Large-Scale Production of Human iPSC-Derived Macrophages for Drug Screening

 , , , ,
, , , , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
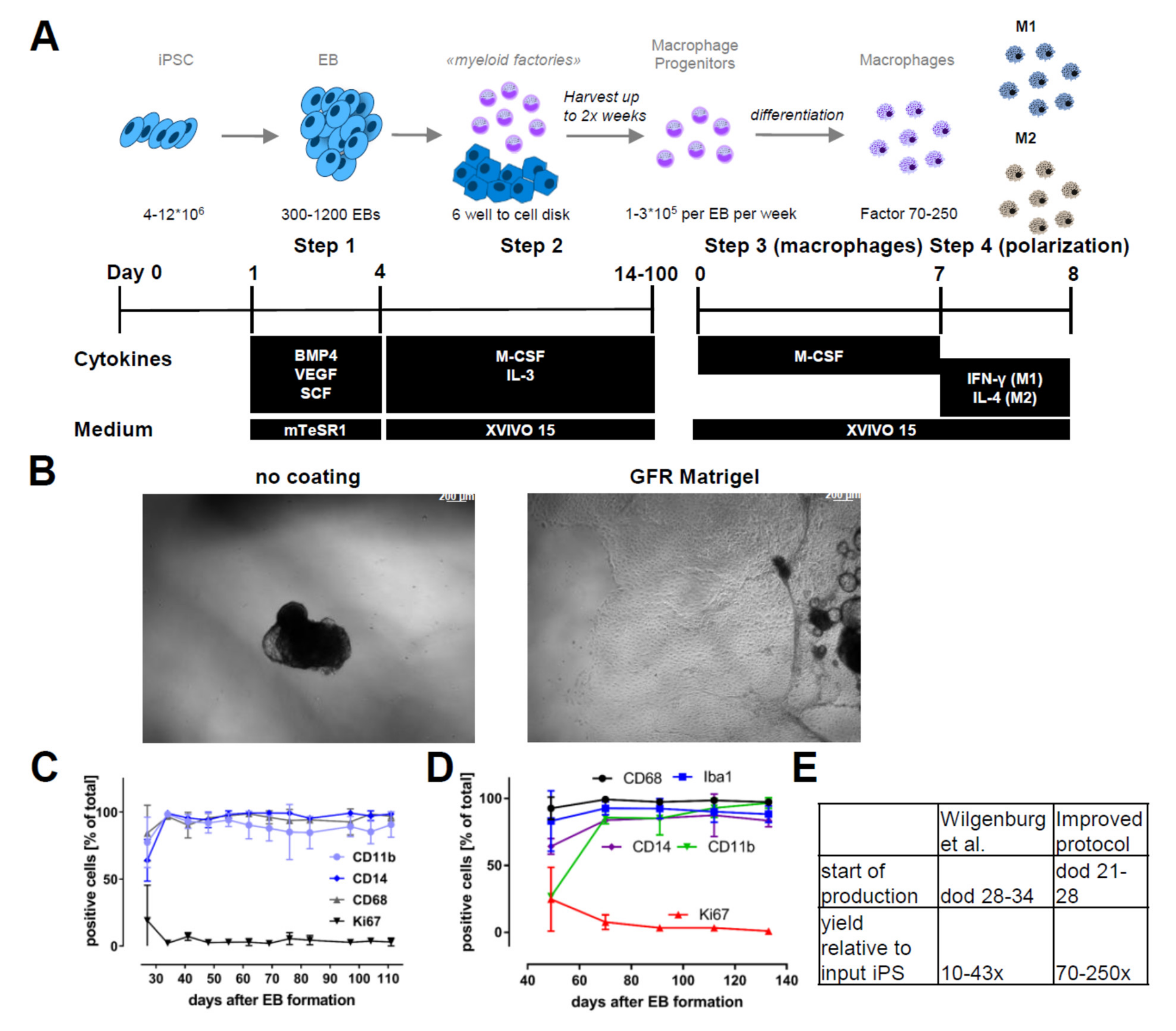
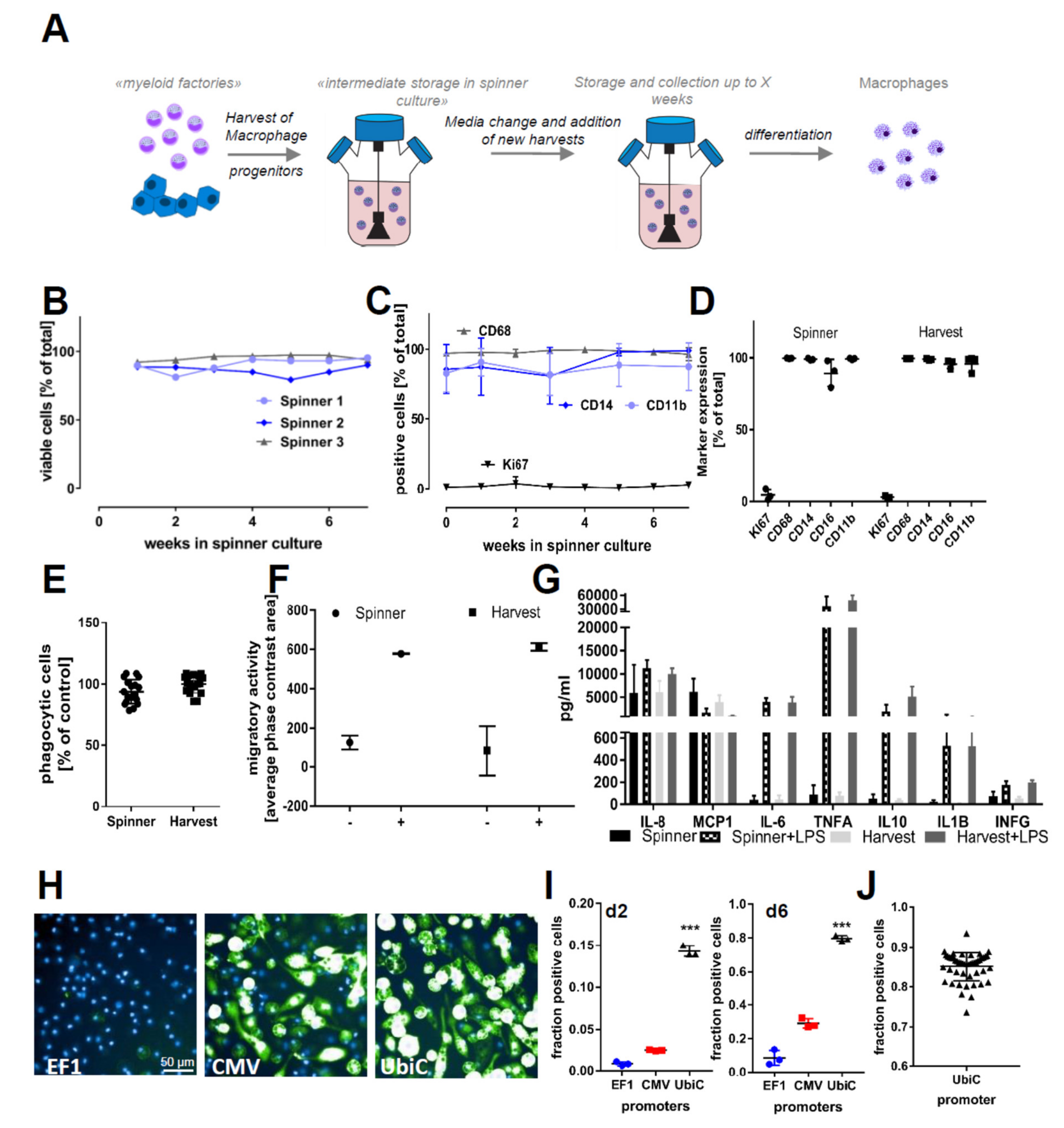
2.1. Differentiation and Storage of Macrophage Progenitors from iPSCs
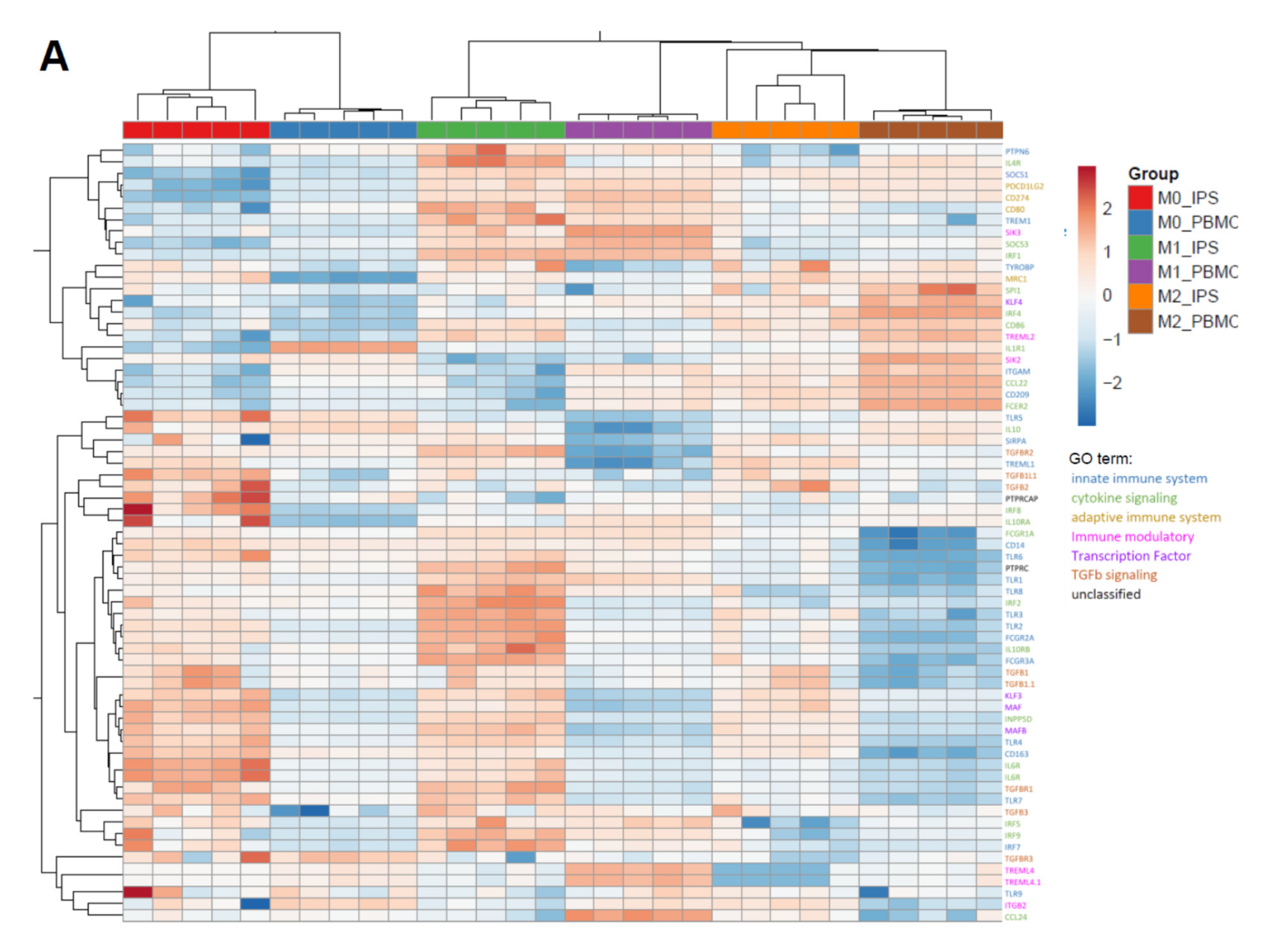
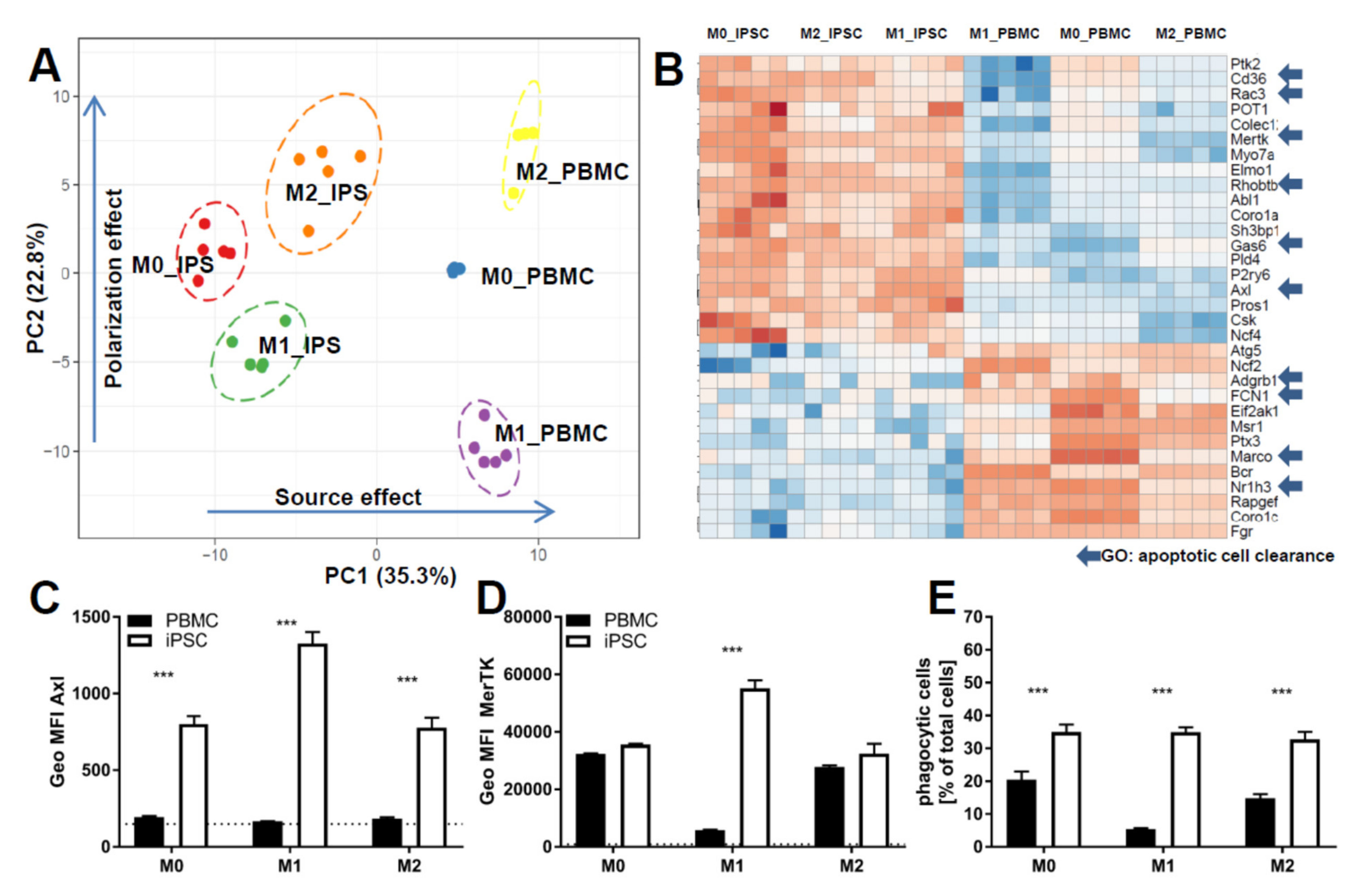
2.2. iPSC-Derived Macrophages Polarize Comparably to Primary Macrophages
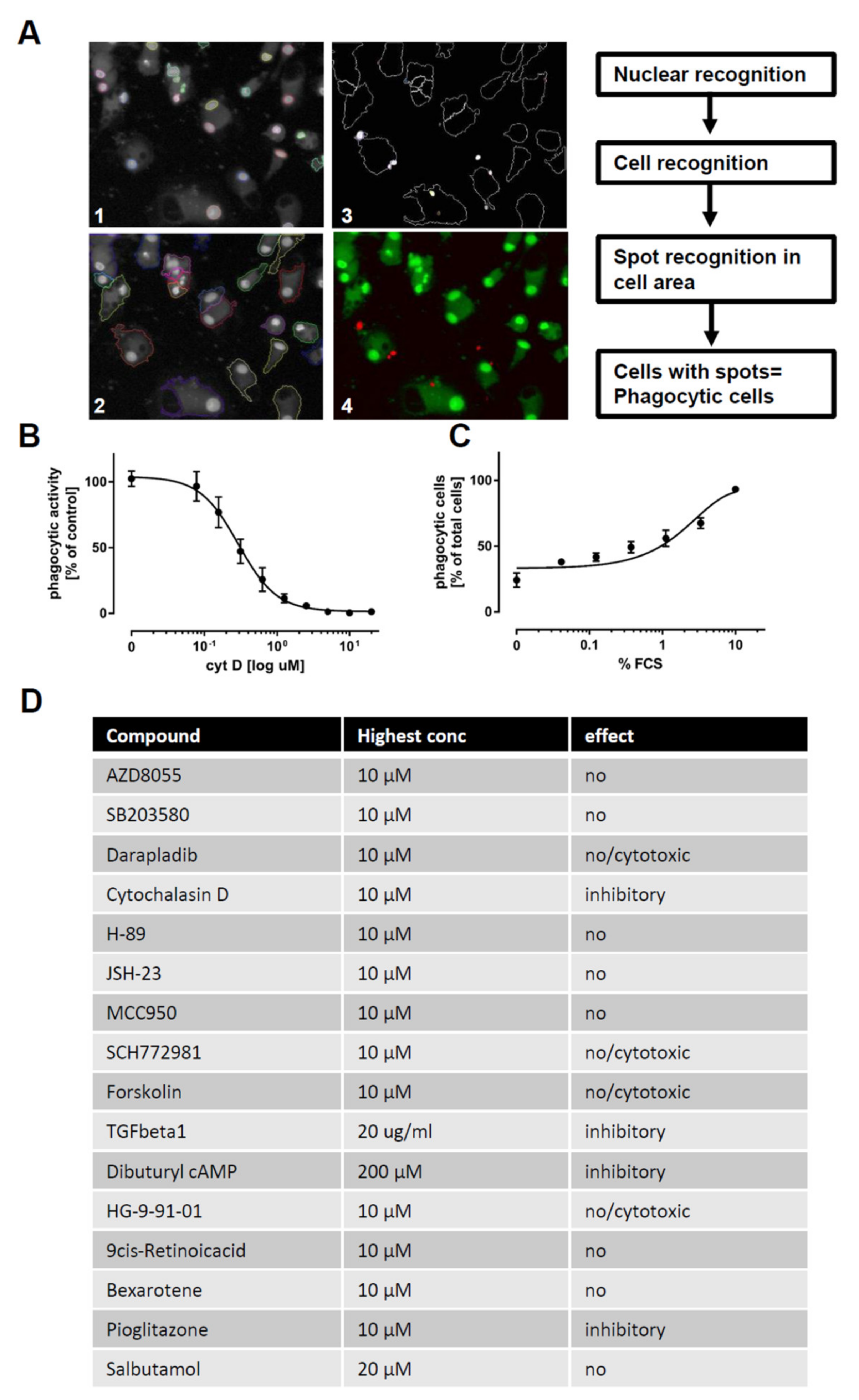
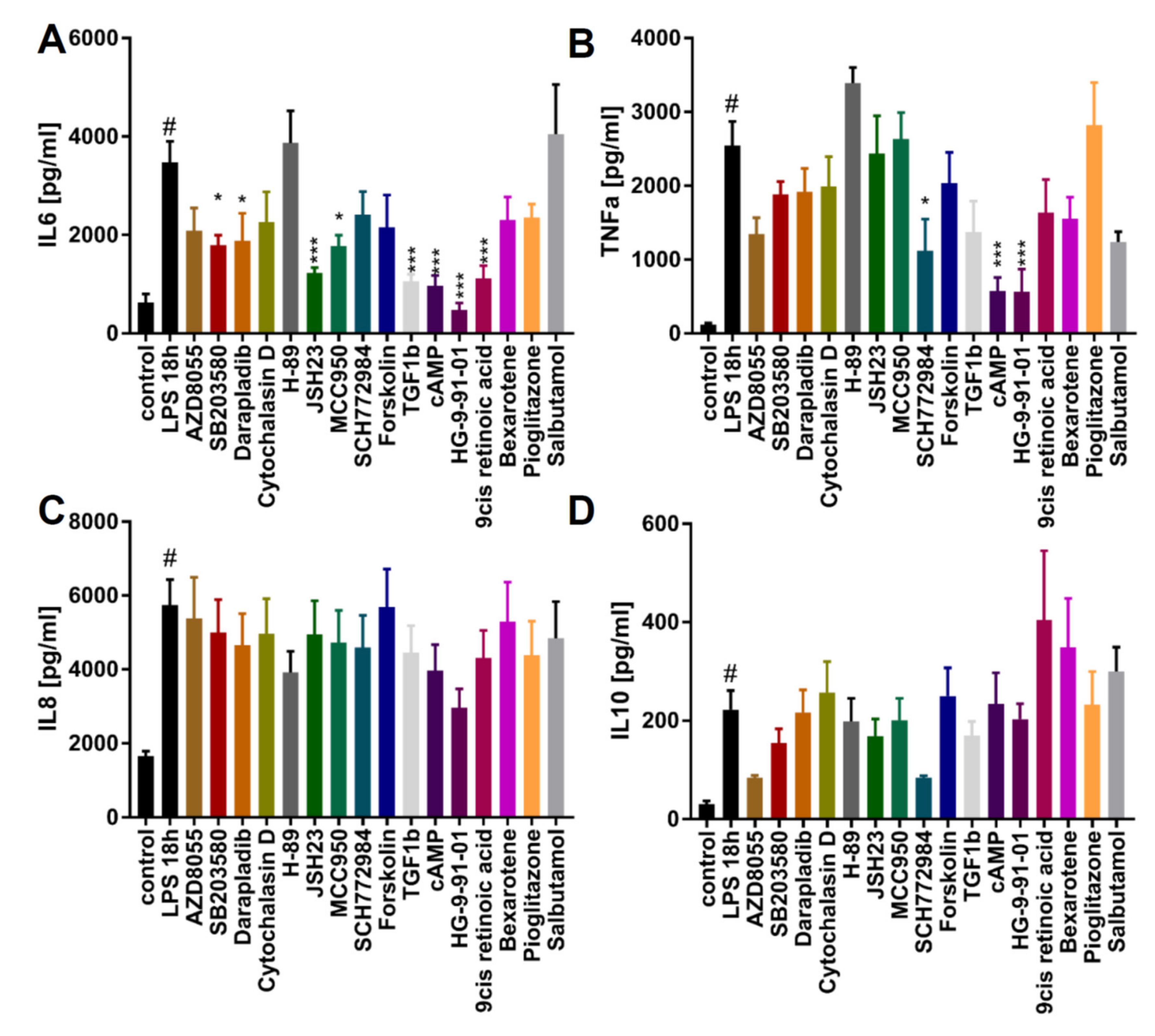
2.3. The Use of iPSC-Derived Macrophages to Identify Modulators of Phagocytosis and Cytokine Release
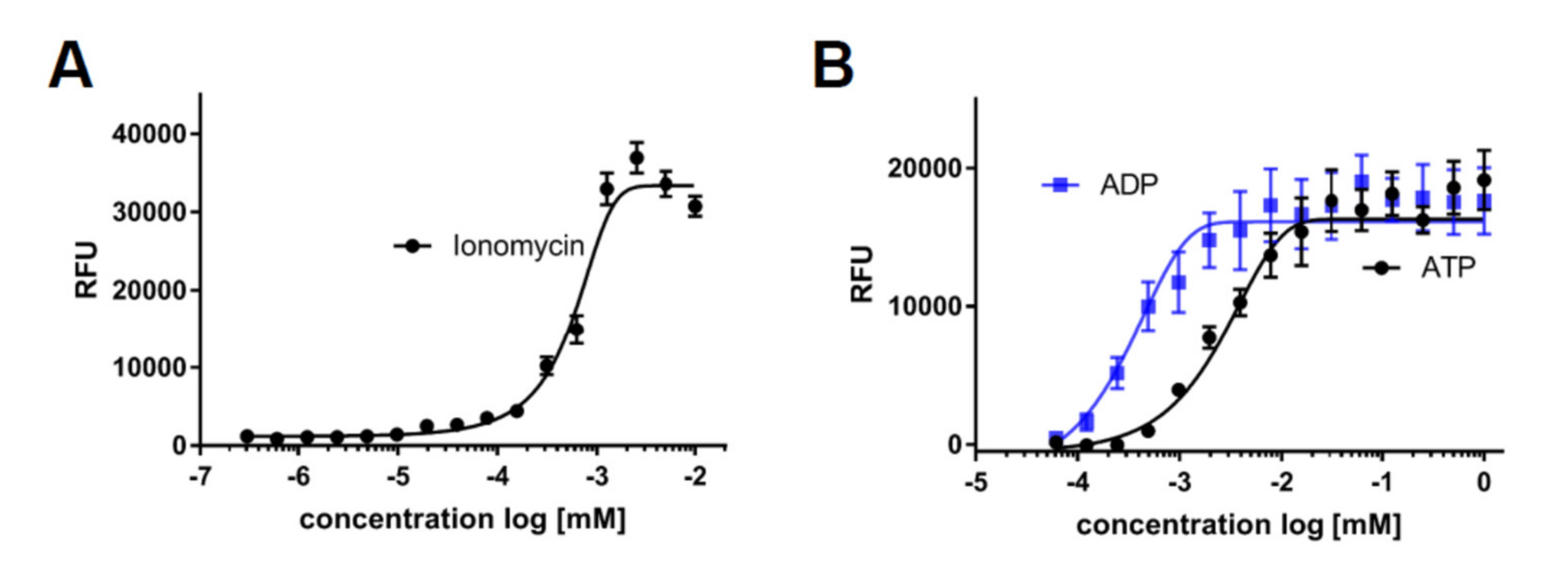
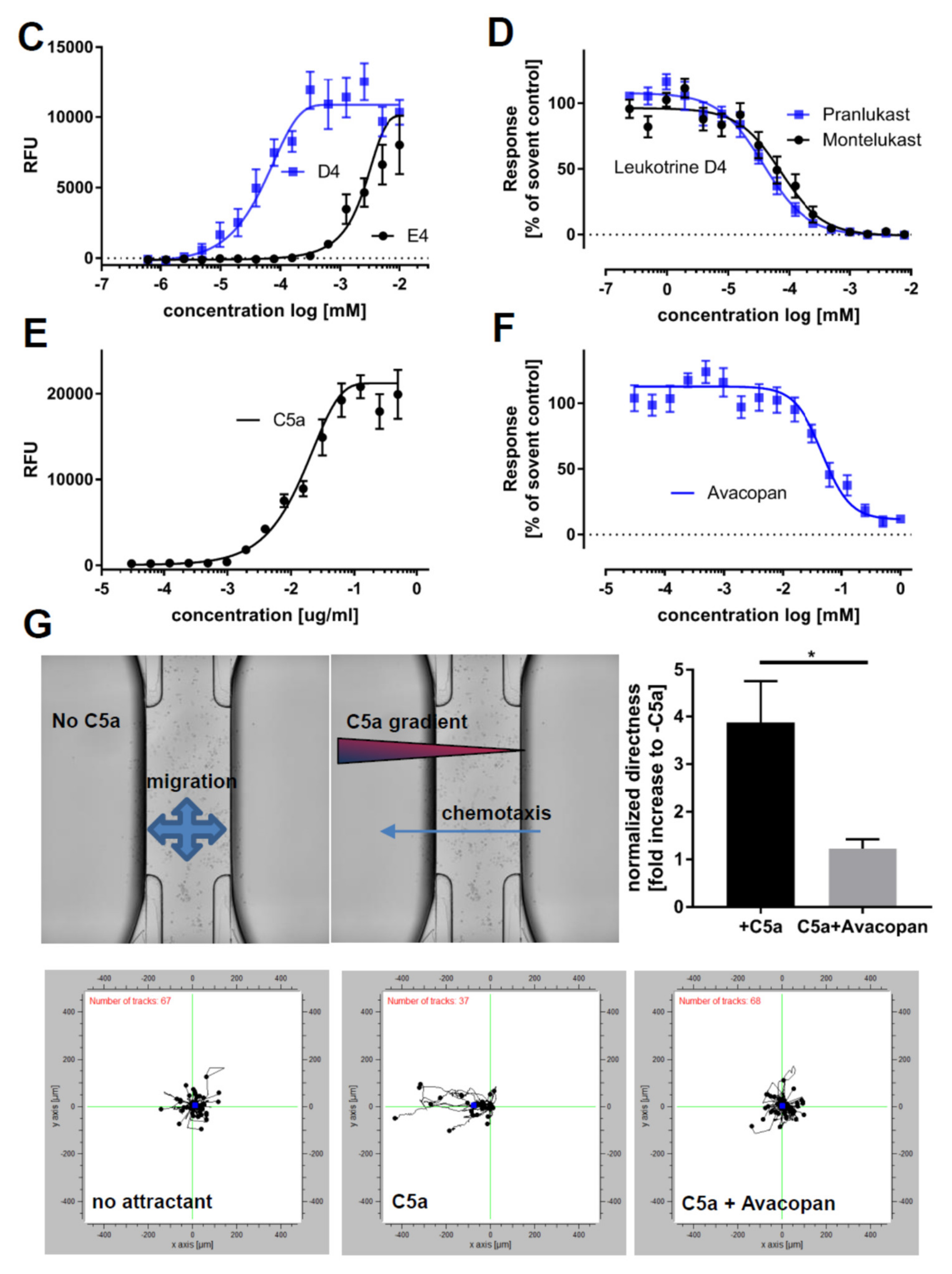
2.4. The Use of iPSC-Derived Macrophages to Screen for Modulators of Calcium Release and Chemotaxis
3. Discussion
4. Materials and Methods
4.1. iPSC Maintenance
4.2. Embryoid Body Generation
4.3. Plating of EBs and Continued Maturation along the Myeloid Lineage
4.4. Macrophage Progenitor Harvesting and Macrophage Differentiation
4.5. Polarization of Macrophages
4.6. Prolonged Cultivation in Suspension Cultures
4.7. Isolation of Monocytes from PBMC-and Monocyte-Derived Macrophage Differentiation
4.8. Culture and Polarization of CD14-Positive Cells
4.9. Quality Control and Marker Expression by Flow Cytometry
4.10. Comparative Marker Expression by Flow Cytometry
4.11. Characterization by RNAseq
4.12. RNAseq Analysis
4.13. Phagocytosis Assay
4.14. Efferocytosis Assay
4.15. Cytokine Release
4.16. Transwell Migration
4.17. Chemotaxis
4.18. Calcium Assay
4.19. Adenovirus Infection
4.20. Data Handling and Statistics
5. Patents
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| BMP4 | Bone morphogenetic protein 4 |
| cAMP | Cyclic adenosine monophosphate |
| EB | Embryoid body |
| FCS | Fetal calf serum |
| GFP | Green fluorescent protein |
| GFR | Growth factor reduced |
| GM-CSF | Granulocyte-macrophage colony-stimulating factor |
| GO | Gene ontology |
| IL-3 | Interleukin 3 |
| IFNγ | Interferon gamma |
| iPSC | Induced pluripotent stem cells |
| LPS | Lipopolysaccharide |
| M-CSF | Macrophage colony-stimulating factor |
| PBMC | Peripheral blood mononuclear cell |
| SCF | Stem cell factor |
| VEGF | Vascular endothelial growth factor |
References
- Wynn, T.A.; Chawla, A.; Pollard, J.W. Macrophage biology in development, homeostasis and disease. Nature 2013, 496, 445–455. [Google Scholar] [CrossRef] [PubMed]
