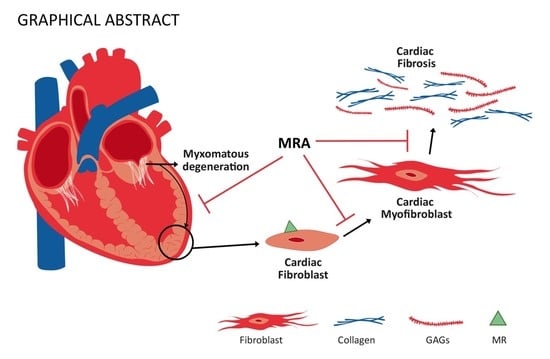
Beneficial Effects of Mineralocorticoid Receptor Antagonism on Myocardial Fibrosis in an Experimental Model of the Myxomatous Degeneration of the Mitral Valve

 , , ,
, , , 
Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
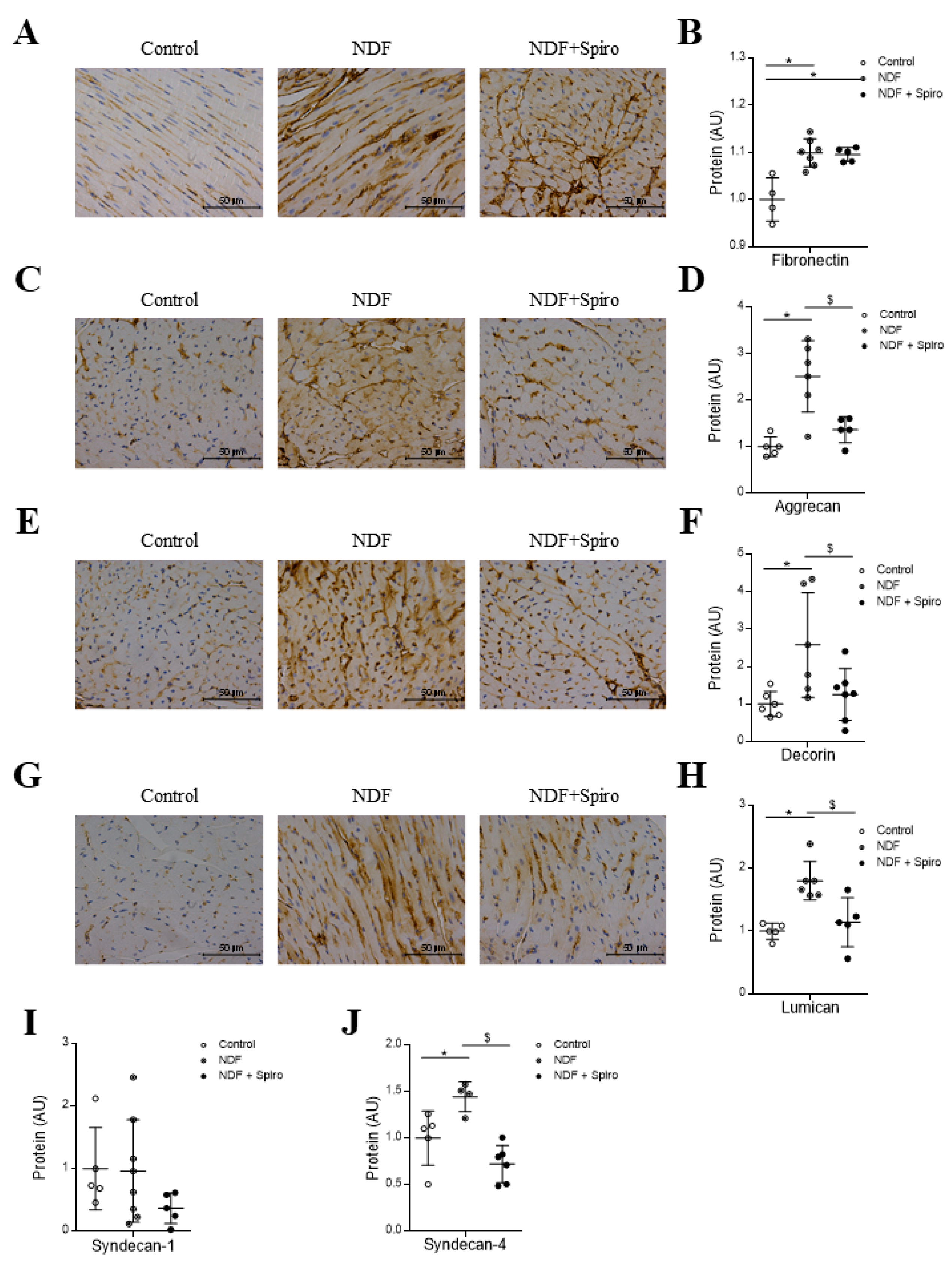
2.1. Effects of Spironolactone on Cardiac Fibrillar Proteins and Fibrotic Markers in a Murine Experimental Model of Fibromyxomatous Degeneration of the Mitral Valve
2.2. Effects of Spironolactone on Cardiac Non-Fibrillar Proteins in A Murine Experimental Model of the Fibromyxomatous Degeneration of the Mitral Valve
2.3. Mineralocorticoid Receptor Mediates the Profibrotic Response of Human Cardiac Fibroblasts to NDF
3. Discussion
4. Materials and Methods
4.1. In Vivo Studies
4.2. Cell Culture
4.3. Real-Time Reverse Transcription PCR
4.4. Western Blot Analysis
4.5. Immunohistological Evaluation
4.6. ELISA
4.7. Statistical Analyses
5. Conclusions
5.1. Translational Perspective
5.2. Limitations
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| MVP | Mitral valve prolapse |
| NDF | Nordexfenfluramine |
| MR | Mineralocorticoid receptor |
| MRA | Mineralocorticoid receptor antagonist |
| ECM | Extracellular matrix |
| HF | Heart failure |
References
- Delling, F.N.; Vasan, R.S. Epidemiology and pathophysiology of mitral valve prolapse: New insights into disease progression, genetics, and molecular basis. Circulation 2014, 129, 2158–2170. [Google Scholar] [CrossRef] [Green Version]
- Fuster, V.; Danielson, M.A.; Robb, R.A.; Broadbent, J.C.; Brown, A.L.J.; Elveback, L.R. Quantitation of left ventricular myocardial fiber hypertrophy and interstitial tissue in human hearts with chronically increased volume and pressure overload. Circulation 1977, 55, 504–508. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Edwards, N.C.; Moody, W.E.; Yuan, M.; Weale, P.; Neal, D.; Townend, J.N.; Steeds, R.P. Quantification of left ventricular interstitial fibrosis in asymptomatic chronic primary degenerative mitral regurgitation. Circ. Cardiovasc. Imaging 2014, 7, 946–953. [Google Scholar] [CrossRef] [Green Version]
- Szymczyk, E.; Wierzbowska-Drabik, K.; Drozdz, J.; Krzemińska-Pakuła, M. Mitral valve regurgitation is a powerful factor of left ventricular hypertrophy. Pol. Arch. Med. Wewn. 2008, 118, 478–483. [Google Scholar] [CrossRef] [PubMed]
- Kitkungvan, D.; Nabi, F.; Kim, R.J.; Bonow, R.O.; Khan, M.A.; Xu, J.; Little, S.H.; Quinones, M.A.; Lawrie, G.M.; Zoghbi, W.A.; et al. Myocardial Fibrosis in Patients With Primary Mitral Regurgitation With and Without Prolapse. J. Am. Coll. Cardiol. 2018, 72, 823–834. [Google Scholar] [CrossRef] [PubMed]
