Expanding the Toolkit of Fluorescent Biosensors for Studying Mitogen Activated Protein Kinases in Plants

Abstract
:1. Introduction
2. Results
2.1. Developing a FRET-Based Plant MAPK Docking Domain Trap
2.2. Testing the Plant MAPK Docking Domain Trap
2.3. Developing a Translocation-Based Biosensor for Use in Arabidopsis
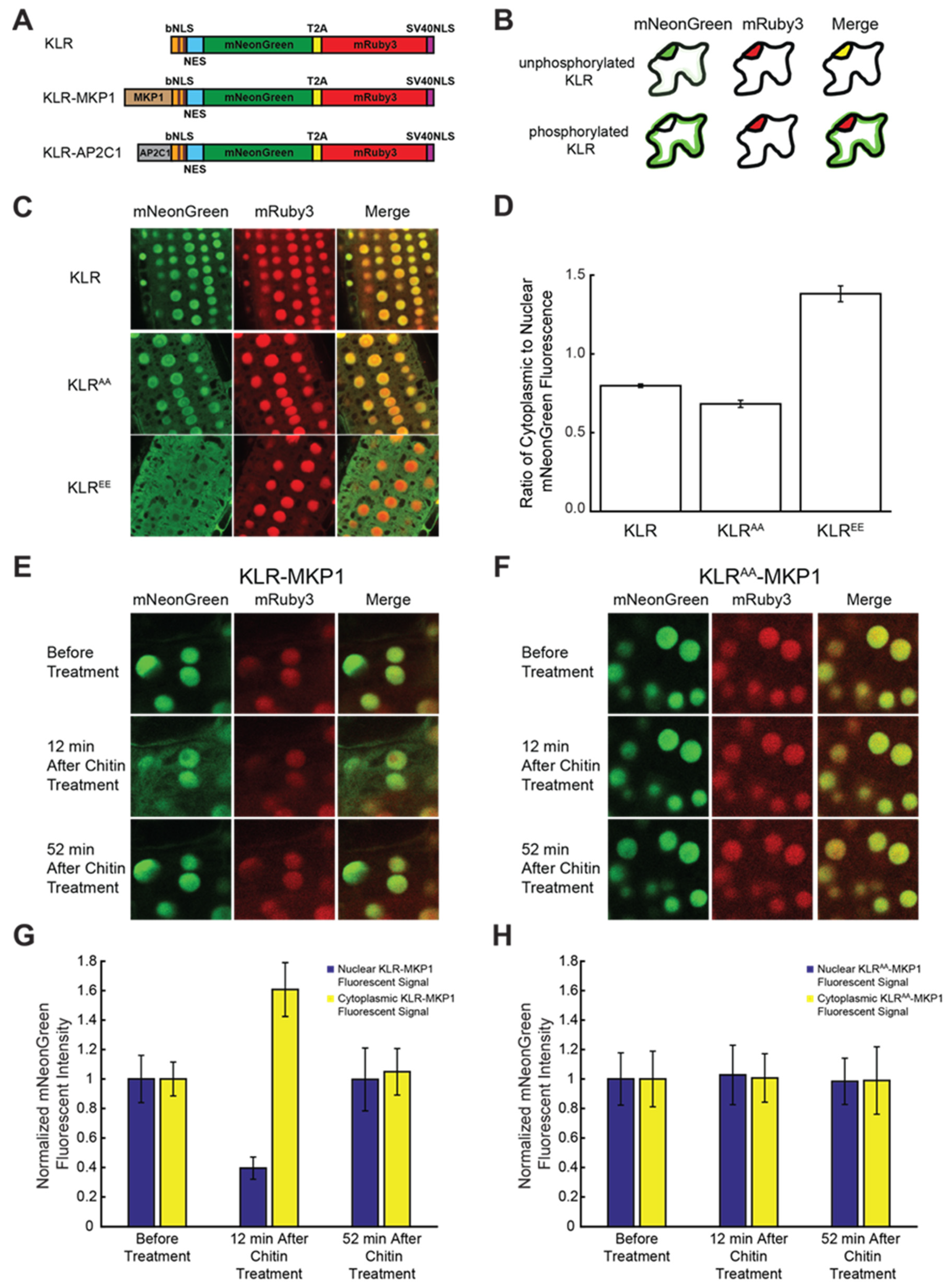
2.4. Addition of a Plant MAPK Docking Domain to KLR Facilitates Subcellular Localization Shifts in Response to Chitin
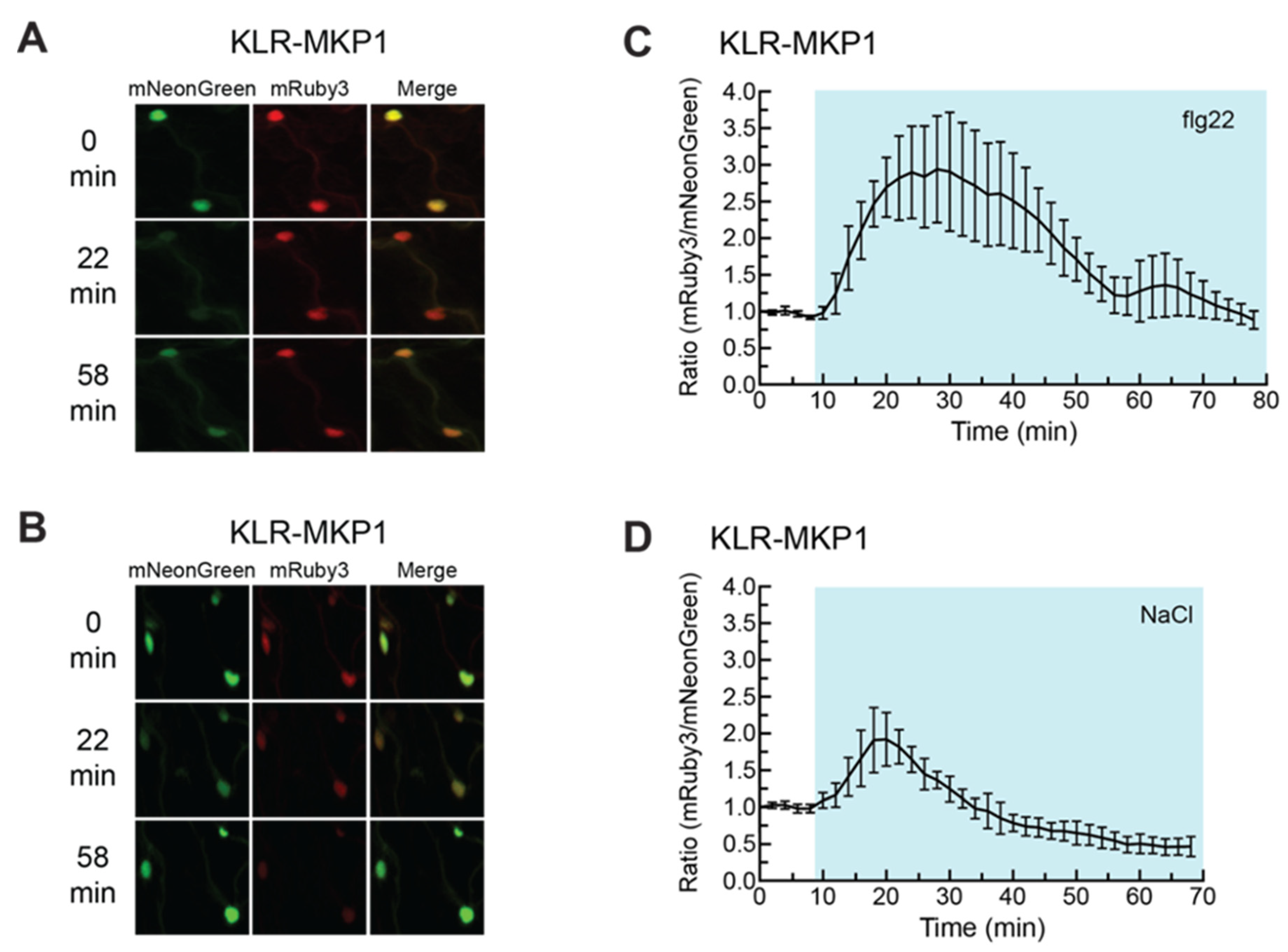
2.5. KLR-MKP1 Exhibited Shifts in Localization in Arabidopsis Cotyledons in Response to Different Chemical Treatments Known to Trigger MAPK Activation
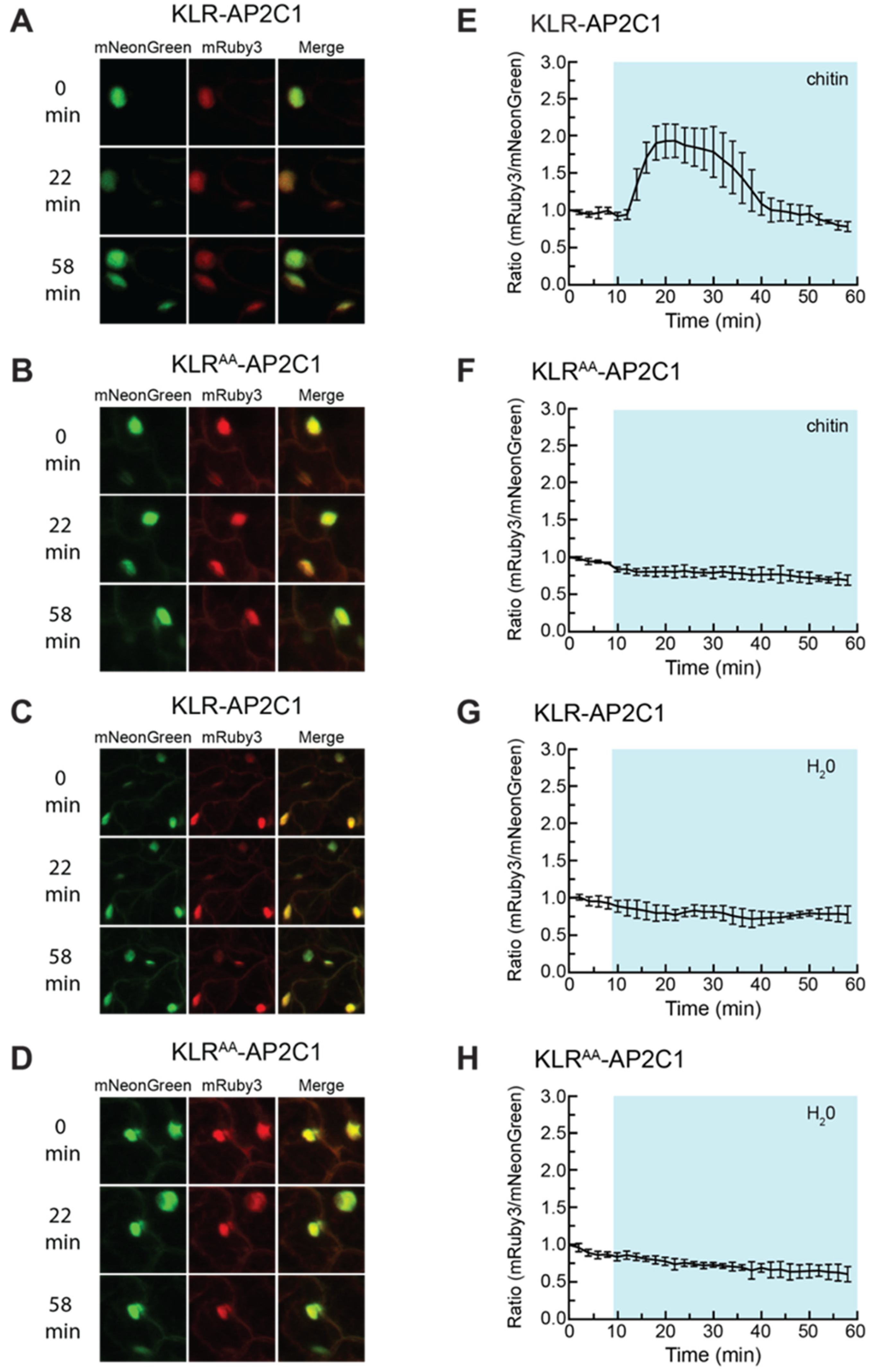
2.6. Incorporating the AP2C1 Docking Domain into KLR Produces a Biosensor That Changes Localization in Response to Chitin
3. Discussion
4. Materials and Methods
4.1. Plasmid Construction
4.2. In Vitro FRET Assay
4.3. Transgenic Lines
4.4. Plant Material and Growth Conditions
4.5. Confocal Microscopy
4.6. Image Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| MAPK | Mitogen-activated protein kinase |
| FRET | Förster resonance energy transfer |
| SOMA | Sensor of MAPK in Arabidopsis |
| NLS | Nuclear localization signal |
| NES | Nuclear exclusion signal |
| ERK | Extracellular signal-regulated kinases |
| MKP1 | MAPK phosphotase 1 |
| CA-MPK6 | Constitutively active MAPK 6 |
| CA-MKP4 | Constitutively active MAPK 4 |
| ATP | Adenosine triphosphate |
| BASL | Breaking of asymmetry in the stomatal lineage |
| AP2C1 | Arabidopsis PP2C-type phosphatase |
| KTR | Kinase translocation reporter |
| bNLS | Bipartite nuclear localization signal |
| nKTR | Nuclear kinase translocation reporter |
| KLR | Kinase translocation reporter |
| T2A | Thosea asigna virus 2A peptide |
| SV40NLS | Simian virus 40 nuclear localization signal |
| ROI | Region of interest |
| Flg22 | Flagellin 22 |
References
- Kültz, D. Phylogenetic and functional classification of mitogen-and stress-activated protein kinases. J. Mol. Evol. 1998, 46, 571–588. [Google Scholar] [CrossRef] [PubMed]
- Müller, J.; Beck, M.; Mettbach, U.; Komis, G.; Hause, G.; Menzel, D.; Šamaj, J. Arabidopsis MPK6 is involved in cell division plane control during early root development, and localizes to the pre-prophase band, phragmoplast, trans-Golgi network and plasma membrane. Plant J. 2010, 61, 234–248. [Google Scholar] [CrossRef]
- Xu, J.; Zhang, S. Mitogen-activated protein kinase cascades in signaling plant growth and development. Trends Plant Sci. 2015, 20, 56–64. [Google Scholar] [CrossRef] [PubMed]
- Mizoguchi, T.; Irie, K.; Hirayama, T.; Hayashida, N.; Yamaguchi-Shinozaki, K.; Matsumoto, K.; Shinozaki, K. A gene encoding a mitogen-activated protein kinase kinase kinase is induced simultaneously with genes for a mitogen-activated protein kinase and an S6 ribosomal protein kinase by touch, cold, and water stress in Arabidopsis thaliana. Proc. Natl. Acad. Sci. USA 1996, 93, 765–769. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ichimura, K.; Mizoguchi, T.; Yoshida, R.; Yuasa, T.; Shinozaki, K. Various abiotic stresses rapidly activate Arabidopsis MAP kinases ATMPK4 and ATMPK6. Plant J. 2000, 24, 655–665. [Google Scholar] [CrossRef] [PubMed]