- Sica, A.; Larghi, P.; Mancino, A.; Rubino, L.; Porta, C.; Totaro, M.G.; Rimoldi, M.; Biswas, S.K.; Allavena, P.; Mantovani, A. Macrophage polarization in tumour progression. Semin. Cancer Biol. 2008, 18, 349–355. [Google Scholar] [CrossRef] [PubMed]
- Mantovani, A.; Biswas, S.K.; Galdiero, M.R.; Sica, A.; Locati, M. Macrophage plasticity and polarization in tissue repair and remodelling. J. Pathol. 2012, 229, 176–185. [Google Scholar] [CrossRef] [PubMed]
- Biswas, S.K.; Chittezhath, M.; Shalova, I.N.; Lim, J.-Y. Macrophage polarization and plasticity in health and disease. Immunol. Res. 2012, 53, 11–24. [Google Scholar] [CrossRef] [PubMed]
- Shapouri-Moghaddam, A.; Mohammadian, S.; Vazini, H.; Taghadosi, M.; Esmaeili, S.-A.; Mardani, F.; Seifi, B.; Mahommadi, A.; Afshari, J.T.; Sahebkar, A. Macrophage plasticity, polarization, and function in health and disease. J. Cell Physiol. 2018, 233, 6425–6440. [Google Scholar] [CrossRef] [PubMed]
- Tiwari, R.; Singh, V.; Barthwal, M.K. Macrophages: An elusive yet emerging therapeutic target of atherosclerosis. Med. Res. Rev. 2008, 28, 483–544. [Google Scholar] [CrossRef] [PubMed]
- Ginhoux, F.; Jung, S. Monocytes and macrophages: Developmental pathways and tissue homeostasis. Nat. Rev. Immunol. 2014, 14, 392–404. [Google Scholar] [CrossRef]
- Lee, C.Z.W.; Kozaki, T.; Ginhoux, F. Studying tissue macrophages in vitro: Are iPSC-derived cells the answer? Nat. Rev. Immunol. 2018, 18, 716–725. [Google Scholar] [CrossRef]
- Haenseler, W.; Zambon, F.; Lee, H.; Vowles, J.; Rinaldi, F.; Duggal, G.; Houlden, H.; Gwinn, K.; Wray, S.; Luk, K.C.; et al. Excess alpha-synuclein compromises phagocytosis in iPSC-derived macrophages. Sci. Rep. 2017, 7, 9003. [Google Scholar] [CrossRef]
- Douvaras, P.; Sun, B.; Wang, M.; Kruglikov, I.; Lallos, G.; Zimmer, M.; Terrenoire, C.; Zhang, B.; Gandy, S.; Schadt, E.; et al. Directed Differentiation of Human Pluripotent Stem Cells to Microglia. Stem Cell Rep. 2017, 8, 1516–1524. [Google Scholar] [CrossRef] [Green Version]
- Park, E.K.; Jung, H.S.; Yang, H.I.; Yoo, M.C.; Kim, C.; Kim, K.S. Optimized THP-1 differentiation is required for the detection of responses to weak stimuli. Inflamm. Res. 2007, 56, 45–50. [Google Scholar] [CrossRef] [PubMed]
