Implications of SARS-CoV-2 Mutations for Genomic RNA Structure and Host microRNA Targeting

Abstract
:1. Introduction
2. Results
2.1. Identification of SARS-Cov-2 Recurrence Mutations
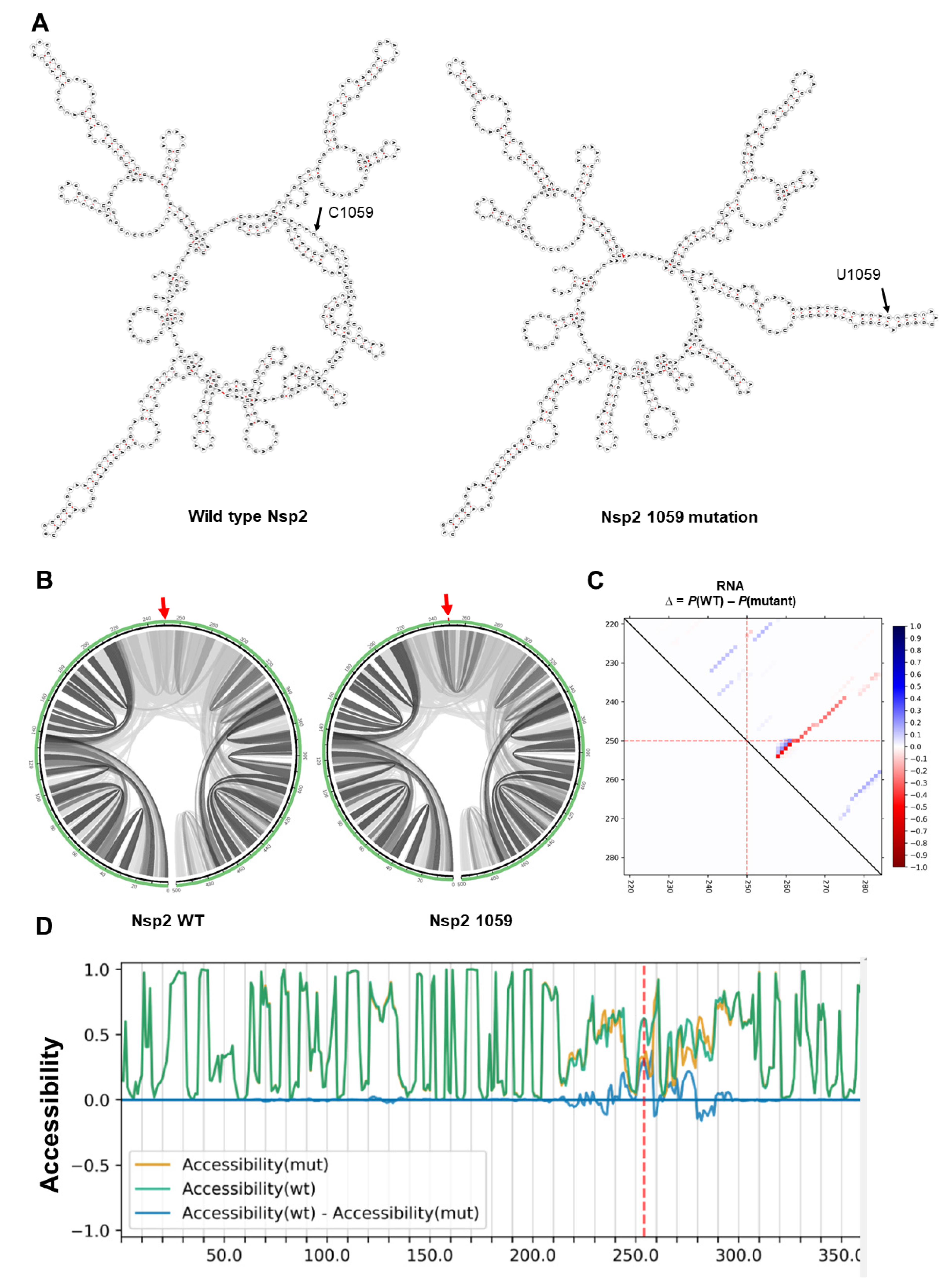
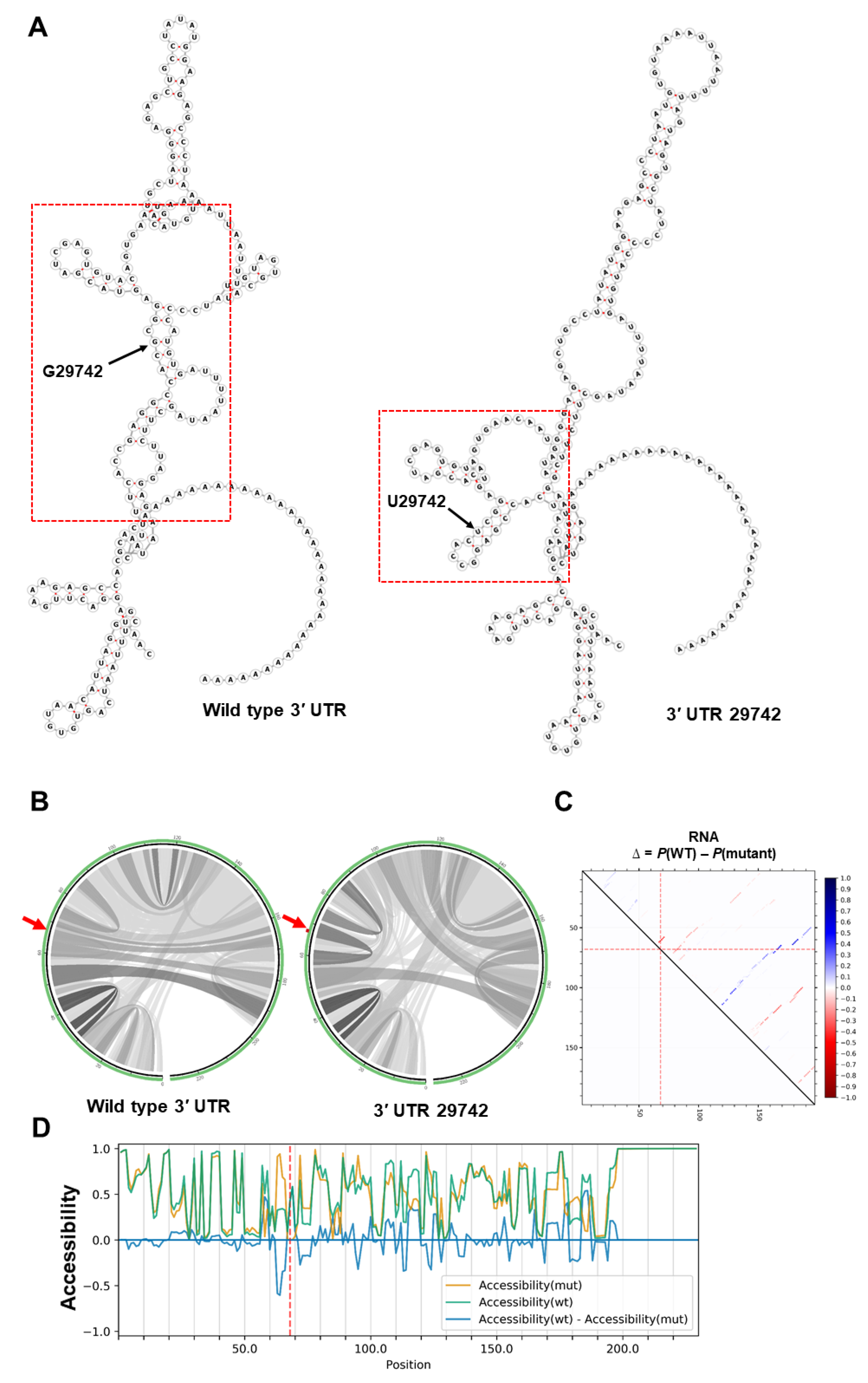
2.2. RNA Secondary Structure
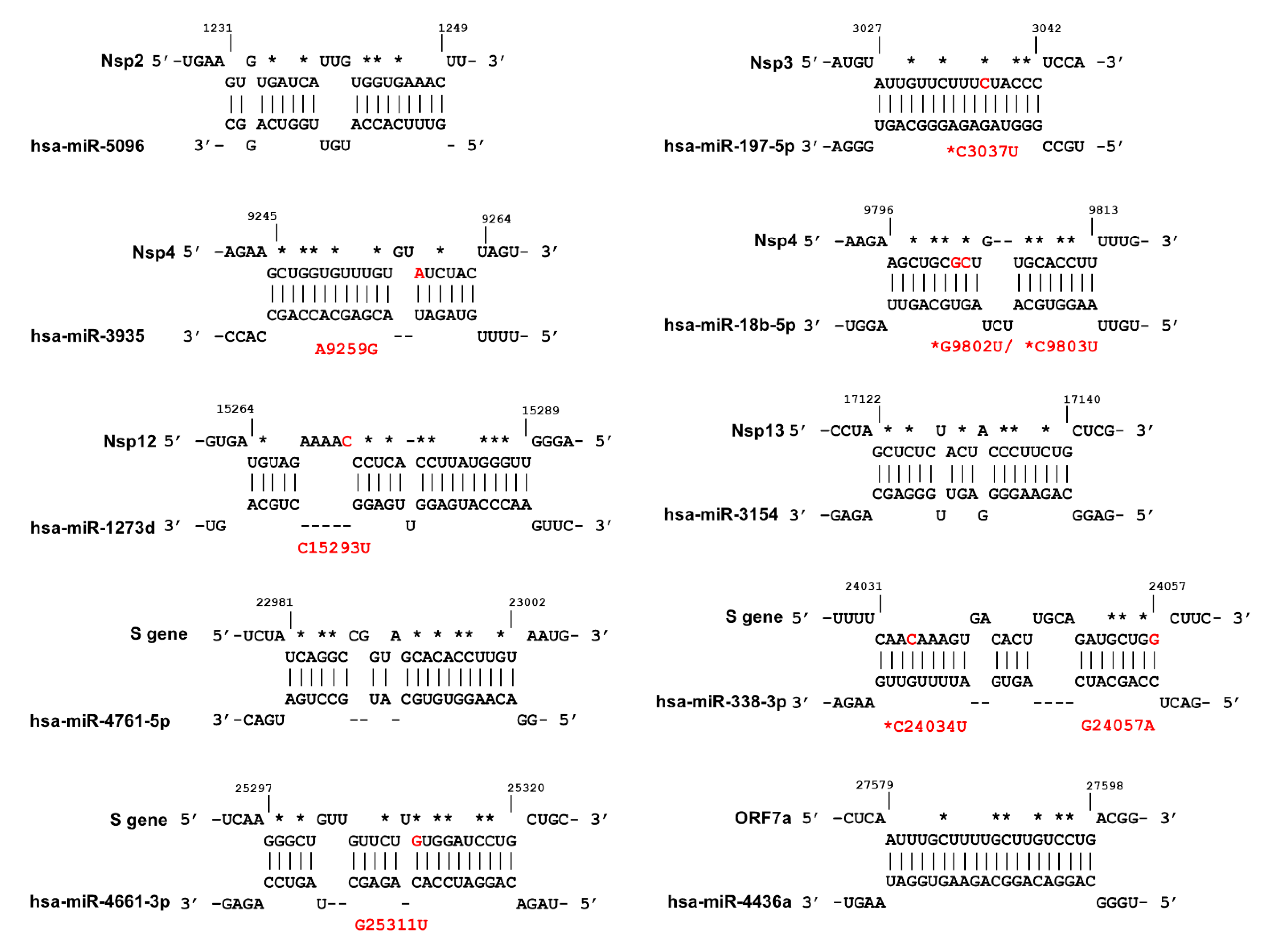
2.3. Potential Interaction of SARS-CoV-2 Transcripts and Human miRNAs
2.4. Possible Impact of Mutations on Cryptic Splice Sites
3. Discussion
4. Methods
4.1. Sequence Alignment
4.2. Mutational Analysis
4.3. RNA Secondary Structure and Base Pair Probability Analysis
4.4. Potential miRNA Binding Site Analysis
4.5. Potential Splice Site Analysis
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Totura, A.L.; Baric, R.S. SARS coronavirus pathogenesis: Host innate immune responses and viral antagonism of interferon. Curr. Opin. Virol. 2012, 2, 264–275. [Google Scholar] [CrossRef] [PubMed]
- Tort, F.L.; Castells, M.; Cristina, J. A comprehensive analysis of genome composition and codon usage patterns of emerging coronaviruses. Virus Res. 2020, 283, 197976. [Google Scholar] [CrossRef] [PubMed]
- MacLean, O.A.; Orton, R.J.; Singer, J.B.; Robertson, D.L. No evidence for distinct types in the evolution of SARS-CoV-2. Virus Evol. 2020, 6, veaa034. [Google Scholar] [CrossRef]
- van Dorp, L.; Richard, D.; Tan, C.C.; Shaw, L.P.; Acman, M.; Balloux, F. No evidence for increased transmissibility from recurrent mutations in SARS-CoV-2. bioRxiv 2020. [Google Scholar]
- Zhang, L.; Jackson, C.B.; Mou, H.; Ojha, A.; Rangarajan, E.S.; Izard, T.; Farzan, M.; Choe, H. The D614G mutation in the SARS-CoV-2 spike protein reduces S1 shedding and increases infectivity. bioRxiv 2020. [Google Scholar] [CrossRef]
- Kusov, Y.; Tan, J.; Alvarez, E.; Enjuanes, L.; Hilgenfeld, R. A G-quadruplex-binding macrodomain within the “SARS-unique domain” is essential for the activity of the SARS-coronavirus replication–transcription complex. Virology 2015, 484, 313–322. [Google Scholar] [CrossRef] [Green Version]
- Chen, C.-Y.; Chang, C.-K.; Chang, Y.-W.; Sue, S.-C.; Bai, H.-I.; Riang, L.; Hsiao, C.-D.; Huang, T.-H. Structure of the SARS coronavirus nucleocapsid protein RNA-binding dimerization domain suggests a mechanism for helical packaging of viral RNA. J. Mol. Biol. 2007, 368, 1075–1086. [Google Scholar] [CrossRef]
- Faure, G.; Ogurtsov, A.Y.; Shabalina, S.A.; Koonin, E.V. Role of mRNA structure in the control of protein folding. Nucleic Acids Res. 2016, 44, 10898–10911. [Google Scholar] [CrossRef] [Green Version]
- Peeri, M.; Tuller, T. High-resolution modeling of the selection on local mRNA folding strength in coding sequences across the tree of life. Genome Biol. 2020, 21, 1–20. [Google Scholar] [CrossRef] [Green Version]
- Rangan, R.; Watkins, A.M.; Kladwang, W.; Das, R. De novo 3D models of SARS-CoV-2 RNA elements and small-molecule-binding RNAs to guide drug discovery. bioRxiv 2020. [Google Scholar] [CrossRef]
- Rangan, R.; Zheludev, I.N.; Das, R. RNA genome conservation and secondary structure in SARS-CoV-2 and SARS-related viruses. bioRxiv 2020. [Google Scholar] [CrossRef] [Green Version]
- Andrews, R.J.; Peterson, J.M.; Haniff, H.F.; Chen, J.; Williams, C.; Greffe, M.; Disney, M.D.; Moss, W.N. An in silico map of the SARS-CoV-2 RNA Structurome. bioRxiv 2020. [Google Scholar] [CrossRef] [Green Version]
