Apocynin Treatment Prevents Cardiac Connexin 43 Hemichannels Hyperactivity by Reducing Nitroso-Redox Stress in Mdx Mice



Abstract
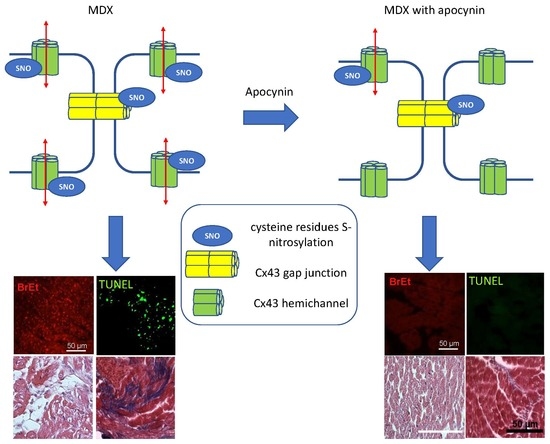
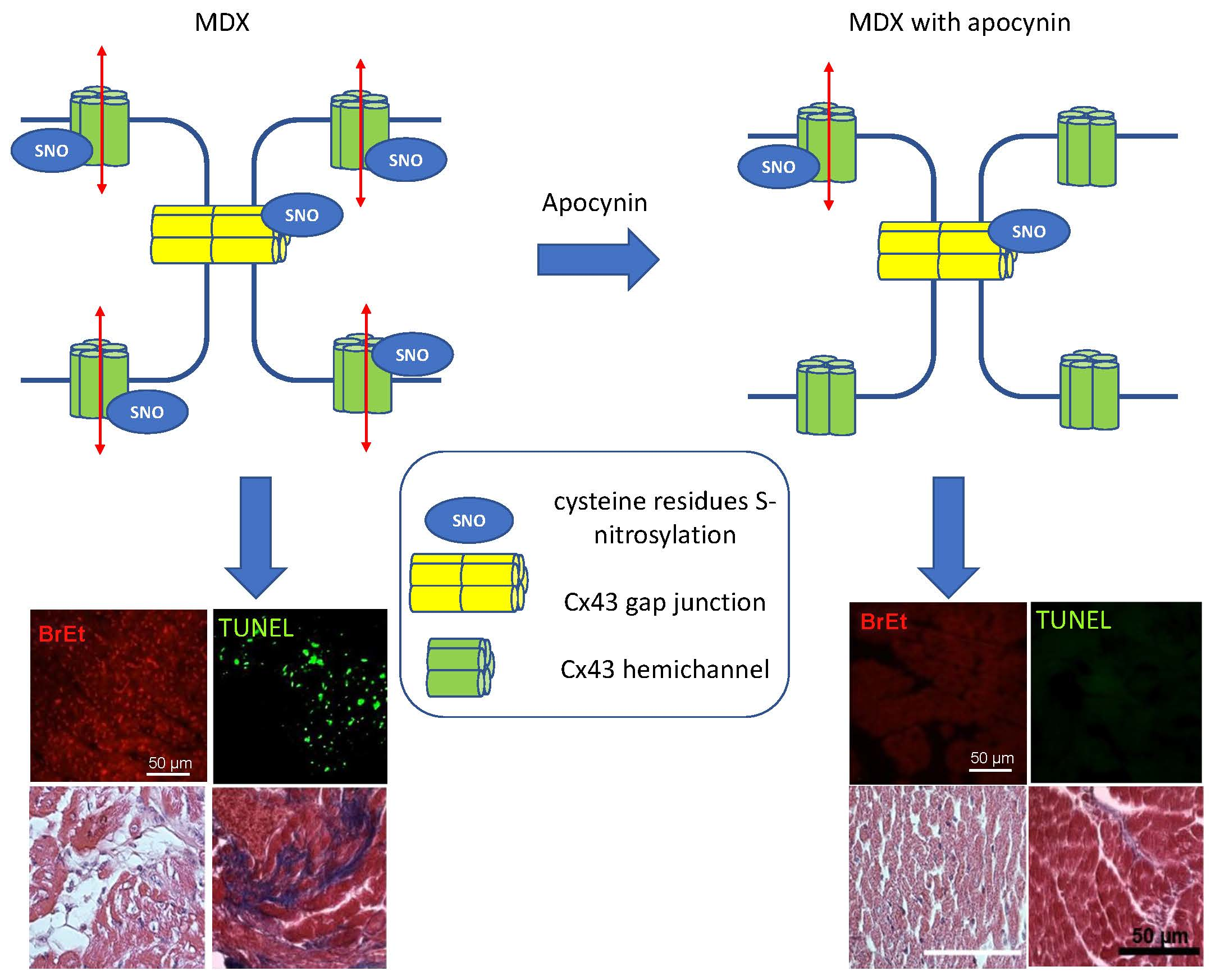
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