- Schildberger, A.; Rossmanith, E.; Eichhorn, T.; Strassl, K.; Weber, V. Monocytes, Peripheral Blood Mononuclear Cells, and THP-1 Cells Exhibit Different Cytokine Expression Patterns following Stimulation with Lipopolysaccharide. Mediat. Inflamm. 2013, 2013, 1–10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tsuchiya, S.; Yamabe, M.; Yamaguchi, Y.; Kobayashi, Y.; Konno, T.; Tada, K. Establishment and characterization of a human acute monocytic leukemia cell line (THP-1). Int. J. Cancer 1980, 26, 171–176. [Google Scholar] [CrossRef] [PubMed]
- Takamatsu, K.; Ikeda, T.; Haruta, M.; Matsumura, K.; Ogi, Y.; Nakagata, N.; Uchino, M.; Ando, Y.; Nishimura, Y.; Senju, S. Degradation of amyloid beta by human induced pluripotent stem cell-derived macrophages expressing Neprilysin-2. Stem Cell Res. 2014, 13, 442–453. [Google Scholar] [CrossRef] [Green Version]
- Senju, S.; Haruta, M.; Matsumura, K.; Matsunaga, Y.; Fukushima, S.; Ikeda, T.; Takamatsu, K.; Irie, A.; Nishimura, Y. Generation of dendritic cells and macrophages from human induced pluripotent stem cells aiming at cell therapy. Gene Ther. 2011, 18, 874–883. [Google Scholar] [CrossRef] [Green Version]
- Karlsson, K.R.; Cowley, S.; Martinez, F.O.; Shaw, M.; Minger, S.L.; James, W.S. Homogeneous monocytes and macrophages from human embryonic stem cells following coculture-free differentiation in M-CSF and IL-3. Exp. Hematol. 2008, 36, 1167–1175. [Google Scholar] [CrossRef] [Green Version]
- Hong, D.; Ding, J.; Li, O.; He, Q.; Ke, M.; Zhu, M.; Liu, L.; Ou, W.-B.; He, Y.; Wu, Y. Human-induced pluripotent stem cell-derived macrophages and their immunological function in response to tuberculosis infection. Stem Cell Res. Ther. 2018, 9, 49. [Google Scholar] [CrossRef] [Green Version]
- Haenseler, W.; Sansom, S.N.; Buchrieser, J.; Newey, S.E.; Moore, C.S.; Nicholls, F.J.; Chintawar, S.; Schnell, C.; Antel, J.P.; Allen, N.D.; et al. A Highly Efficient Human Pluripotent Stem Cell Microglia Model.Model Displays a Neuronal-Co-culture-Specific Expression Profile and Inflammatory Response. Stem Cell Rep. 2017, 8, 1727–1742. [Google Scholar] [CrossRef] [Green Version]
- Ackermann, M.; Kempf, H.; Hetzel, M.; Hesse, C.; Hashtchin, A.R.; Brinkert, K.; Schott, J.W.; Haake, K.; Kühnel, M.P.; Glage, S.; et al. Bioreactor-based mass production of human iPSC-derived macrophages enables immunotherapies against bacterial airway infections. Nat. Commun. 2018, 9, 5088. [Google Scholar] [CrossRef]