- Droillard, M.J.; Boudsocq, M.; Barbier-Brygoo, H.; Laurière, C. Different protein kinase families are activated by osmotic stresses in Arabidopsis thaliana cell suspensions: Involvement of the MAP kinases AtMPK3 and AtMPK6. FEBS Lett. 2002, 527, 43–50. [Google Scholar] [CrossRef]
- Ahlfors, R.; Macioszek, V.; Rudd, J.; Brosché, M.; Schlichting, R.; Scheel, D.; Kangasjärvi, J. Stress hormone-independent activation and nuclear translocation of mitogen-activated protein kinases in Arabidopsis thaliana during ozone exposure. Plant J. 2004, 40, 512–522. [Google Scholar] [CrossRef]
- Droillard, M.J.; Boudsocq, M.; Barbier-Brygoo, H.; Laurière, C. Involvement of MPK4 in osmotic stress response pathways in cell suspensions and plantlets of Arabidopsis thaliana: Activation by hypoosmolarity and negative role in hyperosmolarity tolerance. FEBS Lett. 2004, 574, 42–48. [Google Scholar] [CrossRef]
- Teige, M.; Scheikl, E.; Eulgem, T.; Dóczi, R.; Ichimura, K.; Shinozaki, K.; Dangl, J.L.; Hirt, H. The MKK2 pathway mediates cold and salt stress signaling in Arabidopsis. Mol Cell. 2004, 15, 141–152. [Google Scholar] [CrossRef]
- Felix, G.; Duran, J.D.; Volko, S.; Boller, T. Plants have a sensitive perception system for the most conserved domain of bacterial flagellin. Plant J. 1999, 18, 265–276. [Google Scholar] [CrossRef]
- Gómez-Gómez, L.; Boller, T. FLS2: An LRR receptor–like kinase involved in the perception of the bacterial elicitor flagellin in Arabidopsis. Mol. Cell 2000, 5, 1003–1011. [Google Scholar] [CrossRef]
- Petersen, M.; Brodersen, P.; Næsted, H.; Andreasson, E.; Lindhart, U.; Johansen, B.; Nielsen, H.B.; Lacy, M.; Austin, M.J.; Parker, J.E.; et al. Arabidopsis MAP kinase 4 negatively regulates systemic acquired resistance. Cell 2000, 103, 1111–1120. [Google Scholar] [CrossRef] [Green Version]
- Frye, C.A.; Tang, D.; Innes, R.W. Negative regulation of defense responses in plants by a conserved MAPKK kinase. Proc. Nat. Acad. of Sci. USA 2001, 98, 373–378. [Google Scholar] [CrossRef]
- Asai, T.; Tena, G.; Plotnikova, J.; Willmann, M.R.; Chiu, W.L.; Gomez-Gomez, L.; Boller, T.; Ausubel, F.M.; Sheen, J. MAP kinase signaling cascade in Arabidopsis innate immunity. Nature 2002, 415, 977–983. [Google Scholar] [CrossRef] [PubMed]
- Miya, A.; Albert, P.; Shinya, T.; Desaki, Y.; Ichimura, K.; Shirasu, K.; Narusaka, Y.; Kawakami, N.; Kaku, H.; Shibuya, N. CERK1, a LysM receptor kinase, is essential for chitin elicitor signaling in Arabidopsis. Proc. Nat. Acad. of Sci. USA 2007, 104, 19613–19618. [Google Scholar] [CrossRef] [Green Version]
- Rasmussen, M.W.; Roux, M.; Petersen, M.; Mundy, J. MAP kinase cascades in Arabidopsis innate immunity. Front. Plant Sci. 2012, 3, 169. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meng, X.; Zhang, S. MAPK cascades in plant disease resistance signaling. Annu. Rev. Phytopathol. 2013, 51, 245–266. [Google Scholar] [CrossRef]
- Shinkai, Y.; Satoh, H.; Takeda, N.; Fukuda, M.; Chiba, E.; Kato, T.; Kuramochi, T.; Araki, Y. A testicular germ cell-associated serine-threonine kinase, MAK, is dispensable for sperm formation. Mol. Cell. Biol. 2002, 22, 3276–3280. [Google Scholar] [CrossRef] [Green Version]
