperglycemia further activates the SGK1–NKCC1 pathway, increases expression of NKCC1 and AM1 activation that consequently results in more severe pulmonary edema, and lung inflammation. (C) The NKCC1 inhibitor can inhibit WNK4–SPAK–NKCC1 and SGK1–NKCC1 pathway that leads to decreased AM1 and pro-inflammatory cytokines, increased AM2 and anti-inflammatory cytokines, reduction in water influx. These result in decreasing pulmonary edema, and inflammation. Abbreviation: ALI = acute lung injury, AM1 = alveolar macrophage 1, AM2 = alveolar macrophage 2, WNK4 = with-no-lysine kinases 4, SPAK = STE20/SPS1-related proline/alanine-rich kinase, SGK1 = Serum-glucocorticoid kinase 1, NKCC1 = sodium-potassium-chloride co-transporter one, TNF-α = tumor necrosis factor alpha, IL-1β = interleukin 1 beta, CINC-1 = cytokine-induced neutrophil chemoattractant 1, IL-10 = interleukin 10.
Figure 7.
Mechanism underlying the effect of acute hyperglycemia on SGK1–NKCC1 pathway in acute lung injury. (A) The WNK4–SPAK–NKCC1 pathway is activated in ALI. Higher expression of NKCC1 results in the activation of AM1, increased expression of pro-inflammatory cytokines and impaired alveolar fluid clearance, which result in pulmonary edema and inflammation. (B) Acute hyperglycemia further activates the SGK1–NKCC1 pathway, increases expression of NKCC1 and AM1 activation that consequently results in more severe pulmonary edema, and lung inflammation. (C) The NKCC1 inhibitor can inhibit WNK4–SPAK–NKCC1 and SGK1–NKCC1 pathway that leads to decreased AM1 and pro-inflammatory cytokines, increased AM2 and anti-inflammatory cytokines, reduction in water influx. These result in decreasing pulmonary edema, and inflammation. Abbreviation: ALI = acute lung injury, AM1 = alveolar macrophage 1, AM2 = alveolar macrophage 2, WNK4 = with-no-lysine kinases 4, SPAK = STE20/SPS1-related proline/alanine-rich kinase, SGK1 = Serum-glucocorticoid kinase 1, NKCC1 = sodium-potassium-chloride co-transporter one, TNF-α = tumor necrosis factor alpha, IL-1β = interleukin 1 beta, CINC-1 = cytokine-induced neutrophil chemoattractant 1, IL-10 = interleukin 10.
