Direct On-Chip Differentiation of Intestinal Tubules from Induced Pluripotent Stem Cells


 , ,
, , 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
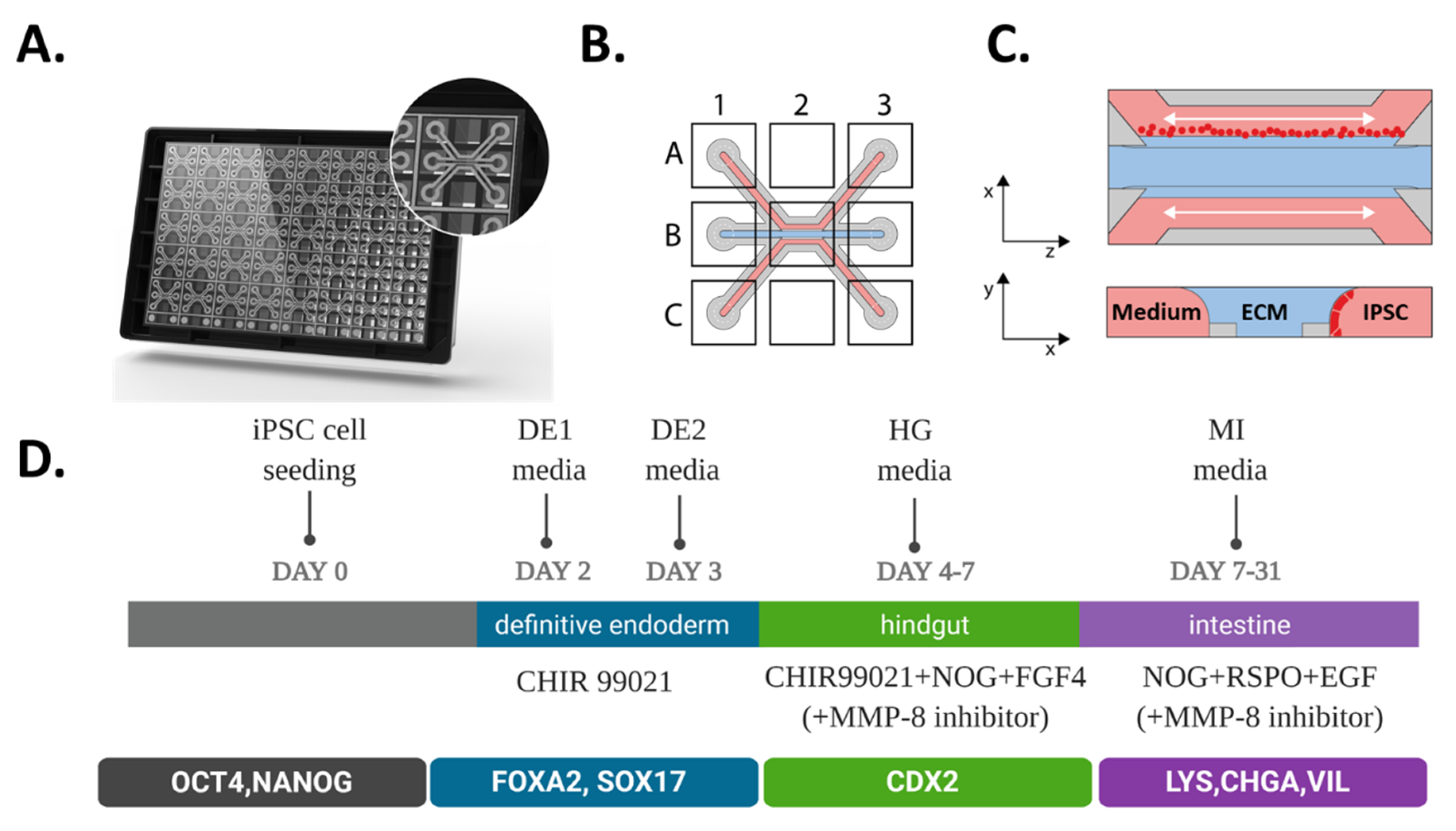
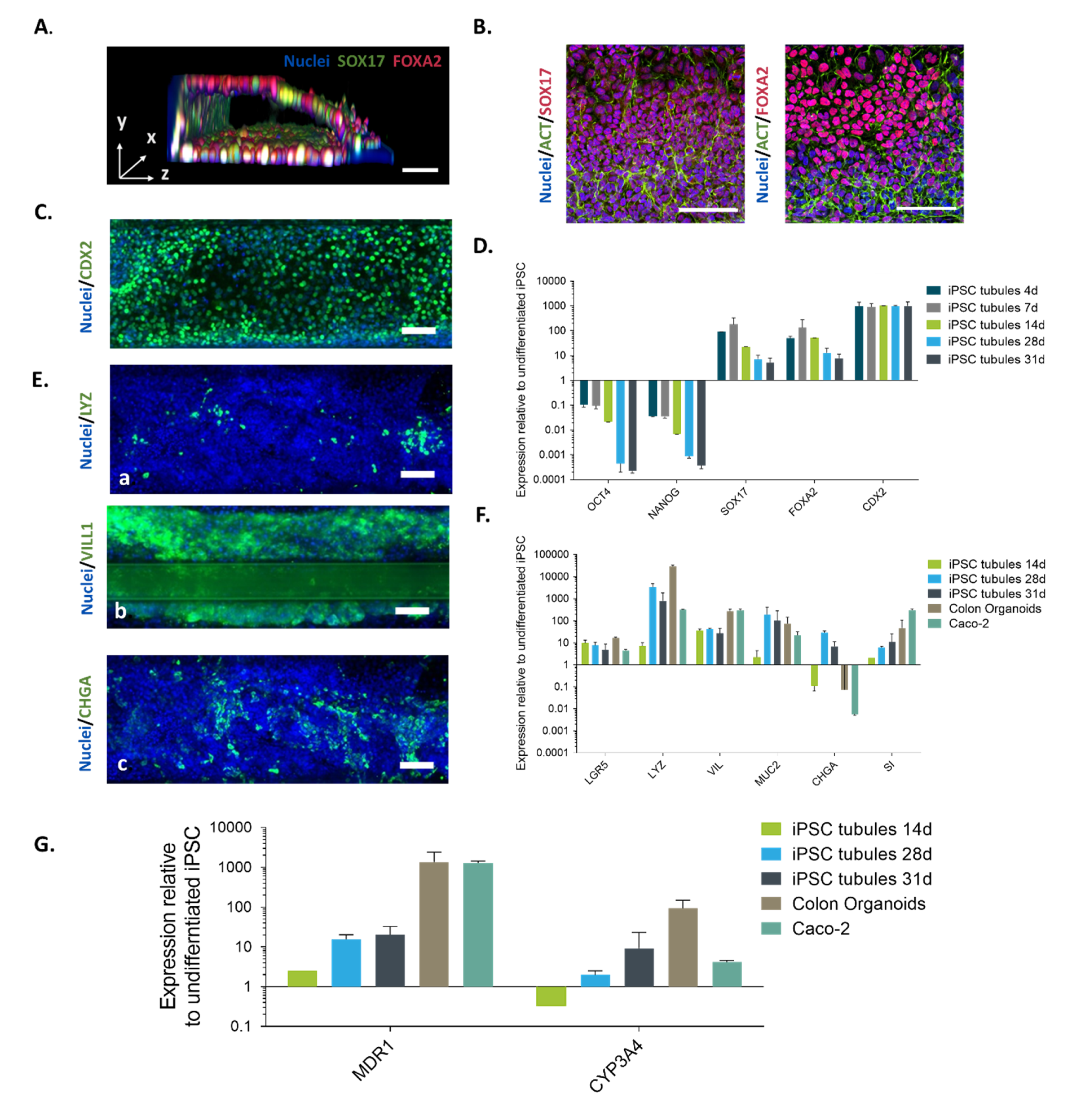
2.1. Directed on Organoplate Differentiation of iPSC Towards Gut-on-a-Chip
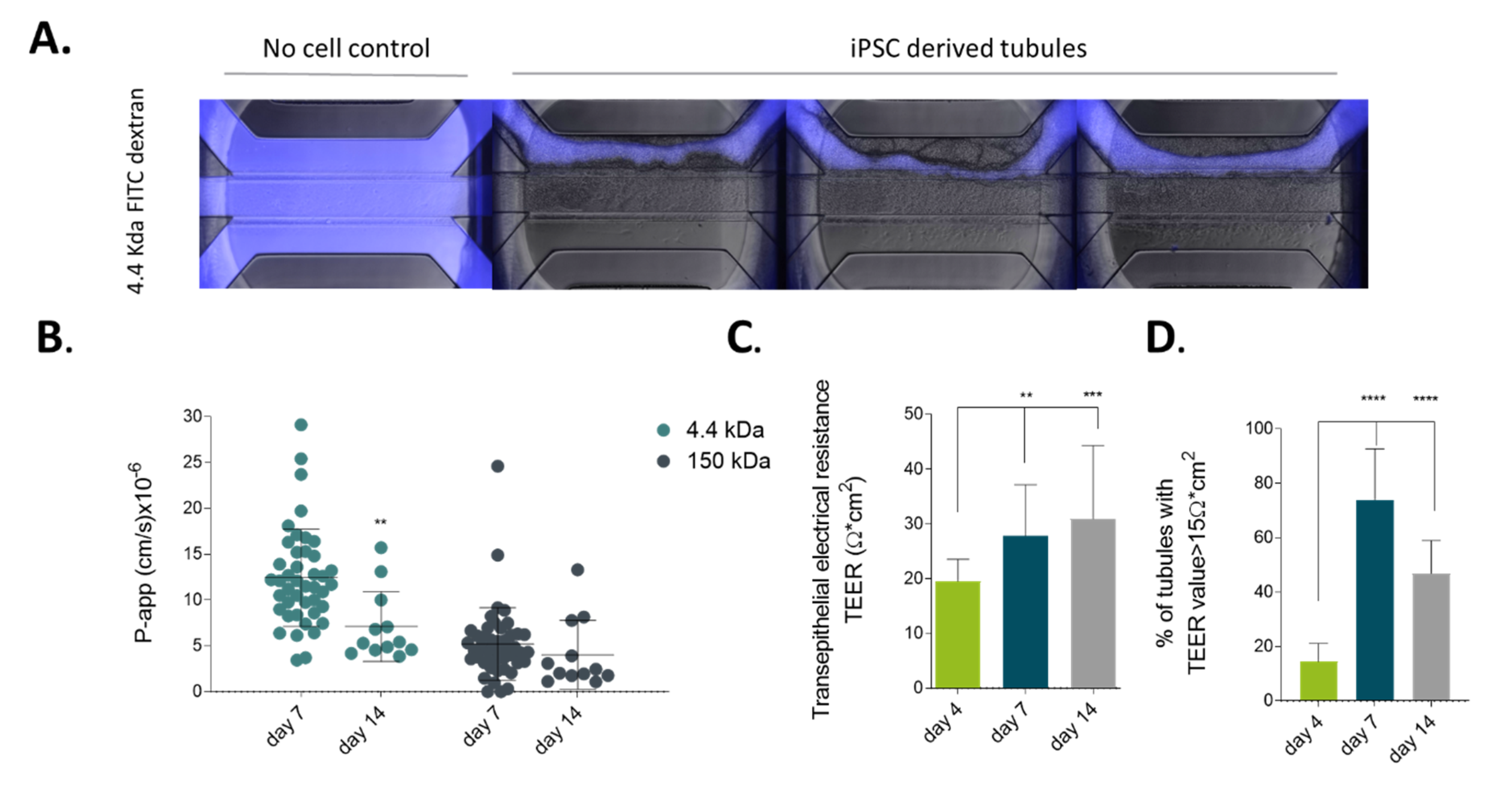
2.2. iPSC-derived Gut-Like Tubules Develop Barrier Properties
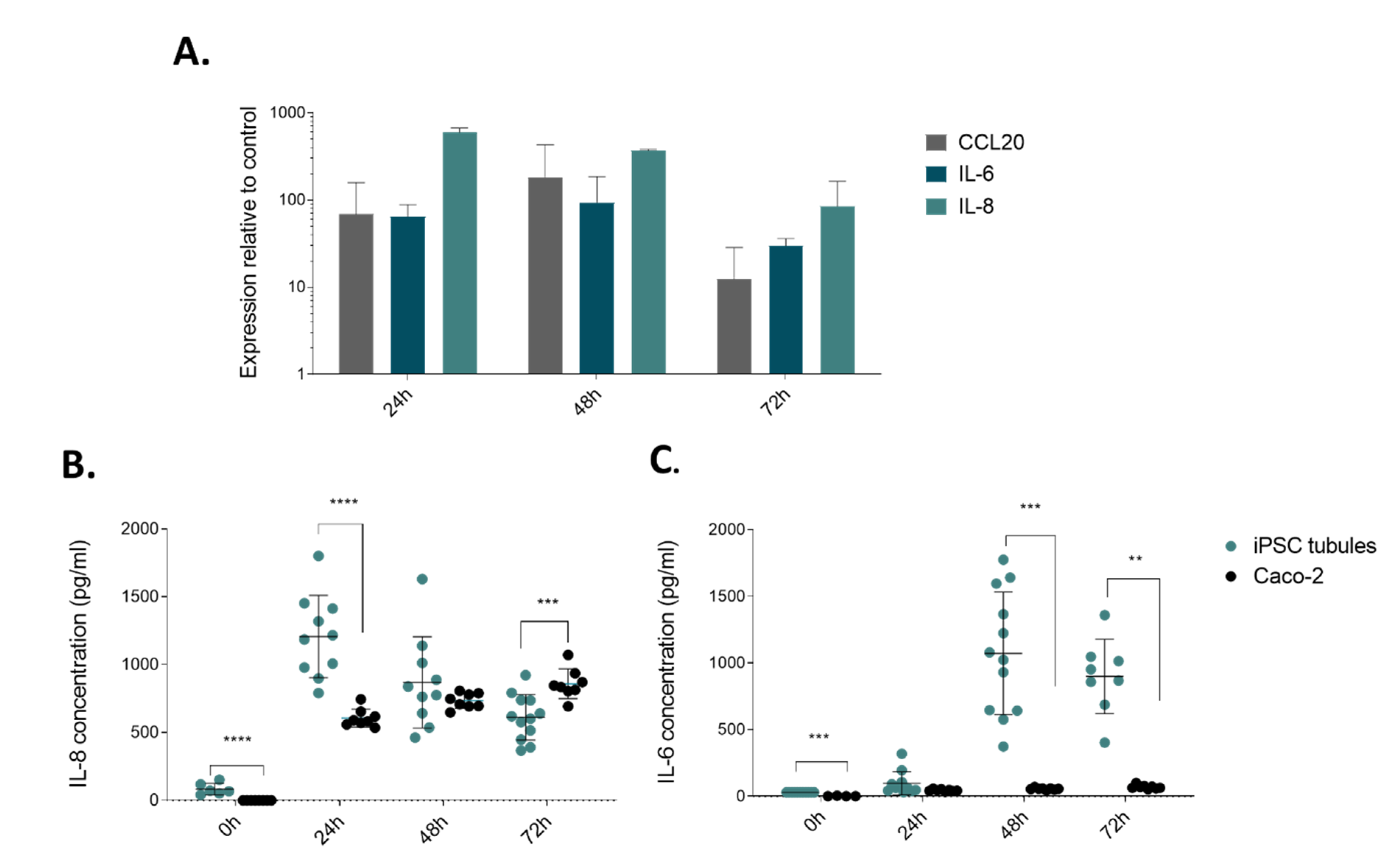
2.3. Modelling Infammation in iPSC-derived Tubules
3. Discussion
4. Materials and Methods
4.1. Ethics Statement
4.2. Cells
4.3. OrganoPlate Seeding and Tubule Formation
4.4. Directed on Plate Differentiation of iPSC
4.5. Barrier Intergrity Assay (BI Assay) and Calculation of Aperent Permeability (P-App)
4.6. TEER Measurements
4.7. Cytokine Trigger and ELISA for Activation Molecules
4.8. Immunocytochemistry (ICC)
4.9. Gene Expression Anaysis
4.10. Statistics and Data Analysis
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| CD | Crohn’s disease |
| CYP | Cytochrome P450 |
| DE | Definitive endoderm |
| EGF | Epidermal growth factor |
| ESCs | Embryonic stem cells |
| HG | Hindgut |
| HIOs | Human Intestinal Organoids |
| FGF4 | Fibroblast growth factor 4 |
| IBD | Inflammatory bowel disease |
| iPSC | induced pluripotent stem cells |
| LGR5 | Leucine-rich-repeat-containing G-protein-coupled receptor 5 |
| MI | Mature intestine |
| MMP | Matrix metalloproteinase |
| P-gp | P-glycoprotein |
| RNase | Ribonuclease |
| ROCK | Rho-associated protein kinase |
| TEER | Trans epithelial electrical resistance |
References
- Astashkina, A.; Mann, B.; Grainger, D.W. A critical evaluation of in vitro cell culture models for high-throughput drug screening and toxicity. Pharmacol. Ther. 2012, 134, 82–106. [Google Scholar] [CrossRef] [PubMed]
- Abraham, V.C.; Towne, D.L.; Waring, J.F.; Warrior, U.; Burns, D.J. Application of a high-content multiparameter cytotoxicity assay to prioritize compounds based on toxicity potential in humans. J. Biomol. Screen. 2008, 13, 527–537. [Google Scholar] [CrossRef] [PubMed]
- Mattei, G.; Giusti, S.; Ahluwalia, A. Design criteria for generating physiologically relevant in vitro models in bioreactors. Processes 2014, 2, 548–569. [Google Scholar] [CrossRef] [Green Version]
- Jain, K.K. Drug delivery systems—An overview. In Methods in Molecular Biology 1–50; Humana Press: Totowa, NJ, USA, 2008. [Google Scholar] [CrossRef]