- Ginhoux, F.; Greter, M.; Leboeuf, M.; Nandi, S.; See, P.; Gokhan, S.; Mehler, M.F.; Conway, S.J.; Gascoigne, N.R.J.; Stanley, E.R.; et al. Fate Mapping Analysis Reveals That Adult Microglia Derive from Primitive Macrophages. Science 2010, 330, 841–845. [Google Scholar] [CrossRef] [Green Version]
- Gomez-Perdiguero, E.; Klapproth, K.; Schulz, C.; Busch, K.; Azzoni, E.; Crozet, L.; Garner, H.; Trouillet, C.; De Bruijn, M.; Geissmann, F.; et al. Tissue-resident macrophages originate from yolk sac-derived erythro-myeloid progenitors. Exp. Hematol. 2015, 43, S64. [Google Scholar] [CrossRef]
- Hoeffel, G.; Ginhoux, F. Ontogeny of Tissue-Resident Macrophages. Front. Immunol. 2015, 6, 413. [Google Scholar] [CrossRef] [Green Version]
- Kierdorf, K.; Erny, D.; Goldmann, T.; Sander, V.; Schulz, C.; Gomez-Perdiguero, E.; Wieghofer, P.; Heinrich, A.; Riemke, P.; Hölscher, C.; et al. Microglia emerge from erythromyeloid precursors via Pu.1- and Irf8-dependent pathways. Nat. Neurosci. 2013, 16, 273–280. [Google Scholar] [CrossRef] [PubMed]
- Schulz, C.; Gomez-Perdiguero, E.; Chorro, L.; Szabo-Rogers, H.; Cagnard, N.; Kierdorf, K.; Prinz, M.; Wu, B.; Jacobsen, S.E.W.; Pollard, J.W.; et al. A Lineage of Myeloid Cells Independent of Myb and Hematopoietic Stem Cells. Science 2012, 336, 86–90. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sheng, J.; Ruedl, C.; Karjalainen, K. Most Tissue-Resident Macrophages Except Microglia Are Derived from Fetal Hematopoietic Stem Cells. Immunity 2015, 43, 382–393. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Epelman, S.; LaVine, K.J.; Beaudin, A.E.; Sojka, R.K.; Carrero, J.A.; Calderon, B.; Brija, T.; Gautier, E.L.; Ivanov, S.; Satpathy, A.T.; et al. Embryonic and adult-derived resident cardiac macrophages are maintained through distinct mechanisms at steady state and during inflammation. Immunity 2014, 40, 91–104. [Google Scholar] [CrossRef] [Green Version]
- Hashimoto, D.; Chow, A.; Noizat, C.; Teo, P.; Beasley, M.B.; Leboeuf, M.; Becker, C.D.; See, P.; Price, J.; Lucas, D.; et al. Tissue-resident macrophages self-maintain locally throughout adult life with minimal contribution from circulating monocytes. Immunity 2013, 38, 792–804. [Google Scholar] [CrossRef] [Green Version]
- Buchrieser, J.; James, W.; Moore, M.D. Human Induced Pluripotent Stem Cell-Derived Macrophages Share Ontogeny with MYB-Independent Tissue-Resident Macrophages. Stem Cell Rep. 2017, 8, 334–345. [Google Scholar] [CrossRef] [Green Version]
- Ginhoux, F.; Guilliams, M. Tissue-Resident Macrophage Ontogeny and Homeostasis. Immunity 2016, 44, 439–449. [Google Scholar] [CrossRef]