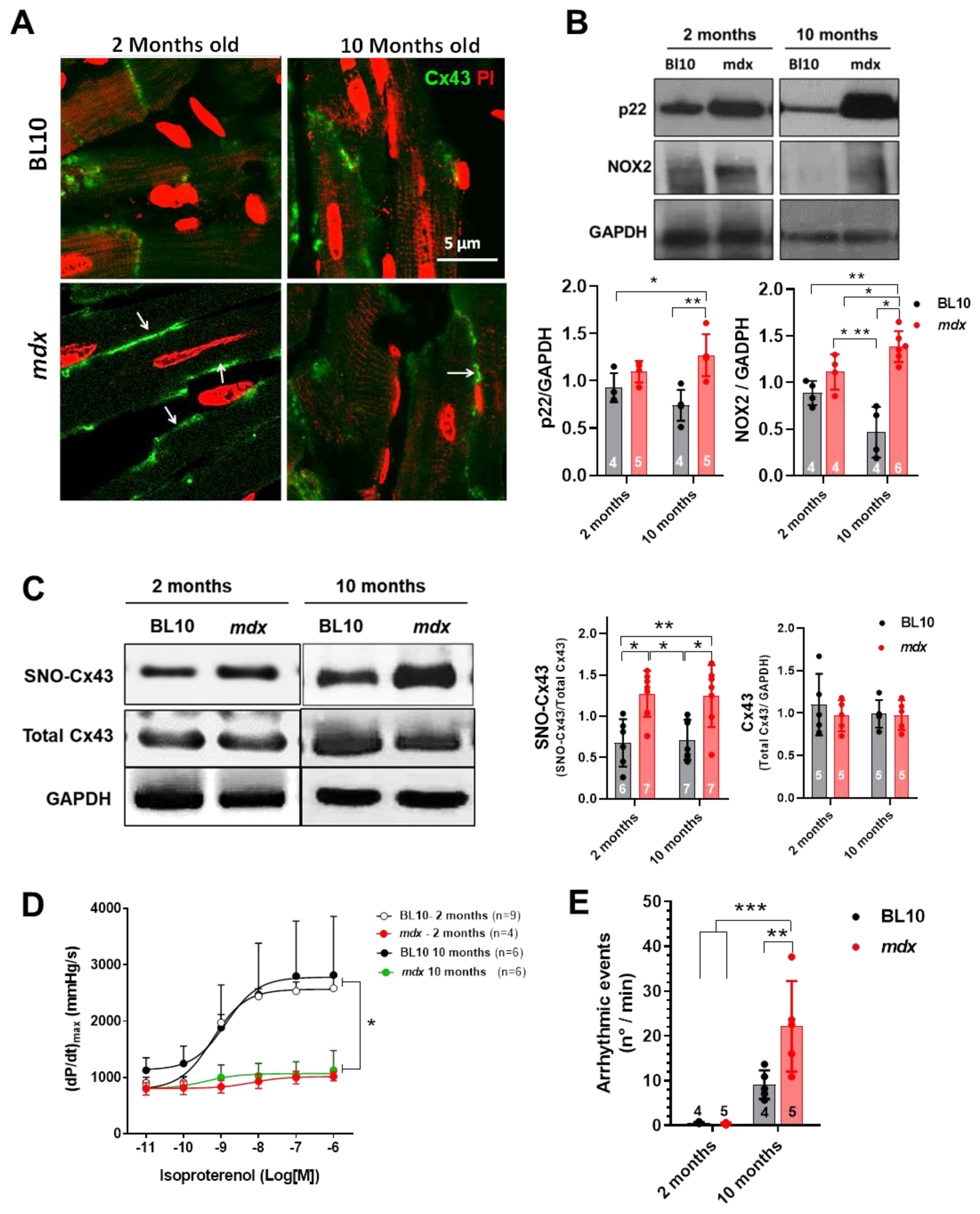
2.1. Cx43 is Lateralized and S-Nitrosylated in Mdx Hearts
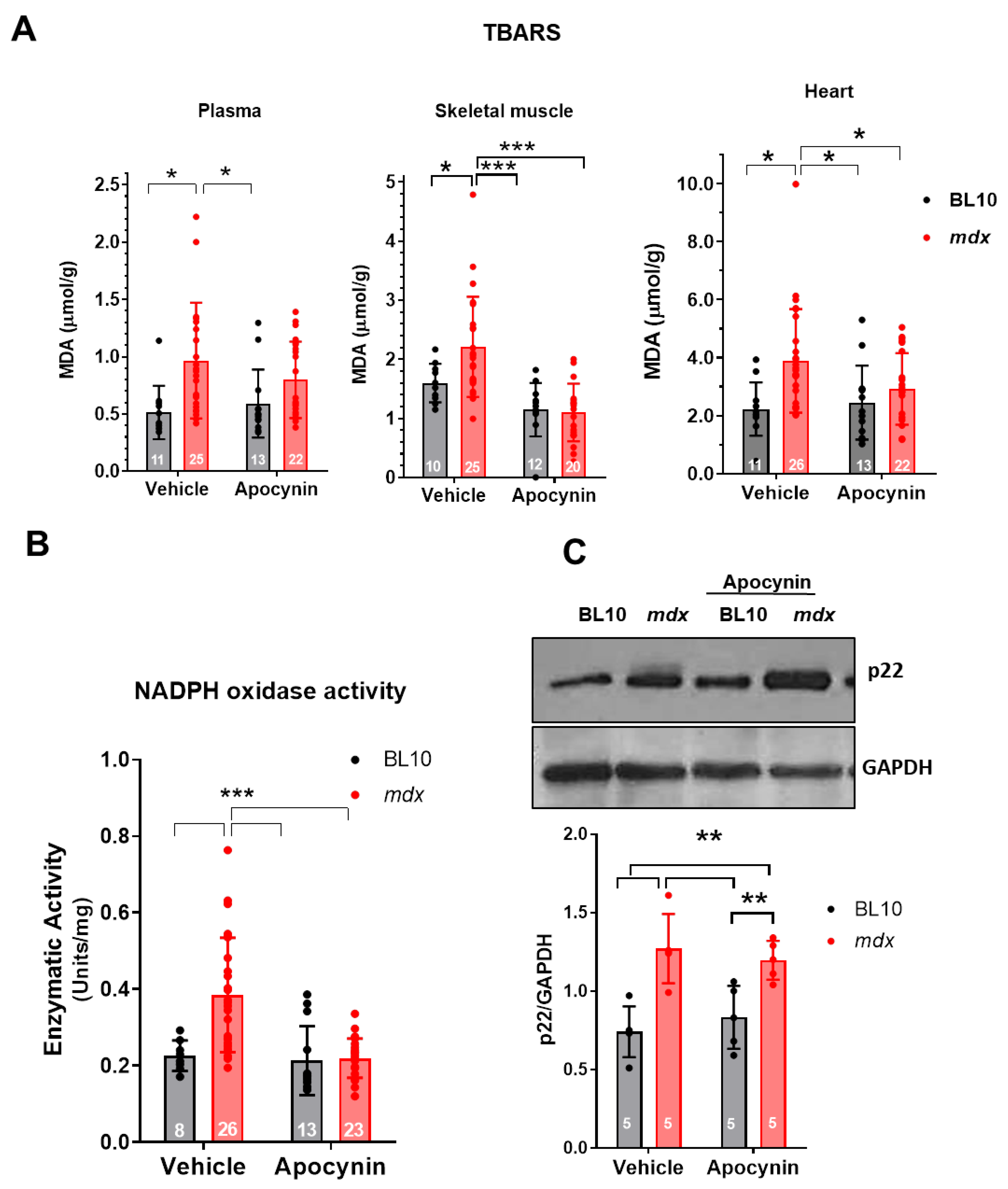