- Akazawa, T.; Yoshida, S.; Ohnishi, S.; Kanazu, T.; Kawai, M.; Takahashi, K. Application of intestinal epithelial cells differentiated from human induced pluripotent stem cells for studies of prodrug hydrolysis and drug absorption in the small intestine. Drug Metab. Dispos. 2018, 46, 1497–1506. [Google Scholar] [CrossRef] [PubMed]
- Beaurivage, C.; Naumovska, E.; Chang, Y.X.; Elstak, E.D.; Nicolas, A.; Wouters, H.; van Moolenbroek, G.; Lanz, H.L.; Trietsch, S.J.; Joore, J.; et al. Development of a Gut-On-A-Chip model for high throughput disease modeling and drug discovery. Int. J. Mol. Sci. 2019, 20, 5661. [Google Scholar] [CrossRef] [Green Version]
- Moisan, A.; Michielin, F.; Jacob, W.; Kronenberg, S.; Wilson, S.; Avignon, B.; Gerard, R.; Benmansour, F.; McIntyre, C.; Meneses-Lorente, G.; et al. Mechanistic investigations of diarrhea toxicity induced by Anti-HER2/3 combination therapy. Mol. Cancer Ther. 2018, 17, 1464–1474. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peters, M.F.; Choy, A.L.; Pin, C.; Leishman, D.J.; Moisan, A.; Ewart, L.; Guzzie-Peck, P.J.; Sura, R.; Keller, D.A.; Scott, C.W.; et al. Developing: In vitro assays to transform gastrointestinal safety assessment: Potential for microphysiological systems. Lab Chip 2020, 20, 1177–1190. [Google Scholar] [CrossRef] [Green Version]
- Elliott, N.T.; Yuan, F. A review of three-dimensional in vitro tissue models for drug discovery and transport studies. J. Pharm. Sci. 2011, 100, 59–74. [Google Scholar] [CrossRef]
- Kraiczy, J.; Zilbauer, M. Intestinal epithelial organoids as tools to study epigenetics in gut health and disease. Stem Cells Int. 2019, 2019, 7242415. [Google Scholar] [CrossRef]
- Lo, B.; Parham, L. Ethical issues in stem cell research. Endocr. Rev. 2009, 30, 204–213. [Google Scholar] [CrossRef]
- Spence, J.R.; Mayhew, C.N.; Rankin, S.A.; Kuhar, M.F.; Vallance, J.E.; Tolle, K.; Hoskins, E.E.; Kalinichenko, V.V.; Wells, S.I.; Zorn, A.M.; et al. Directed differentiation of human pluripotent stem cells into intestinal tissue in vitro. Nature 2011, 470, 105–110. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Takayama, K.; Negoro, R.; Yamashita, T.; Kawai, K.; Ichikawa, M.; Mori, T.; Nakatsu, N.; Harada, K.; Ito, S.; Yamada, H.; et al. Generation of human iPSC–Derived intestinal epithelial cell monolayers by CDX2 transduction. CMGH 2019, 8, 513–526. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McCracken, K.W.; Howell, J.C.; Wells, J.M.; Spence, J.R. Generating human intestinal tissue from pluripotent stem cells in vitro. Nat. Protoc. 2011, 6, 1920–1928. [Google Scholar] [CrossRef] [Green Version]