- Cullen, B.R. Five questions about viruses and microRNAs. PLoS Pathog. 2010, 6. [Google Scholar] [CrossRef] [PubMed]
- Leon-Icaza, S.A.; Zeng, M.; Rosas-Taraco, A.G. microRNAs in viral acute respiratory infections: Immune regulation, biomarkers, therapy, and vaccines. ExRNA 2019, 1, 1. [Google Scholar] [CrossRef] [Green Version]
- Pfeffer, S.; Voinnet, O. Viruses, microRNAs and cancer. Oncogene 2006, 25, 6211–6219. [Google Scholar] [CrossRef] [Green Version]
- Kincaid, R.P.; Burke, J.M.; Sullivan, C.S. RNA virus microRNA that mimics a B-cell oncomiR. Proc. Natl. Acad. Sci. USA 2012, 109, 3077–3082. [Google Scholar] [CrossRef] [Green Version]
- Ha, M.; Pang, M.; Agarwal, V.; Chen, Z.J. Interspecies regulation of microRNAs and their targets. Biochim. Biophys. Acta (BBA) Gene Regul. Mech. 2008, 1779, 735–742. [Google Scholar] [CrossRef] [Green Version]
- Cullen, B.R. Viruses and microRNAs. Nat. Genet. 2006, 38, S25–S30. [Google Scholar] [CrossRef]
- Mahajan, V.S.; Drake, A.; Chen, J. Virus-specific host miRNAs: Antiviral defenses or promoters of persistent infection? Trends Immunol. 2009, 30, 1–7. [Google Scholar] [CrossRef] [Green Version]
- Linsen, S.E.; Tops, B.B.; Cuppen, E. miRNAs: Small changes, widespread effects. Cell Res. 2008, 18, 1157–1159. [Google Scholar] [CrossRef] [Green Version]
- Lai, F.W.; Stephenson, K.B.; Mahony, J.; Lichty, B.D. Human coronavirus OC43 nucleocapsid protein binds microRNA 9 and potentiates NF-κB activation. J. Virol. 2014, 88, 54–65. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mallick, B.; Ghosh, Z.; Chakrabarti, J. MicroRNome analysis unravels the molecular basis of SARS infection in bronchoalveolar stem cells. PLoS ONE 2009, 4. [Google Scholar] [CrossRef] [PubMed]
- Rakhmetullina, A.; Ivashchenko, A.; Akimniyazova, A.; Aisina, D.; Pyrkova, A. The miRNA Complexes Against Coronaviruses COVID-19, SARS-CoV, And MERS-CoV. Preprint 2020. [Google Scholar] [CrossRef]
- Ivashchenko, A.; Rakhmetullina, A.; Aisina, D. How miRNAs can protect humans from coronaviruses COVID-19, SARS-CoV, and MERS-CoV. Preprint 2020. [Google Scholar] [CrossRef] [Green Version]
- Morales, L.; Oliveros, J.C.; Fernandez-Delgado, R.; Robert tenOever, B.; Enjuanes, L.; Sola, I. SARS-CoV-encoded small RNAs contribute to infection-associated lung pathology. Cell Host Microbe 2017, 21, 344–355. [Google Scholar] [CrossRef] [Green Version]
- Demirci, M.D.S.; Adan, A. Computational analysis of microRNA-mediated interactions in SARS-CoV-2 infection. Peerj 2020, 8, e9369. [Google Scholar] [CrossRef]
- Liu, Z.; Wang, J.; Xu, Y.; Guo, M.; Mi, K.; Xu, R.; Pei, Y.; Zhang, Q.; Luan, X.; Hu, Z. Implications of the virus-encoded miRNA and host miRNA in the pathogenicity of SARS-CoV-2. arXiv 2020, arXiv:2004.04874. [Google Scholar]
- Dubois, J.; Terrier, O.; Rosa-Calatrava, M. Influenza viruses and mRNA splicing: Doing more with less. MBio 2014, 5, e00070-14. [Google Scholar] [CrossRef] [Green Version]
- Hardy, W.R.; Sandri-Goldin, R. Herpes simplex virus inhibits host cell splicing, and regulatory protein ICP27 is required for this effect. J. Virol. 1994, 68, 7790–7799. [Google Scholar] [CrossRef] [Green Version]