- Wang, H.; Liu, Y.; Bruffett, K.; Lee, J.; Hause, G.; Walker, J.C.; Zhang, S. Haplo-insufficiency of MPK3 in MPK6 mutant background uncovers a novel function of these two MAPKs in Arabidopsis ovule development. Plant Cell 2008, 20, 602–613. [Google Scholar] [CrossRef] [Green Version]
- López-Bucio, J.S.; Dubrovsky, J.G.; Raya-González, J.; Ugartechea-Chirino, Y.; López-Bucio, J.; de Luna-Valdez, L.A.; Ramos-Vega, M.; León, P.; Guevara-García, A.A. Arabidopsis thaliana mitogen-activated protein kinase 6 is involved in seed formation and modulation of primary and lateral root development. J. Exp. Bot. 2014, 65, 169–183. [Google Scholar] [CrossRef] [Green Version]
- Liu, Y.K.; Liu, Y.B.; Zhang, M.Y.; Li, D.Q. Stomatal development and movement: The roles of MAPK signaling. Plant Signal. Behav. 2010, 5, 1176–1180. [Google Scholar] [CrossRef] [Green Version]
- Lee, Y.; Kim, Y.J.; Kim, M.H.; Kwak, J.M. MAPK cascades in guard cell signal transduction. Front. Plant Sci. 2016, 7, 80. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jonak, C.; Ökrész, L.; Bögre, L.; Hirt, H. Complexity, cross talk and integration of plant MAP kinase signaling. Curr. Opin. Plant Biol. 2002, 5, 415–424. [Google Scholar] [CrossRef]
- Rodriguez, M.C.; Petersen, M.; Mundy, J. Mitogen-activated protein kinase signaling in plants. Ann. Rev. Plant Biol. 2010, 61, 621–649. [Google Scholar]
- Krysan, P.J.; Colcombet, J. Cellular complexity in MAPK signaling in plants: Questions and emerging tools to answer them. Front. Plant Sci. 2018, 9, 1674. [Google Scholar] [CrossRef] [PubMed]
- Harvey, C.D.; Ehrhardt, A.G.; Cellurale, C.; Zhong, H.; Yasuda, R.; Davis, R.J.; Svoboda, K. A genetically encoded fluorescent biosensor of ERK activity. Proc. Nat. Acad. Sci. USA 2008, 105, 19264–19269. [Google Scholar] [CrossRef] [Green Version]
- Oldach, L.; Zhang, J. Genetically encoded fluorescent biosensors for live-cell visualization of protein phosphorylation. Chem. Biol. 2014, 21, 186–197. [Google Scholar] [CrossRef] [Green Version]
- Greenwald, E.C.; Mehta, S.; Zhang, J. Genetically encoded fluorescent biosensors illuminate the spatiotemporal regulation of signaling networks. Chem. Rev. 2018, 118, 11707–11794. [Google Scholar] [CrossRef] [PubMed]
- Sato, M.; Ozawa, T.; Inukai, K.; Asano, T.; Umezawa, Y. Fluorescent indicators for imaging protein phosphorylation in single living cells. Nat. Biotechnol. 2002, 20, 287–294. [Google Scholar] [CrossRef]
- Fosbrink, M.; Aye-Han, N.N.; Cheong, R.; Levchenko, A.; Zhang, J. Visualization of JNK activity dynamics with a genetically encoded fluorescent biosensor. Proc. Nat.l Acad. Sci. USA 2010, 107, 5459–5464. [Google Scholar] [CrossRef] [Green Version]
- Kamioka, Y.; Sumiyama, K.; Mizuno, R.; Sakai, Y.; Hirata, E.; Kiyokawa, E.; Matsuda, M. Live imaging of protein kinase activities in transgenic mice expressing FRET biosensors. Cell Struct. Funct. 2012, 37, 65–73. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aoki, K.; Komatsu, N.; Hirata, E.; Kamioka, Y.; Matsuda, M. Stable expression of FRET biosensors: A new light in cancer research. Cancer Sci. 2012, 103, 614–619. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aoki, K.; Kumagai, Y.; Sakurai, A.; Komatsu, N.; Fujita, Y.; Shionyu, C.; Matsuda, M. Stochastic ERK activation induced by noise and cell-to-cell propagation regulates cell density-dependent proliferation. Mol. Cell 2013, 52, 529–540. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sample, V.; Mehta, S.; Zhang, J. Genetically encoded molecular probes to visualize and perturb signaling dynamics in living biological systems. J. Cell Sci. 2014, 127, 1151–1160. [Google Scholar] [CrossRef] [Green Version]