- Lukovac, S.; Roeselers, G. Intestinal crypt organoids as experimental models. In The Impact of Food Bioactives on Health: In Vitro and Ex Vivo, Models; Verhoeckx, K., Cotter, P., López-Expósito, I., Kleiveland, C., Lea, T., Mackie, A., Requena, T., Swiatecka, D., Wichers, H., Eds.; Springer: Cham, The Netherland, 2015; pp. 245–253. ISBN 9783319157917. [Google Scholar]
- Wallach, T.E.; Bayrer, J.R. Intestinal organoids: New frontiers in the study of intestinal disease and physiology. J. Pediatr. Gastroenterol. Nutr. 2017, 64, 180–185. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Workman, M.J.; Gleeson, J.P.; Troisi, E.J.; Estrada, H.Q.; Kerns, S.J.; Hinojosa, C.D.; Hamilton, G.A.; Targan, S.R.; Svendsen, C.N.; Barrett, R.J. Enhanced utilization of induced pluripotent stem cell–derived human intestinal organoids using microengineered chips. CMGH 2018, 5, 669–677. [Google Scholar] [CrossRef] [Green Version]
- D’Amour, K.A.; Agulnick, A.D.; Eliazer, S.; Kelly, O.G.; Kroon, E.; Baetge, E.E. Efficient differentiation of human embryonic stem cells to definitive endoderm. Nat. Biotechnol. 2005, 23, 1534–1541. [Google Scholar] [CrossRef]
- Siller, R.; Naumovska, E.; Mathapati, S.; Lycke, M.; Greenhough, S.; Sullivan, G.J. Development of a rapid screen for the endodermal differentiation potential of human pluripotent stem cell lines. Sci. Rep. 2016, 6, 37178–37192. [Google Scholar] [CrossRef] [Green Version]
- Hansson, M.; Olesen, D.R.; Peterslund, J.M.L.; Engberg, N.; Kahn, M.; Winzi, M.; Klein, T.; Maddox-Hyttel, P.; Serup, P. A late requirement for Wnt and FGF signaling during activin-induced formation of foregut endoderm from mouse embryonic stem cells. Dev. Biol. 2009, 330, 286–304. [Google Scholar] [CrossRef] [Green Version]
- Davenport, C.; Diekmann, U.; Budde, I.; Detering, N.; Naujok, O. Anterior–Posterior patterning of definitive endoderm generated from human embryonic stem cells depends on the differential signaling of retinoic acid, wnt-, and bmp-signaling. Stem Cells 2016, 34, 2635–2647. [Google Scholar] [CrossRef]