- Purcell, D.; Martin, M.A. Alternative splicing of human immunodeficiency virus type 1 mRNA modulates viral protein expression, replication, and infectivity. J. Virol. 1993, 67, 6365–6378. [Google Scholar] [CrossRef] [Green Version]
- Schneider, P.A.; Schneemann, A.; Lipkin, W.I. RNA splicing in Borna disease virus, a nonsegmented, negative-strand RNA virus. J. Virol. 1994, 68, 5007–5012. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, Y.-C.; Kuo, R.-L.; Lin, J.-Y.; Huang, P.-N.; Huang, Y.; Liu, H.; Arnold, J.J.; Chen, S.-J.; Wang, R.Y.-L.; Cameron, C.E. Cytoplasmic viral RNA-dependent RNA polymerase disrupts the intracellular splicing machinery by entering the nucleus and interfering with Prp8. PLoS Pathog. 2014, 10, e1004119. [Google Scholar] [CrossRef] [PubMed]
- Lai, M.; Baric, R.S.; Brayton, P.R.; Stohlman, S.A. Characterization of leader RNA sequences on the virion and mRNAs of mouse hepatitis virus, a cytoplasmic RNA virus. Proc. Natl. Acad. Sci. USA 1984, 81, 3626–3630. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, D.; Lee, J.-Y.; Yang, J.-S.; Kim, J.W.; Kim, V.N.; Chang, H. The architecture of SARS-CoV-2 transcriptome. Cell 2020, 181, 914–921.e10. [Google Scholar] [CrossRef]
- Barnes, D.; Kunitomi, M.; Vignuzzi, M.; Saksela, K.; Andino, R. Harnessing endogenous miRNAs to control virus tissue tropism as a strategy for developing attenuated virus vaccines. Cell Host Microbe 2008, 4, 239–248. [Google Scholar] [CrossRef] [Green Version]
- Kelly, E.J.; Hadac, E.M.; Greiner, S.; Russell, S.J. Engineering microRNA responsiveness to decrease virus pathogenicity. Nat. Med. 2008, 14, 1278–1283. [Google Scholar] [CrossRef]
- Langlois, R.A.; Albrecht, R.A.; Kimble, B.; Sutton, T.; Shapiro, J.S.; Finch, C.; Angel, M.; Chua, M.A.; Gonzalez-Reiche, A.S.; Xu, K. MicroRNA-based strategy to mitigate the risk of gain-of-function influenza studies. Nat. Biotechnol. 2013, 31, 844–847. [Google Scholar] [CrossRef]
- Perez, J.T.; Pham, A.M.; Lorini, M.H.; Chua, M.A.; Steel, J. MicroRNA-mediated species-specific attenuation of influenza A virus. Nat. Biotechnol. 2009, 27, 572–576. [Google Scholar] [CrossRef]
- Pachetti, M.; Marini, B.; Benedetti, F.; Giudici, F.; Mauro, E.; Storici, P.; Masciovecchio, C.; Angeletti, S.; Ciccozzi, M.; Gallo, R.C. Emerging SARS-CoV-2 mutation hot spots include a novel RNA-dependent-RNA polymerase variant. J. Transl. Med. 2020, 18, 179. [Google Scholar] [CrossRef] [Green Version]
- Li, X.; Giorgi, E.E.; Marichannegowda, M.H.; Foley, B.; Xiao, C.; Kong, X.-P.; Chen, Y.; Gnanakaran, S.; Korber, B.; Gao, F. Emergence of SARS-CoV-2 through recombination and strong purifying selection. Sci. Adv. 2020, 6, eabb9153. [Google Scholar] [CrossRef]
- MacLean, O.A.; Lytras, S.; Singer, J.B.; Weaver, S.; Pond, S.L.K.; Robertson, D.L. Evidence of significant natural selection in the evolution of SARS-CoV-2 in bats, not humans. bioRxiv 2020. [Google Scholar] [CrossRef]
- Khrustalev, V.V.; Giri, R.; Khrustaleva, T.A.; Kapuganti, S.K.; Stojarov, A.N.; Poboinev, V.V. Translation-associated mutational U-pressure in the first ORF of SARS-CoV-2 and other coronaviruses. bioRxiv 2020. [Google Scholar] [CrossRef]
- Farkas, C.; Fuentes-Villalobos, F.; Garrido, J.L.; Haigh, J.J.; Barría, M.I. Insights on early mutational events in SARS-CoV-2 virus reveal founder effects across geographical regions. bioRxiv 2020, 8, e9255. [Google Scholar]