- Tomida, T.; Takekawa, M.; Saito, H. Oscillation of p38 activity controls efficient pro-inflammatory gene expression. Nat. Comm. 2015, 6, 1–9. [Google Scholar] [CrossRef] [Green Version]
- Sari, D.W.K.; Akiyama, R.; Naoki, H.; Ishijima, H.; Bessho, Y.; Matsui, T. Time-lapse observation of stepwise regression of Erk activity in zebrafish presomitic mesoderm. Sci. Rep. 2018, 8, 1–10. [Google Scholar] [CrossRef] [Green Version]
- Zhang, J.; Allen, M.D. FRET-based biosensors for protein kinases: Illuminating the kinome. Mol. Biosyst. 2007, 3, 759–765. [Google Scholar] [CrossRef]
- Kumagai, Y.; Naoki, H.; Nakasyo, E.; Kamioka, Y.; Kiyokawa, E.; Matsuda, M. Heterogeneity in ERK activity as visualized by in vivo FRET imaging of mammary tumor cells developed in MMTV-Neu mice. Oncogene 2015, 34, 1051–1057. [Google Scholar] [CrossRef] [Green Version]
- Durandau, E.; Aymoz, D.; Pelet, S. Dynamic single cell measurements of kinase activity by synthetic kinase activity relocation biosensors. BMC Biol. 2015, 13, 55. [Google Scholar] [CrossRef] [Green Version]
- Sato, M.; Kawai, Y.; Umezawa, Y. Genetically encoded fluorescent indicators to visualize protein phosphorylation by extracellular signal-regulated kinase in single living cells. Anal. Chem. 2007, 79, 2570–2575. [Google Scholar] [CrossRef]
- Ryu, H.; Chung, M.; Song, J.; Lee, S.S.; Pertz, O.; Jeon, N.L. Integrated platform for monitoring single-cell MAPK kinetics in computer-controlled temporal stimulations. Sci. Rep. 2018, 8, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Van, T.N.N.; Morris, M.C. Fluorescent biosensors of protein kinases: From basics to biomedical applications. In Progress in Molecular Biology and Translational Science; Academic Press: Montpellier, France, 2013; Volume 113, pp. 217–274. [Google Scholar]
- Lin, W.; Mehta, S.; Zhang, J. Genetically encoded fluorescent biosensors illuminate kinase signaling in cancer. J. Biol. Chem. 2019, 294, 14814–14822. [Google Scholar] [CrossRef] [Green Version]
- Zaman, N.; Seitz, K.; Kabir, M.; George-Schreder, L.S.; Shepstone, I.; Liu, Y.; Zhang, S.; Krysan, P.J. A Förster resonance energy transfer sensor for live-cell imaging of mitogen-activated protein kinase activity in Arabidopsis. Plant J. 2019, 97, 970–983. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Campbell, R.E.; Ting, A.Y.; Tsien, R.Y. Creating new fluorescent probes for cell biology. Nature Rev. Mol. Cell Biol. 2002, 3, 906–918. [Google Scholar] [CrossRef]
- Aye-Han, N.N.; Ni, Q.; Zhang, J. Fluorescent biosensors for real-time tracking of post-translational modification dynamics. Curr. Opin. Chem. Biol. 2009, 13, 392–397. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ibraheem, A.; Campbell, R.E. Designs and applications of fluorescent protein-based biosensors. Curr. Opin. Chem. Biol. 2010, 14, 30–36. [Google Scholar] [CrossRef] [PubMed]