- Dessimoz, J.; Opoka, R.; Kordich, J.J.; Grapin-Botton, A.; Wells, J.M. FGF signaling is necessary for establishing gut tube domains along the anterior-posterior axis in vivo. Mech. Dev. 2006, 123, 42–55. [Google Scholar] [CrossRef]
- Kretzschmar, K.; Clevers, H. Developmental cell review organoids: Modeling development and the stem cell niche in a dish. Dev. Cell 2016, 38, 590–600. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kasendra, M.; Tovaglieri, A.; Sontheimer-Phelps, A.; Jalili-Firoozinezhad, S.; Bein, A.; Chalkiadaki, A.; Scholl, W.; Zhang, C.; Rickner, H.; Richmond, C.A.; et al. Development of a primary human Small Intestine-on-a-Chip using biopsy-derived organoids. Sci. Rep. 2018, 8, 2871–2885. [Google Scholar] [CrossRef] [PubMed]
- Marx, U. Biology-inspired microphysiological system approaches to solve the prediction dilemma of substance testing. ALTEX 2016, 33, 272. [Google Scholar] [CrossRef] [PubMed]
- Chan, C.Y.; Huang, P.H.; Guo, F.; Ding, X.; Kapur, V.; Mai, J.D.; Yuen, P.K.; Huang, T.J. Accelerating drug discovery via organs-on-chips. Lab Chip 2013, 13, 4697–4710. [Google Scholar] [CrossRef] [Green Version]
- Yum, K.; Hong, S.G.; Healy, K.E.; Lee, L.P. Physiologically relevant organs on chips. Biotechnol. J. 2014, 9, 16–27. [Google Scholar] [CrossRef]
- Huh, D.; Hamilton, G.A.; Ingber, D.E. From 3D cell culture to organs-on-chips. Trends Cell Biol. 2011, 21, 745–754. [Google Scholar] [CrossRef] [Green Version]
- Kaarj, K.; Yoon, J.Y. Methods of delivering mechanical stimuli to Organ-on-a-Chip. Micromachines 2019, 10, 700. [Google Scholar] [CrossRef] [Green Version]
- Vulto, P.; Podszun, S.; Meyer, P.; Hermann, C.; Manz, A.; Urban, G.A. Phaseguides: A paradigm shift in microfluidic priming and emptying. Lab Chip 2011, 11, 1596–1602. [Google Scholar] [CrossRef]
- Sato, T.; Vries, R.G.; Snippert, H.J.; van de Wetering, M.; Barker, N.; Stange, D.E.; van Es, J.H.; Abo, A.; Kujala, P.; Peters, P.J.; et al. Single Lgr5 stem cells build crypt-villus structures in vitro without a mesenchymal niche. Nature 2009, 459, 262–265. [Google Scholar] [CrossRef]
- Grainger, S.; Savory, J.G.A.; Lohnes, D. Cdx2 regulates patterning of the intestinal epithelium. Dev. Biol. 2010, 339, 155–165. [Google Scholar] [CrossRef] [Green Version]