- Kong, P.; Christia, P.; Frangogiannis, N.G. The pathogenesis of cardiac fibrosis. Cell. Mol. Life Sci. 2014, 71, 549–574. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rodriguez, P.; Sassi, Y.; Troncone, L.; Benard, L.; Ishikawa, K.; Gordon, R.E.; Lamas, S.; Laborda, J.; Hajjar, R.J.; Lebeche, D. Deletion of delta-like 1 homologue accelerates fibroblast-myofibroblast differentiation and induces myocardial fibrosis. Eur. Heart J. 2019, 40, 967–978. [Google Scholar] [CrossRef]
- Furtado, M.B.; Costa, M.W.; Rosenthal, N.A. The cardiac fibroblast: Origin, identity and role in homeostasis and disease. Differentiation 2016, 92, 93–101. [Google Scholar] [CrossRef]
- Porter, K.E.; Turner, N.A. Cardiac fibroblasts: At the heart of myocardial remodeling. Pharmacol. Ther. 2009, 123, 255–278. [Google Scholar] [CrossRef]
- Chute, M.; Aujla, P.; Jana, S.; Kassiri, Z. The Non-Fibrillar Side of Fibrosis: Contribution of the Basement Membrane, Proteoglycans, and Glycoproteins to Myocardial Fibrosis. J. Cardiovasc. Dev. Dis. 2019, 6, 35. [Google Scholar] [CrossRef] [Green Version]
- MacFadyen, R.J.; Barr, C.S.; Struthers, A.D. Aldosterone blockade reduces vascular collagen turnover, improves heart rate variability and reduces early morning rise in heart rate in heart failure patients. Cardiovasc. Res. 1997, 35, 30–34. [Google Scholar] [CrossRef] [Green Version]
- Zannad, F.; Alla, F.; Dousset, B.; Perez, A.; Pitt, B. Limitation of excessive extracellular matrix turnover may contribute to survival benefit of spironolactone therapy in patients with congestive heart failure: Insights from the randomized aldactone evaluation study (RALES). Circulation 2000, 102, 2700–2706. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Iraqi, W.; Rossignol, P.; Angioi, M.; Fay, R.; Nuée, J.; Ketelslegers, J.M.; Vincent, J.; Pitt, B.; Zannad, F. Extracellular cardiac matrix biomarkers in patients with acute myocardial infarction complicated by left ventricular dysfunction and heart failure: Insights from the Eplerenone Post-Acute Myocardial Infarction Heart Failure Efficacy and Survival Study (EPHESUS) study. Circulation 2009, 119, 2471–2479. [Google Scholar] [PubMed] [Green Version]
- Zannad, F.; McMurray, J.J.V.; Krum, H.; van Veldhuisen, D.J.; Swedberg, K.; Shi, H.; Vincent, J.; Pocock, S.J.; Pitt, B. EMPHASIS-HF Study Group Eplerenone in Patients with Systolic Heart Failure and Mild Symptoms. N. Engl. J. Med. 2011, 364, 11–21. [Google Scholar] [CrossRef] [Green Version]
- Elangbam, C.S.; Job, L.E.; Zadrozny, L.M.; Barton, J.C.; Yoon, L.W.; Gates, L.D.; Slocum, N. 5-hydroxytryptamine (5HT)-induced valvulopathy: Compositional valvular alterations are associated with 5HT2B receptor and 5HT transporter transcript changes in Sprague-Dawley rats. Exp. Toxicol. Pathol. 2008, 60, 253–262. [Google Scholar] [CrossRef]
- Fitzgerald, L.W.; Burn, T.C.; Brown, B.S.; Patterson, J.P.; Corjay, M.H.; Valentine, P.A.; Sun, J.H.; Link, J.R.; Abbaszade, I.; Hollis, J.M.; et al. Possible role of valvular serotonin 5-HT(2B) receptors in the cardiopathy associated with fenfluramine. Mol. Pharmacol. 2000, 57, 75–81. [Google Scholar]