2.2. Reduction of Oxidative Stress in Mdx Mice after NOX Inhibition Treatment
2.3. Cx43 Hemichannels Activity in Mdx Hearts
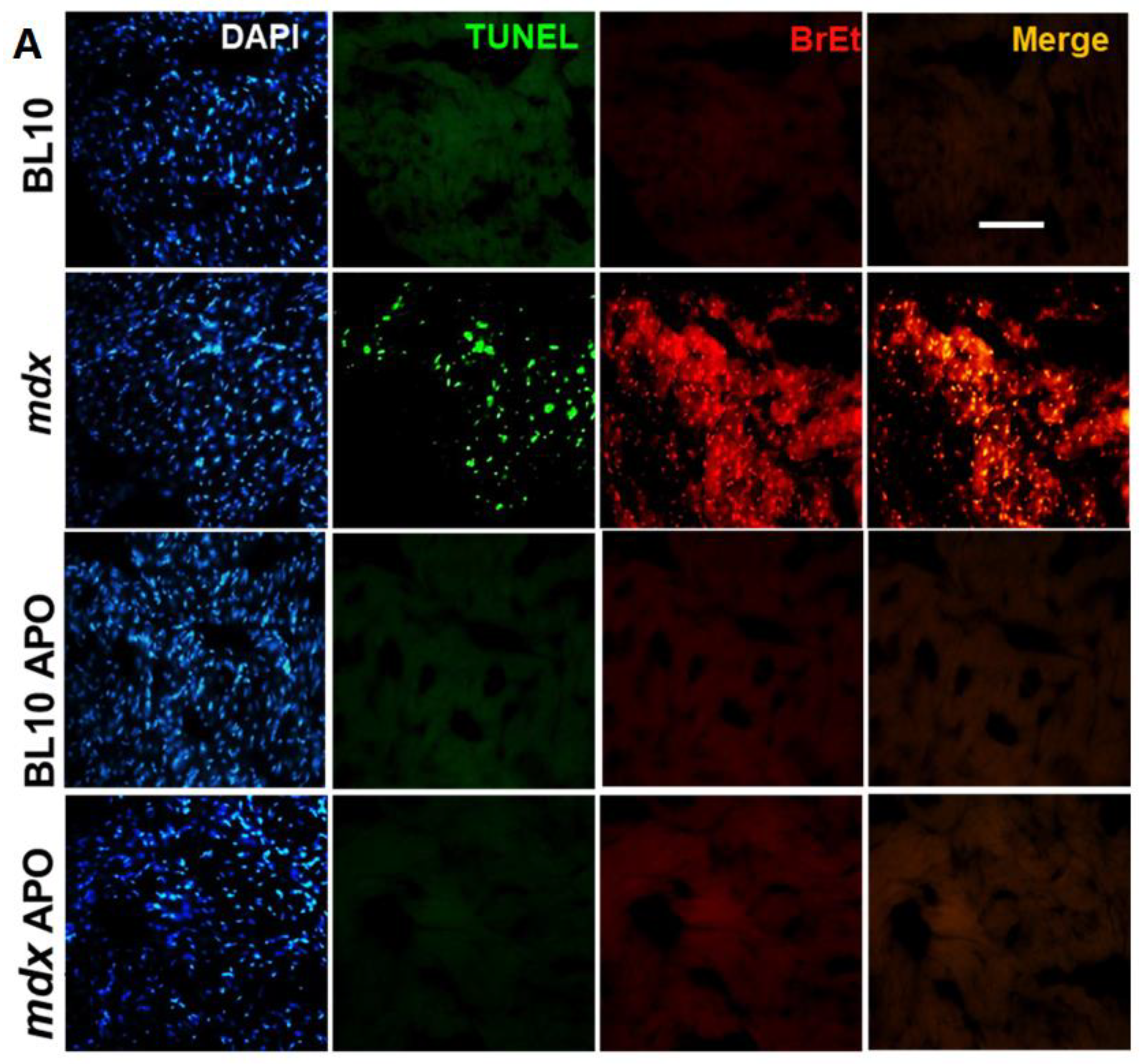
2.4. Apoptosis is Associated to Cx43 Hemichannels Activity