- Komatsu, N.; Aoki, K.; Yamada, M.; Yukinaga, H.; Fujita, Y.; Kamioka, Y.; Matsuda, M. Development of an optimized backbone of FRET biosensors for kinases and GTPases. Mol. Biol. Cell 2011, 22, 4647–4656. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morris, M.C. Fluorescence-Based Biosensors: From Concepts to Applications; Academic Press: Cambridge, MA, USA, 2012. [Google Scholar]
- Lindenburg, L.; Merkx, M. Engineering genetically encoded FRET biosensors. Sensors 2014, 14, 11691–11713. [Google Scholar] [CrossRef] [Green Version]
- Regot, S.; Hughey, J.J.; Bajar, B.T.; Carrasco, S.; Covert, M.W. High-sensitivity measurements of multiple kinase activities in live single cells. Cell 2014, 157, 1724–1734. [Google Scholar] [CrossRef] [Green Version]
- Sparta, B.; Pargett, M.; Minguet, M.; Distor, K.; Bell, G.; Albeck, J.G. Receptor level mechanisms are required for epidermal growth factor (EGF)-stimulated extracellular signal-regulated kinase (ERK) activity pulses. J. Biol. Chem. 2015, 290, 24784–24792. [Google Scholar] [CrossRef] [Green Version]
- De la Cova, C.; Townley, R.; Regot, S.; Greenwald, I. A real-time biosensor for ERK activity reveals signaling dynamics during C. elegans cell fate specification. Dev. Cell 2017, 42, 542–553. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mayr, V.; Sturtzel, C.; Stadler, M.; Grissenberger, S.; Distel, M. Fast dynamic in vivo monitoring of Erk activity at single cell resolution in DREKA zebrafish. Front. Cell Dev. Biol. 2018, 6, 111. [Google Scholar] [CrossRef]
- Kim, J.; Lee, S.; Jung, K.; Oh, W.C.; Kim, N.; Son, S.; Jo, Y.; Kwon, H.B.; Do Heo, W. Intensiometric biosensors visualize the activity of multiple small GTPases in vivo. Nat. Commun. 2019, 10, 1–11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nguyen, A.W.; Daugherty, P.S. Evolutionary optimization of fluorescent proteins for intracellular FRET. Nat. Biotechnol. 2005, 23, 355–360. [Google Scholar] [CrossRef]
- Goedhart, J.; Van Weeren, L.; Hink, M.A.; Vischer, N.O.; Jalink, K.; Gadella, T.W., Jr. Bright cyan fluorescent protein variants identified by fluorescence lifetime screening. Nat. Methods 2010, 7, 137. [Google Scholar] [CrossRef]
- Hofmann, K.; Bucher, P. The FHA domain: A putative nuclear signaling domain found in protein kinases and transcription factors. Trends Biochem. Scis. 1995, 20, 347–349. [Google Scholar] [CrossRef]
- Sun, Z.; Hsiao, J.; Fay, D.S.; Stern, D.F. Rad53 FHA domain associated with phosphorylated Rad9 in the DNA damage checkpoint. Science 1998, 281, 272–274. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez, F.A.; Raden, D.L.; Davis, R.J. Identification of substrate recognition determinants for human ERK1 and ERK2 protein kinases. J. Biol. Chem. 1991, 266, 22159–22163. [Google Scholar]
- Jacobs, D.; Glossip, D.; Xing, H.; Muslin, A.J.; Kornfeld, K. Multiple docking sites on substrate proteins form a modular system that mediates recognition by ERK MAP kinase. Genes Dev. 1999, 13, 163–175. [Google Scholar] [CrossRef] [Green Version]
- Berriri, S.; Garcia, A.V.; dit Frey, N.F.; Rozhon, W.; Pateyron, S.; Leonhardt, N.; Montillet, J.L.; Leung, J.; Hirt, H.; Colcombet, J. Constitutively active mitogen-activated protein kinase versions reveal functions of Arabidopsis MPK4 in pathogen defense signaling. Plant Cell 2012, 24, 4281–4293. [Google Scholar] [CrossRef] [Green Version]