- Gao, N.; White, P.; Kaestner, K.H. Establishment of intestinal identity and epithelial-mesenchymal signaling by Cdx2. Dev. Cell 2009, 16, 588–599. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Smith, N.R.; Davies, P.S.; Silk, A.D.; Wong, M.H. Wong epithelial and mesenchymal contribution to the niche: A safeguard for intestinal stem cell homeostasis. Gastroenterology 2012, 143, 1426–1430. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chan, L.M.S.; Lowes, S.; Hirst, B.H. The ABCs of drug transport in intestine and liver: Efflux proteins limiting drug absorption and bioavailability. Eur. J. Pharm. Sci. 2004, 21, 25–51. [Google Scholar] [CrossRef] [PubMed]
- Kaczorowski, S.; Kaczorowska, M. P glycoprotein and multidrug resistance. Patol. Pol. 1991, 42, 119–125. [Google Scholar]
- Xie, F.; Ding, X.; Zhang, Q.Y. An update on the role of intestinal cytochrome P450 enzymes in drug disposition. Acta Pharm. Sin. B 2016, 6, 374–383. [Google Scholar] [CrossRef] [Green Version]
- Lown, K.S.; Ghosh, M.; and Watkins, P.B. Sequences of intestinal and hepatic cytochrome P450 3A4 cDNAs are identical. Drug Detab. Dispos. 1998, 26, 185–187. [Google Scholar]
- Kasendra, M.; Luc, R.; Yin, J.; Manatakis, D.V.; Kulkarni, G.; Lucchesi, C.; Sliz, J.; Apostolou, A.; Sunuwar, L.; Obrigewitch, J.; et al. Duodenum intestine-chip for preclinical drug assessment in a human relevant model. Elife 2020, 9, 50135. [Google Scholar] [CrossRef]
- Tonn, J.C.; Kerkeay, S.; Hanke, A.; Bouterfa, H.; Mueller, J.G.; Wagner, S.; Hamilton, G.; Roosen, K. Effect of synthetic matrix-metalloproteinase inhibitors on invasive capacity and proliferation of human malignant gliomas in vitro. Int. J. Cancer 1999, 80, 764–772. [Google Scholar] [CrossRef]
- Trietsch, S.J.; Naumovska, E.; Kurek, D.; Setyawati, M.C.; Vormann, M.K.; Wilschut, K.J.; Lanz, H.L.; Nicolas, A.; Ng, C.P.; Joore, J.; et al. Membrane-free culture and real-time barrier integrity assessment of perfused intestinal epithelium tubes. Nat. Commun. 2017, 8, 262. [Google Scholar] [CrossRef] [Green Version]
- Sjöberg, Å.; Lutz, M.; Tannergren, C.; Wingolf, C.; Borde, A.; Ungell, A.-L. Comprehensive study on regional human intestinal permeability and prediction of fraction absorbed of drugs using the Ussing chamber technique. Eur. J. Pharm. Sci. 2013, 48, 166–180. [Google Scholar] [CrossRef]
- Andrews, C.; McLean, M.H.; Durum, S.K. Cytokine Tuning of Intestinal Epithelial Function. Front. Immunol. 2018, 9, 1270. [Google Scholar] [CrossRef] [PubMed]