2.5. Mechanisms Involved in Cx43 Hemichannels Permeability
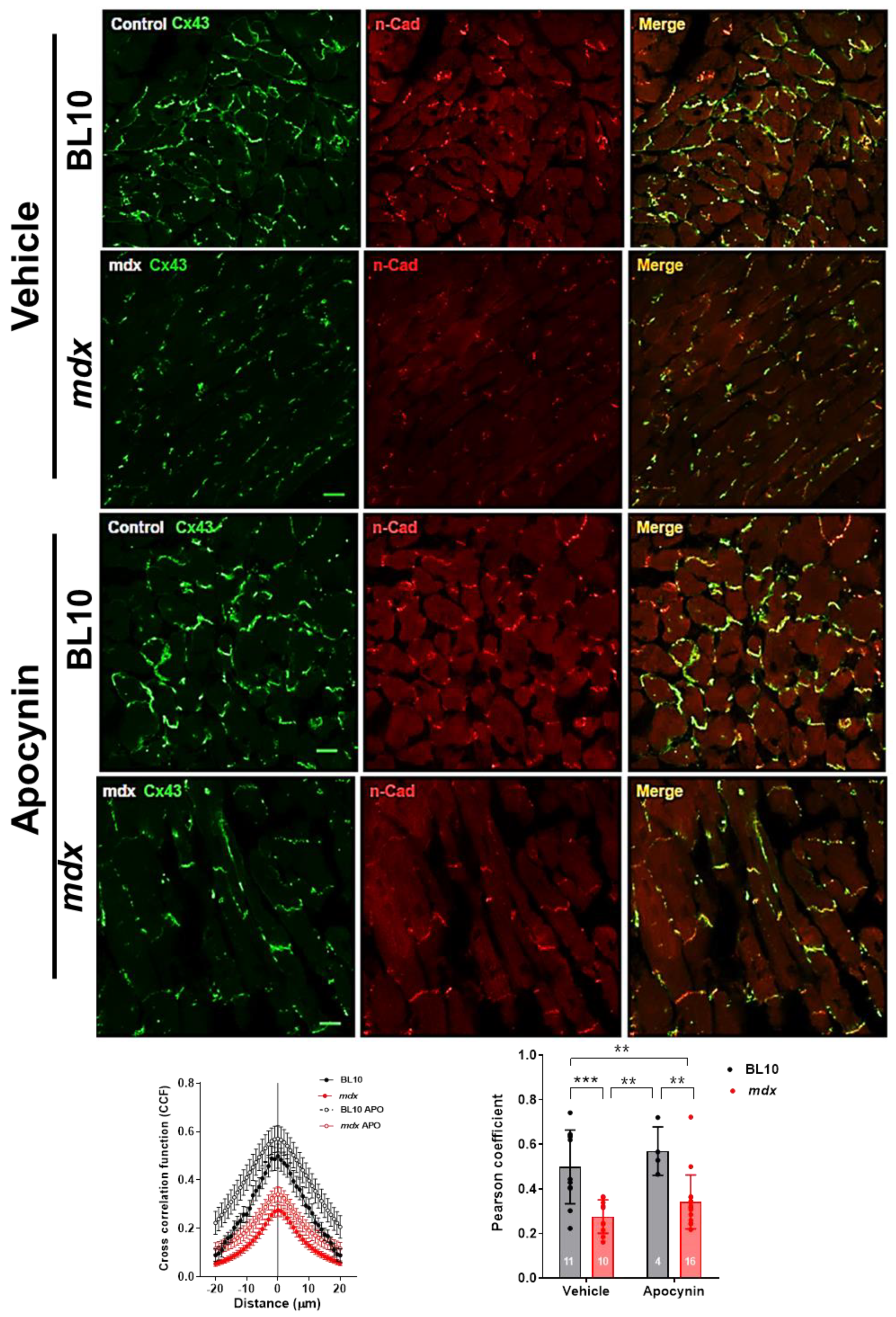
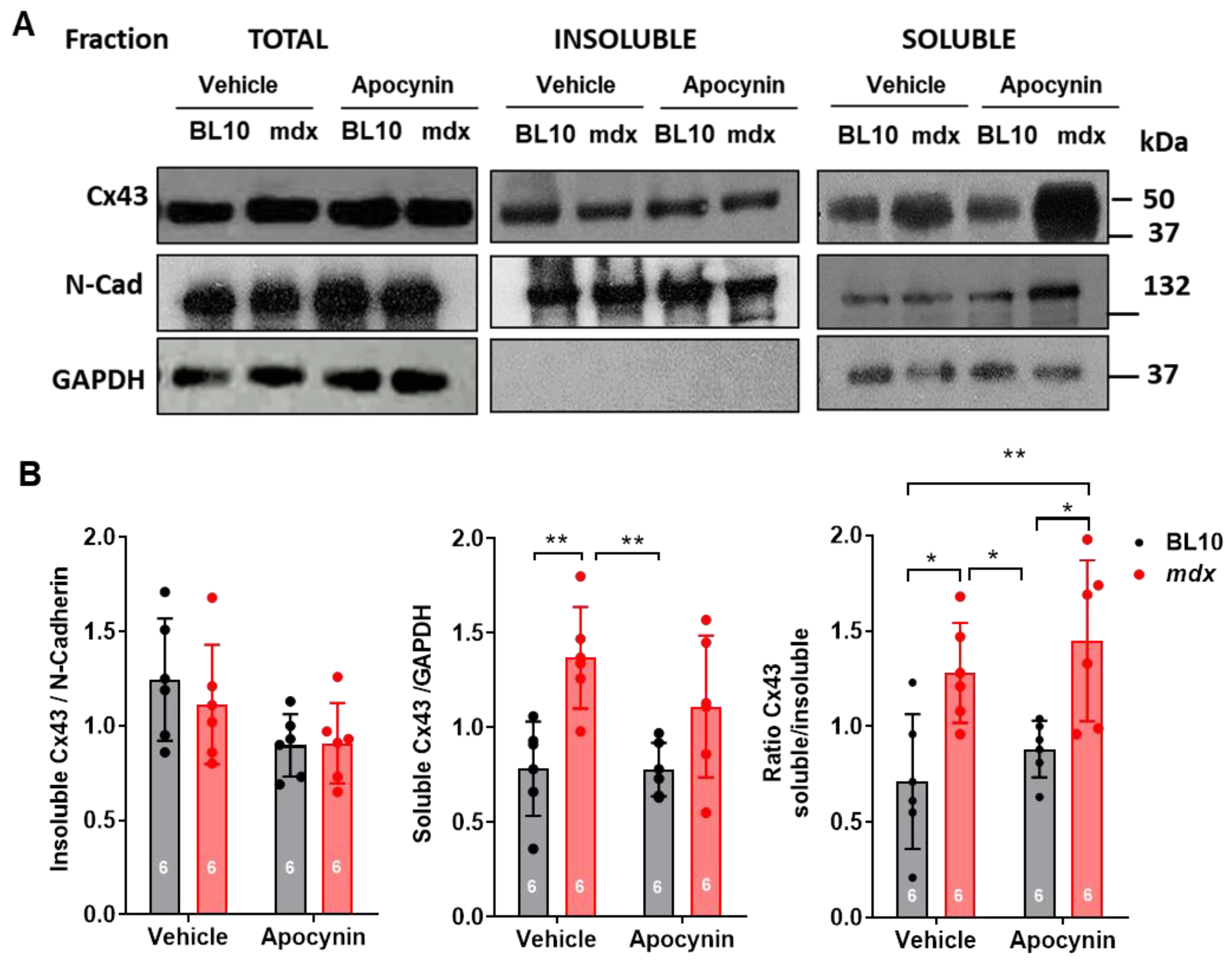