- Dong, J.; MacAlister, C.A.; Bergmann, D.C. BASL controls asymmetric cell division in Arabidopsis. Cell 2009, 137, 1320–1330. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.; Wang, P.; Shao, W.; Zhu, J.K.; Dong, J. The BASL polarity protein controls a MAPK signaling feedback loop in asymmetric cell division. Dev. Cell 2015, 33, 136–149. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.; Guo, X.; Dong, J. Phosphorylation of the polarity protein BASL differentiates asymmetric cell fate through MAPKs and SPCH. Curr. Biol. 2016, 26, 2957–2965. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schweighofer, A.; Kazanaviciute, V.; Scheikl, E.; Teige, M.; Doczi, R.; Hirt, H.; Schwanninger, M.; Kant, M.; Schuurink, R.; Mauch, F.; et al. The PP2C-type phosphatase AP2C1, which negatively regulates MPK4 and MPK6, modulates innate immunity, jasmonic acid, and ethylene levels in Arabidopsis. Plant Cell 2007, 19, 2213–2224. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Park, H.C.; Song, E.H.; Nguyen, X.C.; Lee, K.; Kim, K.E.; Kim, H.S.; Lee, S.M.; Kim, S.H.; Bae, D.W.; Yun, D.J.; et al. Arabidopsis MAP kinase phosphatase 1 is phosphorylated and activated by its substrate AtMPK6. Plant Cell Rep. 2011, 30, 1523–1531. [Google Scholar] [CrossRef]
- Umbrasaite, J.; Schweighofer, A.; Kazanaviciute, V.; Magyar, Z.; Ayatollahi, Z.; Unterwurzacher, V.; Choopayak, C.; Boniecka, J.; Murray, J.A.; Bogre, L.; et al. MAPK phosphatase AP2C3 induces ectopic proliferation of epidermal cells leading to stomata development in Arabidopsis. PLoS ONE 2010, 5, 15357. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shubchynskyy, V.; Boniecka, J.; Schweighofer, A.; Simulis, J.; Kvederaviciute, K.; Stumpe, M.; Mauch, F.; Balazadeh, S.; Mueller-Roeber, B.; Boutrot, F.; et al. Protein phosphatase AP2C1 negatively regulates basal resistance and defense responses to Pseudomonas syringae. J. Exp. Bot. 2017, 68, 1169–1183. [Google Scholar] [PubMed] [Green Version]
- Dalton, S.; Marais, R.; Wynne, J.; Treisman, R. Isolation and characterization of SRF accessory proteins. Philos. Trans. R. Soc. Lond. Ser. B Biol. Sci. 1993, 340, 325–332. [Google Scholar]
- Hipskind, R.A.; Roa, V.N.; Muller, C.G.F.; Raddy, E.S.P.; Nordheim, A. Ets-related protein Elk-1 is homologous to the c-fos regulatory factor p62TCF. Nature 1991, 354, 531–534. [Google Scholar] [CrossRef]
- Lam, A.J.; St-Pierre, F.; Gong, Y.; Marshall, J.D.; Cranfill, P.J.; Baird, M.A.; McKeown, M.R.; Wiedenmann, J.; Davidson, M.W.; Schnitzer, M.J.; et al. Improving FRET dynamic range with bright green and red fluorescent proteins. Nat. Methods 2012, 9, 1005–1012. [Google Scholar] [CrossRef] [Green Version]
- Shaner, N.C.; Lambert, G.G.; Chammas, A.; Ni, Y.; Cranfill, P.J.; Baird, M.A.; Sell, B.R.; Allen, J.R.; Day, R.N.; Israelsson, M.; et al. A bright monomeric green fluorescent protein derived from Branchiostoma lanceolatum. Nat. Methods 2013, 10, 407–409. [Google Scholar] [CrossRef]
- Halpin, C.; Cooke, S.E.; Barakate, A.; Amrani, A.E.; Ryan, M.D. Self-processing 2A-polyproteins–a system for co-ordinate expression of multiple proteins in transgenic plants. Plant. J. 1999, 17, 453–459. [Google Scholar] [CrossRef]