- Kaser, A.; Ludwiczek, O.; Holzmann, S.; Moschen, A.R.; Weiss, G.; Enrich, B.; Graziadei, I.; Dunzendorfer, S.; Wiedermann, C.J.; Mürzl, E.; et al. Increased expression of CCL20 in human inflammatory bowel disease. J. Clin. Immunol. 2004, 24, 74–85. [Google Scholar] [CrossRef] [PubMed]
- O’Brien, S.A.; Zhu, M.; Zhang, W. The importance of IL-6 in the development of LAT-mediated autoimmunity. J. Immunol. 2015, 195, 695–705. [Google Scholar] [CrossRef] [PubMed]
- Mitsuyama, K.; Toyonaga, A.; Sasaki, E.; Watanabe, K.; Tateishi, H.; Nishiyama, T.; Saiki, T.; Ikeda, H.; Tsuruta, O.; Tanikawa, K. IL-8 as an important chemoattractant for neutrophils in ulcerative colitis and Crohn’s disease. Clin. Exp. Immunol. 2008, 96, 432–436. [Google Scholar] [CrossRef] [PubMed]
- Siller, R.; Greenhough, S.; Naumovska, E.; Sullivan, G.J. Small-molecule-driven hepatocyte differentiation of human pluripotent stem cells. Stem Cell Rep. 2015, 4, 939–952. [Google Scholar] [CrossRef] [Green Version]
- Ogaki, S.; Shiraki, N.; Kume, K.; Kume, S. Wnt and Notch signals guide embryonic stem cell differentiation into the intestinal lineages. Stem Cells 2013, 31, 1086–1096. [Google Scholar] [CrossRef]
- Ogaki, S.; Morooka, M.; Otera, K.; Kume, S. A cost-effective system for differentiation of intestinal epithelium from human induced pluripotent stem cells. Sci. Rep. 2015, 5, 1–11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, L.; Nam, S.; Tian, Y.; Yang, F.; Wu, J.; Wang, Y.; Scuto, A.; Polychronopoulos, P.; Magiatis, P.; Skaltsounis, L.; et al. 6-Bromoindirubin-3′-oxime inhibits JAK/STAT3 signaling and induces apoptosis of human melanoma cells. Cancer Res. 2011, 71, 3972–3979. [Google Scholar] [CrossRef] [Green Version]
- Lin, G.; Xu, N.; Xi, R. Paracrine unpaired signaling through the JAK/STAT pathway controls self-renewal and lineage differentiation of drosophila intestinal stem cells. J. Mol. Cell Biol. 2010, 2, 37–49. [Google Scholar] [CrossRef] [Green Version]
- Sato, T.; van Es, J.H.; Snippert, H.J.; Stange, D.E.; Vries, R.G.; van den Born, M.; Barker, N.; Shroyer, N.F.; van de Wetering, M.; Clevers, H. Paneth cells constitute the niche for Lgr5 stem cells in intestinal crypts. Nature 2011, 469, 415–418. [Google Scholar] [CrossRef] [Green Version]