- Lange, S.J.; Maticzka, D.; Möhl, M.; Gagnon, J.N.; Brown, C.M.; Backofen, R. Global or local? Predicting secondary structure and accessibility in mRNAs. Nucleic Acids Res. 2012, 40, 5215–5226. [Google Scholar] [CrossRef] [Green Version]
- Bernhart, S.H.; Mückstein, U.; Hofacker, I.L. RNA Accessibility in cubic time. Algorithms Mol. Biol. 2011, 6, 3. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Robertson, M.P.; Igel, H.; Baertsch, R.; Haussler, D.; Ares, M., Jr.; Scott, W.G. The structure of a rigorously conserved RNA element within the SARS virus genome. PLoS Biol. 2004, 3, e5. [Google Scholar] [CrossRef]
- Li, L.; Kang, H.; Liu, P.; Makkinje, N.; Williamson, S.T.; Leibowitz, J.L.; Giedroc, D.P. Structural lability in stem–loop 1 drives a 5′ UTR–3′ UTR interaction in coronavirus replication. J. Mol. Biol. 2008, 377, 790–803. [Google Scholar] [CrossRef] [PubMed]
- Yang, D.; Leibowitz, J.L. The structure and functions of coronavirus genomic 3′ and 5′ ends. Virus Res. 2015, 206, 120–133. [Google Scholar] [CrossRef]
- Gordon, D.E.; Jang, G.M.; Bouhaddou, M.; Xu, J.; Obernier, K.; O’meara, M.J.; Guo, J.Z.; Swaney, D.L.; Tummino, T.A.; Huttenhain, R. A SARS-CoV-2-human protein-protein interaction map reveals drug targets and potential drug-repurposing. bioRxiv 2020. [Google Scholar] [CrossRef] [Green Version]
- Khan, M.A.-A.-K.; Islam, A.B. SARS-CoV-2 proteins exploit host’s genetic and epigenetic mediators for the annexation of key host signaling pathways that confers its immune evasion and disease pathophysiology. bioRxiv 2020. [Google Scholar] [CrossRef]
- Kleinman, C.L.; Adoue, V.; Majewski, J. RNA editing of protein sequences: A rare event in human transcriptomes. Rna 2012, 18, 1586–1596. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schulte, C.; Molz, S.; Appelbaum, S.; Karakas, M.; Ojeda, F.; Lau, D.M.; Hartmann, T.; Lackner, K.J.; Westermann, D.; Schnabel, R.B. miRNA-197 and miRNA-223 predict cardiovascular death in a cohort of patients with symptomatic coronary artery disease. PLoS ONE 2015, 10. [Google Scholar] [CrossRef] [Green Version]
- Liu, W.; Zheng, J.; Dong, J.; Bai, R.; Song, D.; Ma, X.; Zhao, L.; Yao, Y.; Zhang, H.; Liu, T. Association of miR-197-5p, a circulating biomarker for heart failure, with myocardial fibrosis and adverse cardiovascular events among patients with stage C or D heart failure. Cardiology 2018, 141, 212–225. [Google Scholar] [CrossRef] [PubMed]
- Gupta, S.K.; Bang, C.; Thum, T. Circulating microRNAs as biomarkers and potential paracrine mediators of cardiovascular disease. Circ. Cardiovasc. Genet. 2010, 3, 484–488. [Google Scholar] [CrossRef] [Green Version]
- Xu, Z.; Shi, L.; Wang, Y.; Zhang, J.; Huang, L.; Zhang, C.; Liu, S.; Zhao, P.; Liu, H.; Zhu, L. Pathological findings of COVID-19 associated with acute respiratory distress syndrome. Lancet Respir. Med. 2020, 8, 420–422. [Google Scholar] [CrossRef]
- Korber, B.; Fischer, W.M.; Gnanakaran, S.; Yoon, H.; Theiler, J.; Abfalterer, W.; Hengartner, N.; Giorgi, E.E.; Bhattacharya, T.; Foley, B.; et al. Tracking changes in SARS-CoV-2 Spike: evidence that D614G increases infectivity of the COVID-19 virus. Cell 2020, 20, 30820–308025. [Google Scholar] [CrossRef]