![Ijms 21 04803 g007]()
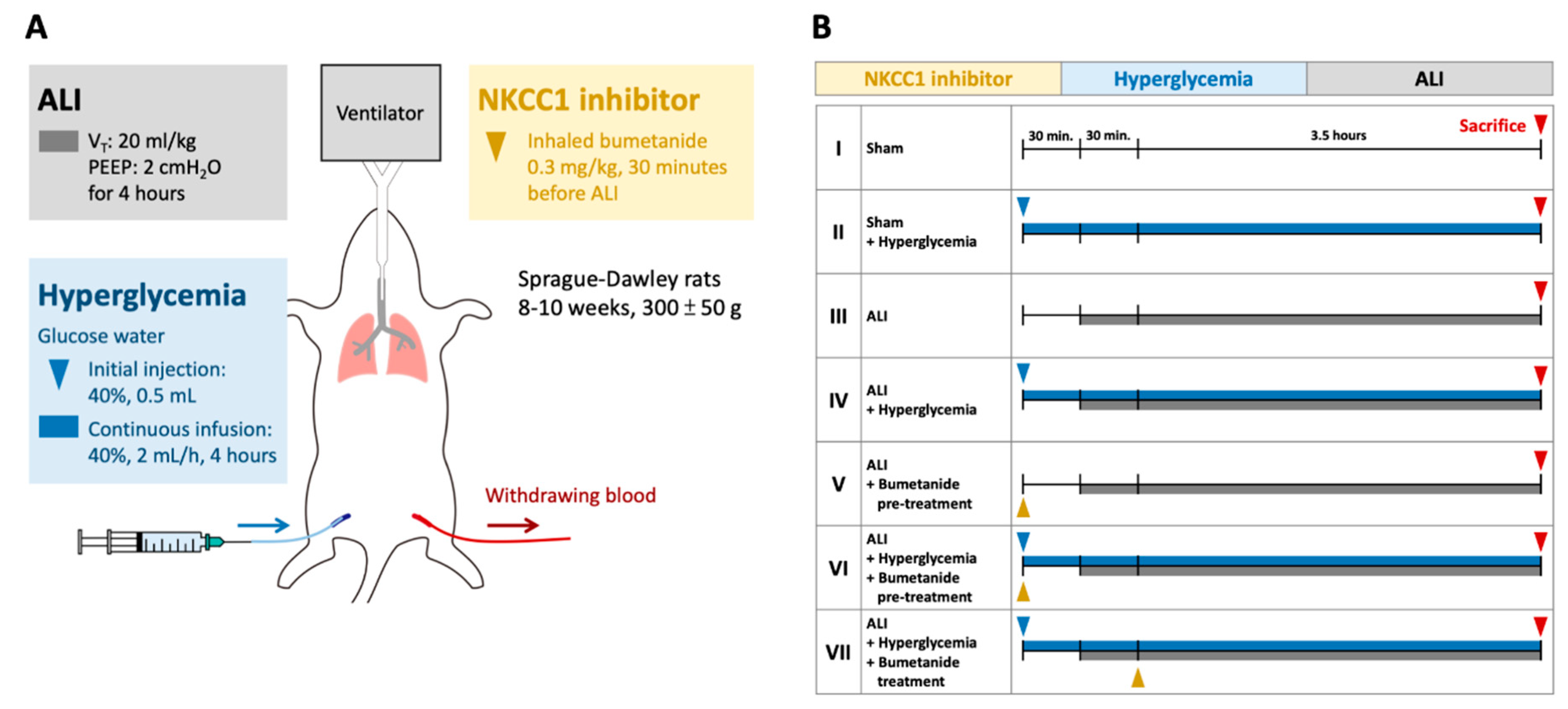
Figure 8.
Protocol for the experiment. (A) The ALI model was induced by a VT of 20 mL/kg for four hours. Acute HG was induced by injection with 0.5 mL 40% glucose solution followed by continuous infusion (2 mL/h). Inhalational bumetanide was administered via tracheostomy. (B) The animals were divided into Group I, sham; Group II, sham+ HG; Group III, ALI; Group IV, ALI + HG; Group V, ALI + pretreatment of inhalational bumetanide (ALI + pre-B); Group VI, ALI+ HG + pretreatment of inhalational bumetanide (ALI + HG + pre-B), and Group VII, ALI + HG + post-ALI inhalational bumetanide (ALI + HG + post-B). Abbreviation: ALI = acute lung injury, VT = tidal volume, NKCC1 = sodium-potassium-chloride co-transporter one, HG = hyperglycemia, pre-B = pretreatment with bumetanide, post-B = post-treatment with bumetanide.
Figure 8.
Protocol for the experiment. (A) The ALI model was induced by a VT of 20 mL/kg for four hours. Acute HG was induced by injection with 0.5 mL 40% glucose solution followed by continuous infusion (2 mL/h). Inhalational bumetanide was administered via tracheostomy. (B) The animals were divided into Group I, sham; Group II, sham+ HG; Group III, ALI; Group IV, ALI + HG; Group V, ALI + pretreatment of inhalational bumetanide (ALI + pre-B); Group VI, ALI+ HG + pretreatment of inhalational bumetanide (ALI + HG + pre-B), and Group VII, ALI + HG + post-ALI inhalational bumetanide (ALI + HG + post-B). Abbreviation: ALI = acute lung injury, VT = tidal volume, NKCC1 = sodium-potassium-chloride co-transporter one, HG = hyperglycemia, pre-B = pretreatment with bumetanide, post-B = post-treatment with bumetanide.


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}