- Srinivasan, B.; Kolli, A.R.; Esch, M.B.; Abaci, H.E.; Shuler, M.L.; Hickman, J.J. TEER Measurement techniques for in vitro barrier model systems. J. Lab. Autom. 2015, 20, 107–126. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Takenaka, T.; Harada, N.; Kuze, J.; Chiba, M.; Iwao, T.; Matsunaga, T. Human small intestinal epithelial cells differentiated from adult intestinal stem cells as a novel system for predicting oral drug absorption in humans. Drug Metab. Dispos. 2014, 42, 1947–1954. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ozawa, T.; Takayama, K.; Okamoto, R.; Negoro, R.; Sakurai, F.; Tachibana, M.; Kawabata, K.; Mizuguchi, H.; Frank, R.; Hargreaves, R.; et al. Generation of enterocyte-like cells from human induced pluripotent stem cells for drug absorption and metabolism studies in human small intestine. Sci. Rep. 2015, 5, 16479. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Iwao, T.; Kodama, N.; Kondo, Y.; Kabeya, T.; Nakamura, K.; Horikawa, T.; Niwa, T.; Kurose, K.; Matsunaga, T. Generation of enterocyte-like cells with pharmacokinetic functions from human induced pluripotent stem cells using small-molecule compounds. Drug Metab. Dispos. 2015, 43, 603–610. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- van de Walle, J.; Hendrickx, A.; Romier, B.; Larondelle, Y.; Schneider, Y.-J. Inflammatory parameters in Caco-2 cells: Effect of stimuli nature, concentration, combination and cell differentiation. Toxicol. Vitr. 2010, 24, 1441–1449. [Google Scholar] [CrossRef]
- Vitkus, S.J.D.; Hanifin, S.A.; McCee, D.W. Factors affecting Caco-2 intestinal epithelial cell interleukin-6 secretion. Vitr. Cell. Dev. Biol. Anim. 1998, 34, 660–664. [Google Scholar] [CrossRef]
- Jeffery, V.; Goldson, A.J.; Dainty, J.R.; Chieppa, M.; Sobolewski, A. IL-6 signaling regulates small intestinal crypt homeostasis. J. Immunol. 2017, 199, 304–311. [Google Scholar] [CrossRef]
- Kuhn, K.A.; Manieri, N.A.; Liu, T.C.; Stappenbeck, T.S. IL-6 stimulates intestinal epithelial proliferation and repair after injury. PLoS ONE 2014, 9. [Google Scholar] [CrossRef] [Green Version]
- Galvez-Llompart, M.; Del Carmen Recio Iglesias, M.; Gálvez, J.; García-Domenech, R. Novel potential agents for ulcerative colitis by molecular topology: Suppression of IL-6 production in Caco-2 and RAW 264.7 cell lines. Mol. Divers. 2013, 17, 573–593. [Google Scholar] [CrossRef]