- Takata, K.; Kozaki, T.; Lee, C.Z.W.; Thion, M.S.; Otsuka, M.; Lim, S.; Utami, K.H.; Fidan, K.; Park, D.S.; Malleret, B.; et al. Induced-Pluripotent-Stem-Cell-Derived Primitive Macrophages Provide a Platform for Modeling Tissue-Resident Macrophage Differentiation and Function. Immunity 2017, 47, 183–198.e6. [Google Scholar] [CrossRef]
- Van Wilgenburg, B.; Browne, C.; Vowles, J.; Cowley, S.A. Efficient, Long Term Production of Monocyte-Derived Macrophages from Human Pluripotent Stem Cells under Partly-Defined and Fully-Defined Conditions. PLoS ONE 2013, 8, e71098. [Google Scholar] [CrossRef] [PubMed]
- Metsalu, T.; Vilo, J. ClustVis: A web tool for visualizing clustering of multivariate data using Principal Component Analysis and heatmap. Nucleic Acids Res. 2015, 43, W566–W570. [Google Scholar] [CrossRef] [PubMed]
- Ernst, O.; Glucksam-Galnoy, Y.; Athamna, M.; Ben-Dror, I.; Ben-Arosh, H.; Levy-Rimler, G.; Fraser, I.D.C.; Zor, T. The cAMP Pathway Amplifies Early MyD88-Dependent and Type I Interferon-Independent LPS-Induced Interleukin-10 Expression in Mouse Macrophages. Mediat. Inflamm. 2019, 2019, 3451461. [Google Scholar] [CrossRef] [PubMed]
- Gobejishvili, L.; Ghare, S.; Khan, R.; Cambon, A.; Barker, D.F.; Barve, S.; McClain, C.; Hill, D. Misoprostol modulates cytokine expression through a cAMP pathway: Potential therapeutic implication for liver disease. Clin. Immunol. 2015, 161, 291–299. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jin, Y.; Liu, Y.; Nelin, L.D. Extracellular Signal-regulated Kinase Mediates Expression of Arginase II but Not Inducible Nitric-oxide Synthase in Lipopolysaccharide-stimulated Macrophages. J. Biol. Chem. 2014, 290, 2099–2111. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leslie, C.C. Cytosolic phospholipase A2: Physiological function and role in disease. J. Lipid Res. 2015, 56, 1386–1402. [Google Scholar] [CrossRef] [Green Version]
- Negreiros-Lima, G.L.; Lima, K.M.; Moreira, I.Z.; Jardim, B.L.O.; Vago, J.; Galvão, I.; Teixeira, L.C.; Pinho, V.; Teixeira, M.M.; Sugimoto, M.A.; et al. Cyclic AMP Regulates Key Features of Macrophages via PKA: Recruitment, Reprogramming and Efferocytosis. Cells 2020, 9, 128. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qian, X.; Zhang, J.; Liu, J. Tumor-secreted PGE2Inhibits CCL5 Production in Activated Macrophages through cAMP/PKA Signaling Pathway. J. Biol. Chem. 2010, 286, 2111–2120. [Google Scholar] [CrossRef] [Green Version]