- Rothman, R.B.; Baumann, M.H.; Savage, J.E.; Rauser, L.; McBride, A.; Hufeisen, S.J.; Roth, B.L. Evidence for possible involvement of 5-HT(2B) receptors in the cardiac valvulopathy associated with fenfluramine and other serotonergic medications. Circulation 2000, 102, 2836–2841. [Google Scholar] [CrossRef] [Green Version]
- Fowles, R.E.; Cloward, T.V.; Yowell, R.L. Endocardial fibrosis associated with fenfluramine-phentermine. N. Engl. J. Med. 1998, 338, 1316–1317. [Google Scholar] [CrossRef]
- Mann, D.A.; Oakley, F. Serotonin paracrine signaling in tissue fibrosis. Biochim. Biophys. Acta 2013, 1832, 905–910. [Google Scholar] [CrossRef] [Green Version]
- Shyu, K.-G.G. Serotonin 5-HT2B receptor in cardiac fibroblast contributes to cardiac hypertrophy: A new therapeutic target for heart failure? Circ. Res. 2009, 104, 1–3. [Google Scholar] [CrossRef] [Green Version]
- Hutcheson, J.D.; Setola, V.; Roth, B.L.; Merryman, W.D. Serotonin receptors and heart valve disease—It was meant 2B. Pharmacol. Ther. 2011, 132, 146–157. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Connolly, J.M.; Bakay, M.A.; Fulmer, J.T.; Gorman, R.C.; Gorman, J.H.; Oyama, M.A.; Levy, R.J. Fenfluramine disrupts the mitral valve interstitial cell response to serotonin. Am. J. Pathol. 2009, 175, 988–997. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yao, W.; Mu, W.; Zeifman, A.; Lofti, M.; Remillard, C.V.; Makino, A.; Perkins, D.L.; Garcia, J.G.N.; Yuan, J.X.J.; Zhang, W. Fenfluramine-Induced gene dysregulation in human pulmonary artery smooth muscle and endothelial cells. Pulm. Circ. 2011, 1, 405–418. [Google Scholar] [CrossRef] [Green Version]
- Ayme-Dietrich, E.; Lawson, R.; Côté, F.; de Tapia, C.; Da Silva, S.; Ebel, C.; Hechler, B.; Gachet, C.; Guyonnet, J.; Rouillard, H.; et al. The role of 5-HT2B receptors in mitral valvulopathy: Bone marrow mobilization of endothelial progenitors. Br. J. Pharmacol. 2017, 174, 4123–4139. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ibarrola, J.; Garcia-Peña, A.; Matilla, L.; Bonnard, B.; Sádaba, R.; Arrieta, V.; Alvarez, V.; Fernández-Celis, A.; Gainza, A.; Navarro, A.; et al. A New Role for the Aldosterone/Mineralocorticoid Receptor Pathway in the Development of Mitral Valve Prolapse. Circ. Res. 2020, 127, e80–e93. [Google Scholar] [CrossRef] [PubMed]
- Jaisser, F.; Farman, N. Emerging Roles of the Mineralocorticoid Receptor in Pathology: Toward New Paradigms in Clinical Pharmacology. Pharmacol. Rev. 2016, 68, 49–75. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- van den Berg, T.N.A.; Rongen, G.A.; Fröhlich, G.M.; Deinum, J.; Hausenloy, D.J.; Riksen, N.P. The cardioprotective effects of mineralocorticoid receptor antagonists. Pharmacol. Ther. 2014, 142, 72–87. [Google Scholar] [CrossRef]