- Ahier, A.; Jarriault, S. Simultaneous expression of multiple proteins under a single promoter in Caenorhabditis elegans via a versatile 2A-based toolkit. Genetics 2014, 196, 605–613. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bajar, B.T.; Wang, E.S.; Lam, A.J.; Kim, B.B.; Jacobs, C.L.; Howe, E.S.; Davidson, M.W.; Lin, M.Z.; Chu, J. Improving brightness and photostability of green and red fluorescent proteins for live cell imaging and FRET reporting. Sci. Rep. 2016, 6, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Kalderon, D. Roberts BL, Richardson WD, Smith AE. A short amino acid sequence able to specify nuclear location. Cell 1984, 39, 499–509. [Google Scholar] [CrossRef]
- Ulm, R.; Revenkova, E.; di Sansebastiano, G.P.; Bechtold, N.; Paszkowski, J. Mitogen-activated protein kinase phosphatase is required for genotoxic stress relief in Arabidopsis. Genes Dev. 2001, 15, 699–709. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vang, S.; Seitz, K.; Krysan, P.J. A simple microfluidic device for live cell imaging of Arabidopsis cotyledons, leaves, and seedlings. Biotechniques 2018, 64, 255–261. [Google Scholar] [CrossRef] [Green Version]
- Wan, J.; Zhang, S.; Stacey, G. Activation of a mitogen-activated protein kinase pathway in Arabidopsis by chitin. Mol. Plant Pathol. 2004, 5, 125–135. [Google Scholar] [CrossRef]
- Yamada, K.; Yamaguchi, K.; Shirakawa, T.; Nakagami, H.; Mine, A.; Ishikawa, K.; Fujiwara, M.; Narusaka, M.; Narusaka, Y.; Ichimura, K.; et al. The Arabidopsis CERK 1-associated kinase PBL 27 connects chitin perception to MAPK activation. EMBO J. 2016, 35, 2468–2483. [Google Scholar] [CrossRef]
- Ichimura, K.; Casais, C.; Peck, S.C.; Shinozaki, K.; Shirasu, K. MEKK1 is required for MPK4 activation and regulates tissue-specific and temperature-dependent cell death in Arabidopsis. J. Biol. Chem. 2006, 281, 36969–36976. [Google Scholar] [CrossRef] [Green Version]
- Mészáros, T.; Helfer, A.; Hatzimasoura, E.; Magyar, Z.; Serazetdinova, L.; Rios, G.; Bardóczy, V.; Teige, M.; Koncz, C.; Peck, S.; et al. The Arabidopsis MAP kinase kinase MKK1 participates in defence responses to the bacterial elicitor flagellin. Plant J. 2006, 48, 485–498. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dóczi, R.; Brader, G.; Pettkó-Szandtner, A.; Rajh, I.; Djamei, A.; Pitzschke, A.; Teige, M.; Hirt, H. The Arabidopsis mitogen-activated protein kinase kinase MKK3 is upstream of group C mitogen-activated protein kinases and participates in pathogen signaling. Plant Cell 2007, 19, 3266–3279. [Google Scholar] [CrossRef] [Green Version]
- Suarez-Rodriguez, M.C.; Adams-Phillips, L.; Liu, Y.; Wang, H.; Su, S.H.; Jester, P.J.; Zhang, S.; Bent, A.F.; Krysan, P.J. MEKK1 is required for flg22-induced MPK4 activation in Arabidopsis plants. Plant Physiol. 2007, 143, 661–669. [Google Scholar] [CrossRef] [Green Version]