- Yang, Z.; Li, J.; Feng, G.; Wang, Y.; Yang, G.; Liu, Y.; Zhang, S.; Feng, J.; Zhang, X. Hepatitis B virus X protein enhances hepatocarcinogenesis by depressing the targeting of NUSAP1 mRNA by miR-18b. Cancer Biol. Med. 2019, 16, 276. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Duy, J.; Koehler, J.W.; Honko, A.N.; Schoepp, R.J.; Wauquier, N.; Gonzalez, J.-P.; Pitt, M.L.; Mucker, E.M.; Johnson, J.C.; O’Hearn, A. Circulating microRNA profiles of Ebola virus infection. Sci. Rep. 2016, 6, 24496. [Google Scholar] [CrossRef]
- Condorelli, G.; Latronico, M.V.; Dorn, G.W. microRNAs in heart disease: Putative novel therapeutic targets? Eur. Heart J. 2010, 31, 649–658. [Google Scholar] [CrossRef] [Green Version]
- Luo, P.; Zhang, W. MicroRNA-18b* induces apoptosis in cardiomyocytes through targeting Topoisomerase 1 (TOP1). Int. J. Clin. Exp. Med. 2017, 10, 6742–6748. [Google Scholar]
- Tijsen, A.J.; Creemers, E.E.; Moerland, P.D.; de Windt, L.J.; van der Wal, A.C.; Kok, W.E.; Pinto, Y.M. MiR423-5p as a circulating biomarker for heart failure. Circ. Res. 2010, 106, 1035. [Google Scholar] [CrossRef] [PubMed]
- Fu, X.; Tan, D.; Hou, Z.; Hu, Z.; Liu, G.; Ouyang, Y.; Liu, F. The effect of miR-338-3p on HBx deletion-mutant (HBx-d382) mediated liver-cell proliferation through CyclinD1 regulation. PLoS ONE 2012, 7, e43204. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liang, Y.; Xu, X.; Wang, T.; Li, Y.; You, W.; Fu, J.; Liu, Y.; Jin, S.; Ji, Q.; Zhao, W. The EGFR/miR-338-3p/EYA2 axis controls breast tumor growth and lung metastasis. Cell Death Dis. 2017, 8, e2928. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, P.; Chen, X.; Su, L.; Li, C.; Zhi, Q.; Yu, B.; Sheng, H.; Wang, J.; Feng, R.; Cai, Q. Epigenetic silencing of miR-338-3p contributes to tumorigenicity in gastric cancer by targeting SSX2IP. PLoS ONE 2013, 8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Winther, T.N.; Bang-Berthelsen, C.H.; Heiberg, I.L.; Pociot, F.; Hogh, B. Differential plasma microRNA profiles in HBeAg positive and HBeAg negative children with chronic hepatitis B. PLoS ONE 2013, 8. [Google Scholar] [CrossRef] [PubMed]
- Yu, K.; Shi, G.; Li, N. The function of MicroRNA in hepatitis B virus-related liver diseases: From Dim to Bright. Ann. Hepatol. 2015, 14, 450–456. [Google Scholar] [CrossRef]
- Hasan, M.; McLean, E.; Bagasra, O. A computational analysis to construct a potential post-Exposure therapy against pox epidemic using miRNAs in silico. J. Bioterror Biodef 2016, 7, 2. [Google Scholar] [CrossRef]
- Chang, T.-H.; Huang, H.-Y.; Hsu, J.B.-K.; Weng, S.-L.; Horng, J.-T.; Huang, H.-D. An enhanced computational platform for investigating the roles of regulatory RNA and for identifying functional RNA motifs. Proc. BMC Bioinf. 2013, 14, 1–8. [Google Scholar] [CrossRef] [Green Version]
- Desmet, F.-O.; Hamroun, D.; Lalande, M.; Collod-Béroud, G.; Claustres, M.; Béroud, C. Human Splicing Finder: An online bioinformatics tool to predict splicing signals. Nucleic Acids Res. 2009, 37, e67. [Google Scholar] [CrossRef] [Green Version]
- Hiller, M.; Zhang, Z.; Backofen, R.; Stamm, S. Pre-mRNA secondary structures influence exon recognition. PLoS Genet. 2007, 3, e204. [Google Scholar] [CrossRef] [Green Version]