- Wang, D.; Liu, Y.H.; Yang, X.P.; Rhaleb, N.E.; Xu, J.; Peterson, E.; Rudolph, A.E.; Carretero, O.A. Role of a selective aldosterone blocker in mice with chronic heart failure. J. Card. Fail. 2004, 10, 67–73. [Google Scholar] [CrossRef]
- Fraccarollo, D.; Galuppo, P.; Sieweke, J.T.; Napp, L.C.; Grobbecker, P.; Bauersachs, J. Efficacy of mineralocorticoid receptor antagonism in the acute myocardial infarction phase: Eplerenone versus spironolactone. ESC Heart Fail. 2015, 2, 150–158. [Google Scholar] [CrossRef]
- Parviz, Y.; Iqbal, J.; Pitt, B.; Adlam, D.; Al-Mohammad, A.; Zannad, F. Emerging cardiovascular indications of mineralocorticoid receptor antagonists. Trends Endocrinol. Metab. 2015, 26, 201–211. [Google Scholar] [CrossRef]
- Engebretsen, K.V.T.T.; Lunde, I.G.; Strand, M.E.; Waehre, A.; Sjaastad, I.; Marstein, H.S.; Skrbic, B.; Dahl, C.P.; Askevold, E.T.; Christensen, G.; et al. Lumican is increased in experimental and clinical heart failure, and its production by cardiac fibroblasts is induced by mechanical and proinflammatory stimuli. FEBS J. 2013, 280, 2382–2398. [Google Scholar] [CrossRef] [PubMed]
- Matsui, Y.; Ikesue, M.; Danzaki, K.; Morimoto, J.; Sato, M.; Tanaka, S.; Kojima, T.; Tsutsui, H.; Uede, T. Syndecan-4 prevents cardiac rupture and dysfunction after myocardial infarction. Circ. Res. 2011, 108, 1328–1339. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fejes-Tóth, G.; Náray-Fejes-Tóth, A. Early Aldosterone-Regulated Genes in Cardiomyocytes: Clues to Cardiac Remodeling? Endocrinology 2007, 148, 1502–1510. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Beeri, R.; Yosefy, C.; Guerrero, J.L.; Nesta, F.; Abedat, S.; Chaput, M.; del Monte, F.; Handschumacher, M.D.; Stroud, R.; Sullivan, S.; et al. Mitral Regurgitation Augments Post-Myocardial Infarction Remodeling: Failure of Hypertrophic Compensation. J. Am. Coll. Cardiol. 2008, 51, 476–486. [Google Scholar] [CrossRef] [Green Version]
- Pu, M.; Gao, Z.; Zhang, X.; Liao, D.; Pu, D.K.; Brennan, T.; Davidson, W.R. Impact of mitral regurgitation on left ventricular anatomic and molecular remodeling and systolic function: Implication for outcome. Am. J. Physiol.-Heart Circ. Physiol. 2009, 296, H1727–H1732. [Google Scholar] [CrossRef]
- Kim, J.; Kochav, J.D.; Gurevich, S.; Afroz, A.; Petashnick, M.; Volo, S.; Diaz, B.; Okin, P.M.; Horn, E.; Devereux, R.B.; et al. Left ventricular geometric remodeling in relation to non-ischemic scar pattern on cardiac magnetic resonance imaging. Int. J. Cardiovasc. Imaging 2014, 30, 1559–1567. [Google Scholar] [CrossRef] [Green Version]
- Schelbert, E.B.; Wong, T.C.; Gheorghiade, M. Think small and examine the constituents of left ventricular hypertrophy and heart failure: Cardiomyocytes versus fibroblasts, collagen, and capillaries in the interstitium. J. Am. Heart Assoc. 2015, 4, e002188. [Google Scholar] [CrossRef] [Green Version]