- van de Wetering, M.; Francies, H.E.; Francis, J.M.; Bounova, G.; Iorio, F.; Pronk, A.; van Houdt, W.; van Gorp, J.; Taylor-Weiner, A.; Kester, L.; et al. Prospective derivation of a living organoid biobank of colorectal cancer patients. Cell 2015, 161, 933–945. [Google Scholar] [CrossRef] [Green Version]
- Lim, M.L.; Jungebluth, P.; Sjöqvist, S.; Nikdin, H.; Kjartansdóttir, K.R.; Unger, C.; Vassliev, I.; Macchiarini, P. Decellularized feeders: An optimized method for culturing pluripotent cells. Stem Cells Transl. Med. 2013, 2, 975–982. [Google Scholar] [CrossRef] [PubMed]
- Gijzen, L.; Marescotti, D.; Raineri, E.; Nicolas, A.; Lanz, H.L.; Guerrera, D.; van Vught, R.; Joore, J.; Vulto, P.; Peitsch, M.C.; et al. An Intestine-on-a-chip model of plug-and-play modularity to study inflammatory processes. Slas Technol. 2020. [Google Scholar] [CrossRef] [PubMed]
- van Duinen, V.; van den Heuvel, A.; Trietsch, S.J.; Lanz, H.L.; van Gils, J.M.; van Zonneveld, A.J.; Vulto, P.; Hankemeier, T. 96 Perfusable blood vessels to study vascular permeability in vitro. Sci. Rep. 2017, 7, 1–11. [Google Scholar] [CrossRef] [PubMed]




© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Naumovska, E.; Aalderink, G.; Wong Valencia, C.; Kosim, K.; Nicolas, A.; Brown, S.; Vulto, P.; Erdmann, K.S.; Kurek, D. Direct On-Chip Differentiation of Intestinal Tubules from Induced Pluripotent Stem Cells. Int. J. Mol. Sci. 2020, 21, 4964. https://doi.org/10.3390/ijms21144964
Naumovska E, Aalderink G, Wong Valencia C, Kosim K, Nicolas A, Brown S, Vulto P, Erdmann KS, Kurek D. Direct On-Chip Differentiation of Intestinal Tubules from Induced Pluripotent Stem Cells. International Journal of Molecular Sciences. 2020; 21(14):4964. https://doi.org/10.3390/ijms21144964
Chicago/Turabian StyleNaumovska, Elena, Germaine Aalderink, Christian Wong Valencia, Kinga Kosim, Arnaud Nicolas, Stephen Brown, Paul Vulto, Kai S. Erdmann, and Dorota Kurek. 2020. "Direct On-Chip Differentiation of Intestinal Tubules from Induced Pluripotent Stem Cells" International Journal of Molecular Sciences 21, no. 14: 4964. https://doi.org/10.3390/ijms21144964
APA StyleNaumovska, E., Aalderink, G., Wong Valencia, C., Kosim, K., Nicolas, A., Brown, S., Vulto, P., Erdmann, K. S., & Kurek, D. (2020). Direct On-Chip Differentiation of Intestinal Tubules from Induced Pluripotent Stem Cells. International Journal of Molecular Sciences, 21(14), 4964. https://doi.org/10.3390/ijms21144964