- Raden, M.; Ali, S.M.; Alkhnbashi, O.S.; Busch, A.; Costa, F.; Davis, J.A.; Eggenhofer, F.; Gelhausen, R.; Georg, J.; Heyne, S. Freiburg RNA tools: A central online resource for RNA-focused research and teaching. Nucleic Acids Res. 2018, 46, W25–W29. [Google Scholar] [CrossRef]
- Sironi, M.; Menozzi, G.; Riva, L.; Cagliani, R.; Comi, G.P.; Bresolin, N.; Giorda, R.; Pozzoli, U. Silencer elements as possible inhibitors of pseudoexon splicing. Nucleic Acids Res. 2004, 32, 1783–1791. [Google Scholar] [CrossRef]
- Xia, X. Extreme genomic CpG deficiency in SARS-CoV-2 and evasion of host antiviral defense. Mol. Biol. Evol. 2020. [Google Scholar] [CrossRef]
- Chen, L.; Li, C.; Peng, Z.; Zhao, J.; Gong, G.; Tan, D. miR-197 expression in peripheral blood mononuclear cells from hepatitis B virus-infected patients. Gut Liver 2013, 7, 335. [Google Scholar] [CrossRef]
- Tang, W.-F.; Huang, R.-T.; Chien, K.-Y.; Huang, J.-Y.; Lau, K.-S.; Jheng, J.-R.; Chiu, C.-H.; Wu, T.-Y.; Chen, C.-Y.; Horng, J.-T. Host microRNA miR-197 plays a negative regulatory role in the enterovirus 71 infectious cycle by targeting the RAN protein. J. Virol. 2016, 90, 1424–1438. [Google Scholar] [CrossRef] [Green Version]
- Wang, H.; Su, X.; Yang, M.; Chen, T.; Hou, J.; Li, N.; Cao, X. Reciprocal control of miR-197 and IL-6/STAT3 pathway reveals miR-197 as potential therapeutic target for hepatocellular carcinoma. Oncoimmunology 2015, 4, e1031440. [Google Scholar] [CrossRef]
- Peng, F.; Loo, J.F.C.; Kong, S.K.; Li, B.; Gu, D. Identification of serum MicroRNAs as diagnostic biomarkers for influenza H7N9 infection. Virology Reports 2017, 7, 1–8. [Google Scholar] [CrossRef]
- Nishikura, K. Functions and regulation of RNA editing by ADAR deaminases. Ann. Rev. Biochem. 2010, 79, 321–349. [Google Scholar] [CrossRef] [Green Version]
- Zhang, L.; Yang, C.-S.; Varelas, X.; Monti, S. Altered RNA editing in 3′ UTR perturbs microRNA-mediated regulation of oncogenes and tumor-suppressors. Sci. Rep. 2016, 6, 1–13. [Google Scholar] [CrossRef] [Green Version]
- Lim, J.; Byun, J.; Guk, K.; Hwang, S.G.; Bae, P.K.; Jung, J.; Kang, T.; Lim, E.-K. Highly Sensitive in Vitro Diagnostic System of Pandemic Influenza A (H1N1) Virus Infection with Specific MicroRNA as a Biomarker. ACS Omega 2019, 4, 14560–14568. [Google Scholar] [CrossRef]
- Zhu, Z.; Qi, Y.; Fan, H.; Cui, L.; Shi, Z. Systematic identification and bioinformatic analysis of microRNAs in response to infections of coxsackievirus A16 and enterovirus 71. BioMed Res. Int. 2016, 2016. [Google Scholar] [CrossRef]
- Arslan, S.; Engin, A.; Aydemir, E.I.; Sahin, N.O.; Bayyurt, B.; Sari, I.; Cosgun, Y.; Bakir, M. Identification of potential microRNA markers related to Crimean-Congo hemorrhagic fever disease. J. Cell. Biochem. 2019, 120, 15506–15517. [Google Scholar] [CrossRef]
- Dong, D.-L.; Yang, B.-F. Role of microRNAs in cardiac hypertrophy, myocardial fibrosis and heart failure. Acta Pharm. Sin. B 2011, 1, 1–7. [Google Scholar] [CrossRef] [Green Version]
- Wang, X. Composition of seed sequence is a major determinant of microRNA targeting patterns. Bioinformatics 2014, 30, 1377–1383. [Google Scholar] [CrossRef] [Green Version]
- Carmel, I.; Shomron, N.; Heifetz, Y. Does base-pairing strength play a role in microRNA repression? Rna 2012, 18, 1947–1956. [Google Scholar] [CrossRef] [Green Version]