- Von Lueder, T.G.; Wang, B.H.; Kompa, A.R.; Huang, L.; Webb, R.; Jordaan, P.; Atar, D.; Krum, H. Angiotensin receptor neprilysin inhibitor LCZ696 attenuates cardiac remodeling and dysfunction after myocardial infarction by reducing cardiac fibrosis and hypertrophy. Circ. Heart Fail. 2015, 8, 71–78. [Google Scholar] [CrossRef] [Green Version]
- Nishimura, R.A.; McGoon, M.D.; Shub, C.; Miller, F.A.; Ilstrup, D.M.; Tajik, A.J. Echocardiographically Documented Mitral-Valve Prolapse: Long-Term Follow-up of 237 Patients. N. Engl. J. Med. 1985, 313, 1305–1309. [Google Scholar] [CrossRef]
- Basso, C.; Perazzolo Marra, M.; Rizzo, S.; De Lazzari, M.; Giorgi, B.; Cipriani, A.; Frigo, A.C.; Rigato, I.; Migliore, F.; Pilichou, K.; et al. Arrhythmic Mitral Valve Prolapse and Sudden Cardiac Death. Circulation 2015, 132, 556–566. [Google Scholar] [CrossRef] [Green Version]
- Zia, M.I.; Valenti, V.; Cherston, C.; Criscito, M.; Uretsky, S.; Wolff, S. Relation of mitral valve prolapse to basal left ventricular hypertrophy as determined by cardiac magnetic resonance imaging. Am. J. Cardiol. 2012, 109, 1321–1325. [Google Scholar] [CrossRef] [PubMed]





© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ibarrola, J.; Garaikoetxea, M.; Garcia-Peña, A.; Matilla, L.; Jover, E.; Bonnard, B.; Cuesta, M.; Fernández-Celis, A.; Jaisser, F.; López-Andrés, N. Beneficial Effects of Mineralocorticoid Receptor Antagonism on Myocardial Fibrosis in an Experimental Model of the Myxomatous Degeneration of the Mitral Valve. Int. J. Mol. Sci. 2020, 21, 5372. https://doi.org/10.3390/ijms21155372
Ibarrola J, Garaikoetxea M, Garcia-Peña A, Matilla L, Jover E, Bonnard B, Cuesta M, Fernández-Celis A, Jaisser F, López-Andrés N. Beneficial Effects of Mineralocorticoid Receptor Antagonism on Myocardial Fibrosis in an Experimental Model of the Myxomatous Degeneration of the Mitral Valve. International Journal of Molecular Sciences. 2020; 21(15):5372. https://doi.org/10.3390/ijms21155372
Chicago/Turabian StyleIbarrola, Jaime, Mattie Garaikoetxea, Amaia Garcia-Peña, Lara Matilla, Eva Jover, Benjamin Bonnard, Maria Cuesta, Amaya Fernández-Celis, Frederic Jaisser, and Natalia López-Andrés. 2020. "Beneficial Effects of Mineralocorticoid Receptor Antagonism on Myocardial Fibrosis in an Experimental Model of the Myxomatous Degeneration of the Mitral Valve" International Journal of Molecular Sciences 21, no. 15: 5372. https://doi.org/10.3390/ijms21155372
APA StyleIbarrola, J., Garaikoetxea, M., Garcia-Peña, A., Matilla, L., Jover, E., Bonnard, B., Cuesta, M., Fernández-Celis, A., Jaisser, F., & López-Andrés, N. (2020). Beneficial Effects of Mineralocorticoid Receptor Antagonism on Myocardial Fibrosis in an Experimental Model of the Myxomatous Degeneration of the Mitral Valve. International Journal of Molecular Sciences, 21(15), 5372. https://doi.org/10.3390/ijms21155372