- Veremeyko, T.; Yung, A.W.Y.; Dukhinova, M.; Kuznetsova, I.S.; Pomytkin, I.; Lyundup, A.; Strekalova, T.; Barteneva, N.S.; Ponomarev, E.D. Cyclic AMP Pathway Suppress Autoimmune Neuroinflammation by Inhibiting Functions of Encephalitogenic CD4 T Cells and Enhancing M2 Macrophage Polarization at the Site of Inflammation. Front. Immunol. 2018, 9, 50. [Google Scholar] [CrossRef] [Green Version]
- Zhou, M.; Xu, W.; Wang, J.; Yan, J.; Shi, Y.; Zhang, C.; Ge, W.; Wu, J.; Du, P.; Chen, Y. Boosting mTOR-dependent autophagy via upstream TLR4-MyD88-MAPK signalling and downstream NF-kappaB pathway quenches intestinal inflammation and oxidative stress injury. EBioMedicine 2018, 35, 345–360. [Google Scholar] [CrossRef] [Green Version]
- Chawla, A. Control of macrophage activation and function by PPARs. Circ. Res. 2010, 106, 1559–1569. [Google Scholar] [CrossRef] [PubMed]
- Coll, R.C.; Robertson, A.A.; Chae, J.J.; Higgins, S.C.; Muñoz-Planillo, R.; Inserra, M.C.; Vetter, I.; Dungan, L.S.; Monks, B.G.; Stutz, A.; et al. A small-molecule inhibitor of the NLRP3 inflammasome for the treatment of inflammatory diseases. Nat. Med. 2015, 21, 248–255. [Google Scholar] [CrossRef] [Green Version]
- Darling, N.J.; Toth, R.; Arthur, J.S.C.; Clark, K. Inhibition of SIK2 and SIK3 during differentiation enhances the anti-inflammatory phenotype of macrophages. Biochem. J. 2017, 474, 521–537. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Heming, M.; Gran, S.; Jauch, S.L.; Fischer-Riepe, L.; Russo, A.; Klotz, L.; Hermann, S.; Schäfers, M.; Roth, J.; Barczyk-Kahlert, K. Peroxisome Proliferator-Activated Receptor-gamma Modulates the Response of Macrophages to Lipopolysaccharide and Glucocorticoids. Front. Immunol. 2018, 9, 893. [Google Scholar] [CrossRef] [PubMed]
- Nuñez, V.; Alameda, D.; Rico, D.; Mota-Blanco, R.A.; Gonzalo, P.; Cedenilla, M.; Fischer, T.; Boscá, L.; Glass, C.K.; Arroyo, A.G.; et al. Retinoid X receptor α controls innate inflammatory responses through the up-regulation of chemokine expression. Proc. Natl. Acad. Sci. USA 2010, 107, 10626–10631. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, S.; Liu, F.; Tan, K.S.; Ser, H.; Tan, L.T.; Lee, L.; Tan, W. Effect of (R)-salbutamol on the switch of phenotype and metabolic pattern in LPS-induced macrophage cells. J. Cell. Mol. Med. 2020, 24, 722–736. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brull, M.; Spreng, A.-S.; Gutbier, S.; Loser, D.; Krebs, A.; Reich, M.; Kraushaar, U.; Britschgi, M.; Patsch, C.; Leist, M. Incorporation of stem cell-derived astrocytes into neuronal organoids to allow neuro-glial interactions in toxicological studies. ALTEX 2020. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hasselmann, J.; Blurton-Jones, M. Human iPSC-derived microglia: A growing toolset to study the brain’s innate immune cells. Glia 2020, 68, 721–739. [Google Scholar] [CrossRef]
- Cao, X.; van den Hil, F.E.; Mummery, C.L.; Orlova, V.V. Generation and Functional Characterization of Monocytes and Macrophages Derived from Human Induced Pluripotent Stem Cells. Curr. Protoc. Stem Cell Biol. 2020, 52, e108. [Google Scholar] [CrossRef]
- Zhang, H.; Xue, C.; Shah, R.; Bermingham, K.; Hinkle, C.C.; Li, W.; Rodrigues, A.; Tabita-Martinez, J.; Millar, J.S.; Cuchel, M.; et al. Functional analysis and transcriptomic profiling of iPSC-derived macrophages and their application in modeling Mendelian disease. Circ. Res. 2015, 117, 17–28. [Google Scholar] [CrossRef] [Green Version]
- Andreone, B.J.; Przybyla, L.; Llapashtica, C.; Rana, A.; Davis, S.S.; Van Lengerich, B.; Lin, K.; Shi, J.; Mei, Y.; Astarita, G.; et al. Alzheimer’s-associated PLCγ2 is a signaling node required for both TREM2 function and the inflammatory response in human microglia. Nat. Neurosci. 2020, 1–12. [Google Scholar] [CrossRef]
- McQuade, A.; Coburn, M.A.; Tu, C.H.; Hasselmann, J.; Davtyan, H.; Blurton-Jones, M. Development and validation of a simplified method to generate human microglia from pluripotent stem cells. Mol. Neurodegener. 2018, 13, 67. [Google Scholar] [CrossRef]
- Fernandes, H.J.R.; Hartfield, E.M.; Christian, H.C.; Emmanoulidou, E.; Zheng, Y.; Booth, H.; Bogetofte, H.; Lang, C.; Ryan, B.; Sardi, S.P.; et al. ER Stress and Autophagic Perturbations Lead to Elevated Extracellular α-Synuclein in GBA-N370S Parkinson’s iPSC-Derived Dopamine Neurons. Stem Cell Rep. 2016, 6, 342–356. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Volpato, V.; Smith, J.; Sandor, C.; Ried, J.S.; Baud, A.; Handel, A.; Newey, S.E.; Wessely, F.; Attar, M.; Whiteley, E.; et al. Reproducibility of Molecular Phenotypes after Long-Term Differentiation to Human iPSC-Derived Neurons: A Multi-Site Omics Study. Stem Cell Rep. 2018, 11, 897–911. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dobin, A.; Davis, C.A.; Schlesinger, F.; Drenkow, J.; Zaleski, C.; Jha, S.; Batut, P.; Chaisson, M.; Gingeras, T.R. STAR: Ultrafast universal RNA-seq aligner. Bioinformatics 2012, 29, 15–21. [Google Scholar] [CrossRef]