- Rolle, K.; Piwecka, M.; Belter, A.; Wawrzyniak, D.; Jeleniewicz, J.; Barciszewska, M.Z.; Barciszewski, J. The sequence and structure determine the function of mature human miRNAs. PLoS ONE 2016, 11. [Google Scholar] [CrossRef]
- O’shea, T.J.; Cryan, P.M.; Cunningham, A.A.; Fooks, A.R.; Hayman, D.T.; Luis, A.D.; Peel, A.J.; Plowright, R.K.; Wood, J.L. Bat flight and zoonotic viruses. Emerg. Infect. Dis. 2014, 20, 741. [Google Scholar] [CrossRef] [Green Version]
- Broughton, J.P.; Lovci, M.T.; Huang, J.L.; Yeo, G.W.; Pasquinelli, A.E. Pairing beyond the seed supports microRNA targeting specificity. Mol. Cell 2016, 64, 320–333. [Google Scholar] [CrossRef] [Green Version]
- Zhang, K.; Zhang, X.; Cai, Z.; Zhou, J.; Cao, R.; Zhao, Y.; Chen, Z.; Wang, D.; Ruan, W.; Zhao, Q. A novel class of microRNA-recognition elements that function only within open reading frames. Nat. Struct. Mol. Biol. 2018, 25, 1019–1027. [Google Scholar] [CrossRef]
- Apcher, S.; Daskalogianni, C.; Lejeune, F.; Manoury, B.; Imhoos, G.; Heslop, L.; Fåhraeus, R. Major source of antigenic peptides for the MHC class I pathway is produced during the pioneer round of mRNA translation. Proc. Natl. Acad. Sci. USA 2011, 108, 11572–11577. [Google Scholar] [CrossRef] [Green Version]
- Granados, D.P.; Yahyaoui, W.; Laumont, C.M.; Daouda, T.; Muratore-Schroeder, T.L.; Côté, C.; Laverdure, J.-P.; Lemieux, S.; Thibault, P.; Perreault, C. MHC I–associated peptides preferentially derive from transcripts bearing miRNA response elements. Blood J. Am. Soc. Hematol. 2012, 119, e181–e191. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wei, J.; Yewdell, J.W. Flu DRiPs in MHC Class I Immunosurveillance. Virol. Sin. 2019, 34, 162–167. [Google Scholar] [CrossRef] [Green Version]
- Korber, B. Computational analysis of HIV molecular sequences. HIV Signat. Seq. Var. Anal. 2000, 55, 55–72. [Google Scholar]
- Zuker, M.; Stiegler, P. Optimal computer folding of large RNA sequences using thermodynamics and auxiliary information. Nucleic Acids Res. 1981, 9, 133–148. [Google Scholar] [CrossRef] [PubMed]
- Ding, Y.; Chan, C.Y.; Lawrence, C.E. RNA secondary structure prediction by centroids in a Boltzmann weighted ensemble. Rna 2005, 11, 1157–1166. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bernhart, S.H.; Hofacker, I.L.; Will, S.; Gruber, A.R.; Stadler, P.F. RNAalifold: Improved consensus structure prediction for RNA alignments. BMC Bioinf. 2008, 9, 474. [Google Scholar] [CrossRef] [Green Version]
- Sharma, Y.; Miladi, M.; Dukare, S.; Boulay, K.; Caudron-Herger, M.; Groß, M.; Backofen, R.; Diederichs, S. A pan-cancer analysis of synonymous mutations. Nat. Commun. 2019, 10, 1–14. [Google Scholar]
- Sabarinathan, R.; Tafer, H.; Seemann, S.E.; Hofacker, I.L.; Stadler, P.F.; Gorodkin, J. RNA snp: Efficient detection of local RNA secondary structure changes induced by SNP s. Hum. Mutat. 2013, 34, 546–556. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, Y.; Wang, X. miRDB: An online database for prediction of functional microRNA targets. Nucleic Acids Res. 2020, 48, D127–D131. [Google Scholar] [CrossRef] [Green Version]