- Yu, L.; Nie, J.; Cao, C.; Jin, Y.; Yan, M.; Wang, F.; Liu, J.; Xiao, Y.; Liang, Y.; Zhang, W. Phosphatidic acid mediates salt stress response by regulation of MPK6 in Arabidopsis thaliana. New Phytol. 2010, 188, 762–773. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Ngwenyama, N.; Liu, Y.; Walker, J.C.; Zhang, S. Stomatal development and patterning are regulated by environmentally responsive mitogen-activated protein kinases in Arabidopsis. Plant Cell 2007, 19, 63–73. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bishop, A.C.; Ubersax, J.A.; Petsch, D.T.; Matheos, D.P.; Gray, N.S.; Blethrow, J.; Shimizu, E.; Tsien, J.Z.; Schultz, P.G.; Rose, M.D.; et al. A chemical switch for inhibitor-sensitive alleles of any protein kinase. Nature 2000, 407, 395–401. [Google Scholar] [CrossRef]
- Xu, J.; Xie, J.; Yan, C.; Zou, X.; Ren, D.; Zhang, S. A chemical genetic approach demonstrates that MPK 3/MPK 6 activation and NADPH oxidase-mediated oxidative burst are two independent signaling events in plant immunity. Plant J. 2014, 77, 222–234. [Google Scholar] [CrossRef]
- Clough, S.J.; Bent, A.F. Floral dip: A simplified method for Agrobacterium-mediated transformation of Arabidopsis thaliana. Plant J. 1998, 16, 735–743. [Google Scholar] [CrossRef] [Green Version]
- Schindelin, J.; Arganda-Carreras, I.; Frise, E.; Kaynig, V.; Longair, M.; Pietzsch, T.; Preibisch, S.; Rueden, C.; Saalfeld, S.; Schmid, B.; et al. Fiji: An open-source platform for biological-image analysis. Nat. Methods 2012, 9, 676–682. [Google Scholar] [CrossRef] [Green Version]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| No Kinase | CA-MPK6 | CA-MPK4 | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Turq | YPet | YPet/Turq | Turq | YPet | Turq | Δ FRET | Turq | YPet | Turq | Δ FRET | |
| EKAREV | 102,547 | 76,365 | 0.74 | 106,420 | 86,561 | 0.81 | 9% | 111,014 | 82,988 | 0.75 | 0% |
| SOMA | 77,325 | 69,725 | 0.90 | 63,494 | 96,300 | 1.52 | 68% | 73,668 | 84,687 | 1.15 | 27% |
| EKAREV-BASL | 81,070 | 58,495 | 0.72 | 56,506 | 87,706 | 1.55 | 115% | 62,386 | 83,222 | 1.33 | 85% |
| EKAREVT48A-BASL | 75,400 | 55,327 | 0.73 | 84,693 | 60,990 | 0.72 | −2% | 83,782 | 59,807 | 0.71 | −3% |
| SOMAT679A | 65,166 | 60,931 | 0.94 | 71,900 | 65,530 | 0.91 | −3% | 67,915 | 62,828 | 0.93 | −1% |
| EKAREV-AP2C1 | 89,861 | 66,267 | 0.74 | 55,116 | 101,647 | 1.84 | 150% | 55,971 | 99,090 | 1.77 | 140% |
| EKAREVT48A-AP2C1 | 87,272 | 66,980 | 0.77 | 97,788 | 74,682 | 0.76 | 0% | 95,430 | 73,391 | 0.77 | 0% |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Seitz, K.; Krysan, P.J. Expanding the Toolkit of Fluorescent Biosensors for Studying Mitogen Activated Protein Kinases in Plants. Int. J. Mol. Sci. 2020, 21, 5350. https://doi.org/10.3390/ijms21155350
Seitz K, Krysan PJ. Expanding the Toolkit of Fluorescent Biosensors for Studying Mitogen Activated Protein Kinases in Plants. International Journal of Molecular Sciences. 2020; 21(15):5350. https://doi.org/10.3390/ijms21155350
Chicago/Turabian StyleSeitz, Kati, and Patrick J. Krysan. 2020. "Expanding the Toolkit of Fluorescent Biosensors for Studying Mitogen Activated Protein Kinases in Plants" International Journal of Molecular Sciences 21, no. 15: 5350. https://doi.org/10.3390/ijms21155350
APA StyleSeitz, K., & Krysan, P. J. (2020). Expanding the Toolkit of Fluorescent Biosensors for Studying Mitogen Activated Protein Kinases in Plants. International Journal of Molecular Sciences, 21(15), 5350. https://doi.org/10.3390/ijms21155350