- Andrews, S. Fastqc: A Quality Control Tool for High Throughput Sequence Data; Babraham Institute: Cambridge, UK, 2010. [Google Scholar]
- Ewels, P.; Magnusson, M.; Lundin, S.; Käller, M. MultiQC: Summarize analysis results for multiple tools and samples in a single report. Bioinformatics 2016, 32, 3047–3048. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, H.; Handsaker, B.; Wysoker, A.; Fennell, T.; Ruan, J.; Homer, N.; Marth, G.; Abecasis, G.R.; Durbin, R.; 1000 Genome Project Data Processing Subgroup. The Sequence Alignment/Map format and SAMtools. Bioinformatics 2009, 25, 2078–2079. [Google Scholar] [CrossRef] [Green Version]
- Mortazavi, A.; A Williams, B.; McCue, K.; Schaeffer, L.; Wold, B. Mapping and quantifying mammalian transcriptomes by RNA-Seq. Nat. Methods 2008, 5, 621–628. [Google Scholar] [CrossRef]








© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gutbier, S.; Wanke, F.; Dahm, N.; Rümmelin, A.; Zimmermann, S.; Christensen, K.; Köchl, F.; Rautanen, A.; Hatje, K.; Geering, B.; et al. Large-Scale Production of Human iPSC-Derived Macrophages for Drug Screening. Int. J. Mol. Sci. 2020, 21, 4808. https://doi.org/10.3390/ijms21134808
Gutbier S, Wanke F, Dahm N, Rümmelin A, Zimmermann S, Christensen K, Köchl F, Rautanen A, Hatje K, Geering B, et al. Large-Scale Production of Human iPSC-Derived Macrophages for Drug Screening. International Journal of Molecular Sciences. 2020; 21(13):4808. https://doi.org/10.3390/ijms21134808
Chicago/Turabian StyleGutbier, Simon, Florian Wanke, Nadine Dahm, Anna Rümmelin, Silke Zimmermann, Klaus Christensen, Fabian Köchl, Anna Rautanen, Klas Hatje, Barbara Geering, and et al. 2020. "Large-Scale Production of Human iPSC-Derived Macrophages for Drug Screening" International Journal of Molecular Sciences 21, no. 13: 4808. https://doi.org/10.3390/ijms21134808
APA StyleGutbier, S., Wanke, F., Dahm, N., Rümmelin, A., Zimmermann, S., Christensen, K., Köchl, F., Rautanen, A., Hatje, K., Geering, B., Zhang, J. D., Britschgi, M., Cowley, S. A., & Patsch, C. (2020). Large-Scale Production of Human iPSC-Derived Macrophages for Drug Screening. International Journal of Molecular Sciences, 21(13), 4808. https://doi.org/10.3390/ijms21134808