- Muus, C.; Luecken, M.D.; Eraslan, G.; Waghray, A.; Heimberg, G.; Sikkema, L.; Kobayashi, Y.; Vaishnav, E.D.; Subramanian, A.; Smilie, C. Integrated analyses of single-cell atlases reveal age, gender, and smoking status associations with cell type-specific expression of mediators of SARS-CoV-2 viral entry and highlights inflammatory programs in putative target cells. bioRxiv 2020. [Google Scholar] [CrossRef] [Green Version]
- Puelles, V.G.; Lütgehetmann, M.; Lindenmeyer, M.T.; Sperhake, J.P.; Wong, M.N.; Allweiss, L.; Chilla, S.; Heinemann, A.; Wanner, N.; Liu, S.; et al. Multiorgan and Renal Tropism of SARS-CoV-2. N. Engl. J. Med. 2020. [Google Scholar] [CrossRef] [PubMed]
- Ludwig, N.; Leidinger, P.; Becker, K.; Backes, C.; Fehlmann, T.; Pallasch, C.; Rheinheimer, S.; Meder, B.; Stähler, C.; Meese, E. Distribution of miRNA expression across human tissues. Nucleic Acids Res. 2016, 44, 3865–3877. [Google Scholar] [CrossRef] [PubMed]
- Palmieri, V.; Backes, C.; Ludwig, N.; Fehlmann, T.; Kern, F.; Meese, E.; Keller, A. IMOTA: An interactive multi-omics tissue atlas for the analysis of human miRNA–target interactions. Nucleic Acids Res. 2018, 46, D770–D775. [Google Scholar] [CrossRef] [Green Version]
- Santos, A.; Tsafou, K.; Stolte, C.; Pletscher-Frankild, S.; O’Donoghue, S.I.; Jensen, L.J. Comprehensive comparison of large-scale tissue expression datasets. PeerJ 2015, 3, e1054. [Google Scholar] [CrossRef] [PubMed]
- Mann, M.; Wright, P.R.; Backofen, R. IntaRNA 2.0: Enhanced and customizable prediction of RNA–RNA interactions. Nucleic Acids Res. 2017, 45, W435–W439. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene | Mutation | Amino Acid Change |
|---|---|---|
| 5′ UTR | C to U—nt241 | - |
| Nsp1 | C to U—nt313 | No (L16) |
| Nsp2 | C to U—nt1059 | T85I |
| G to A—nt1397 | V198I | |
| Deletion 1606–1609 | D268 deletion | |
| Nsp3 | C to U—nt3037 | No (F106) |
| Nsp4 | C to U—nt8782 | No (S76) |
| C to U—9802 | No (A416) | |
| G to U—9803 | No (L417) | |
| Nsp6 | G to U—nt11083 | L37F |
| Nsp12 | C to U—nt14408 | P232L |
| C to U—nt14805 | No (Y455) | |
| Nsp13 | U to C—nt17247 | No (R337) |
| S | A to G—nt23403 | D614G |
| C to U—nt24034 | No (N824) | |
| ORF3a | G to U—nt25563 | Q57H |
| G to U—nt26144 | G251V | |
| ORF8 | C to U—nt27964 | S24L |
| U to C- nt28144 | L84S | |
| N | C to U—nt28311 | P13L |
| U to C—nt28688 | No (L139) | |
| GGG to AAC—nt28881-28884 | R203K and G204R | |
| 3′ UTR | G to U—nt29742 | - |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hosseini Rad SM, A.; McLellan, A.D. Implications of SARS-CoV-2 Mutations for Genomic RNA Structure and Host microRNA Targeting. Int. J. Mol. Sci. 2020, 21, 4807. https://doi.org/10.3390/ijms21134807
Hosseini Rad SM A, McLellan AD. Implications of SARS-CoV-2 Mutations for Genomic RNA Structure and Host microRNA Targeting. International Journal of Molecular Sciences. 2020; 21(13):4807. https://doi.org/10.3390/ijms21134807
Chicago/Turabian StyleHosseini Rad SM, Ali, and Alexander D. McLellan. 2020. "Implications of SARS-CoV-2 Mutations for Genomic RNA Structure and Host microRNA Targeting" International Journal of Molecular Sciences 21, no. 13: 4807. https://doi.org/10.3390/ijms21134807
APA StyleHosseini Rad SM, A., & McLellan, A. D. (2020). Implications of SARS-CoV-2 Mutations for Genomic RNA Structure and Host microRNA Targeting. International Journal of Molecular Sciences, 21(13), 4807. https://doi.org/10.3390/ijms21134807