2.5.1. Lateralization of Cx43
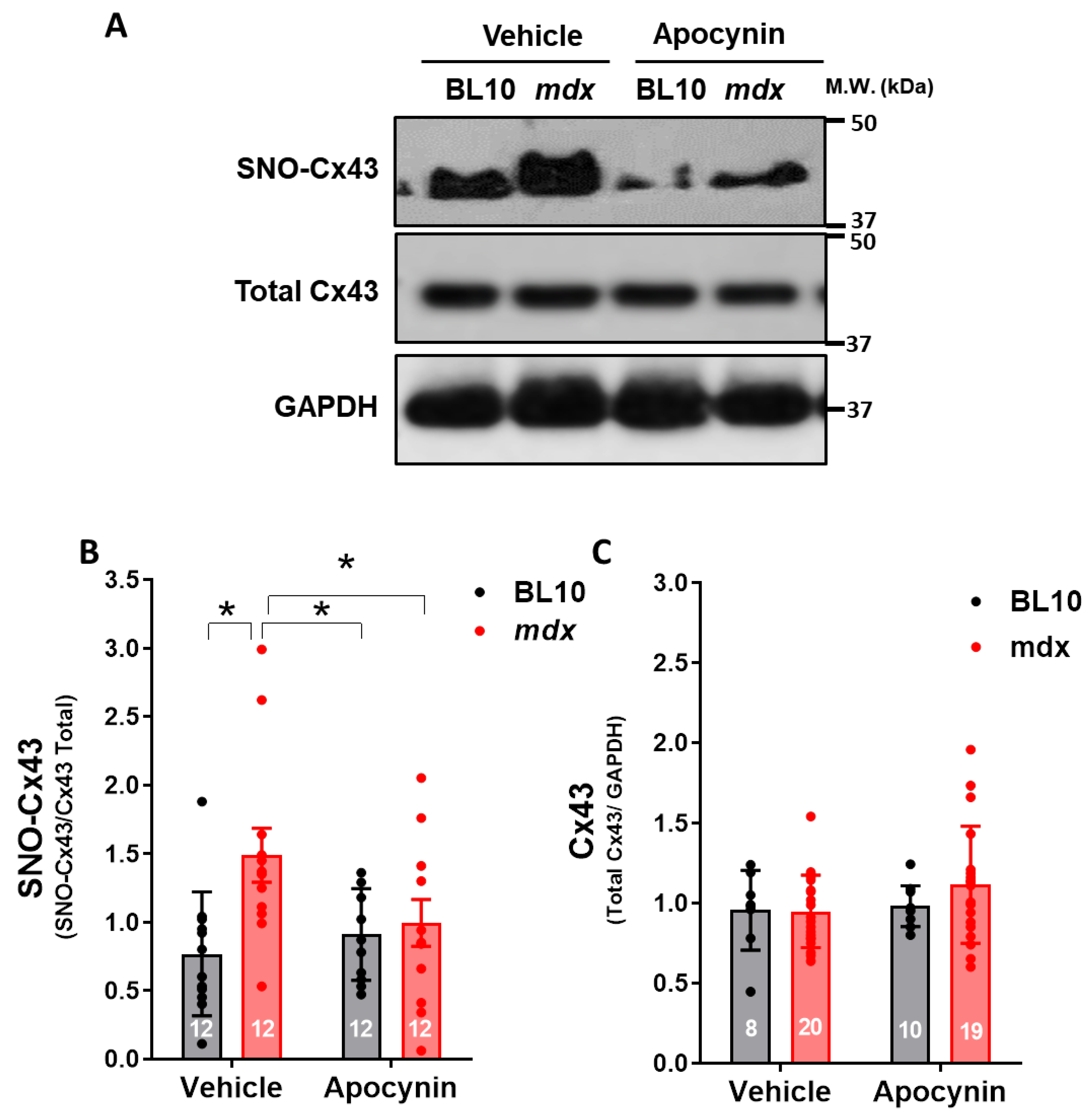
2.5.2. Hyper S-Nitrosylation of Cx43 is Prevented by NOX Inhibition
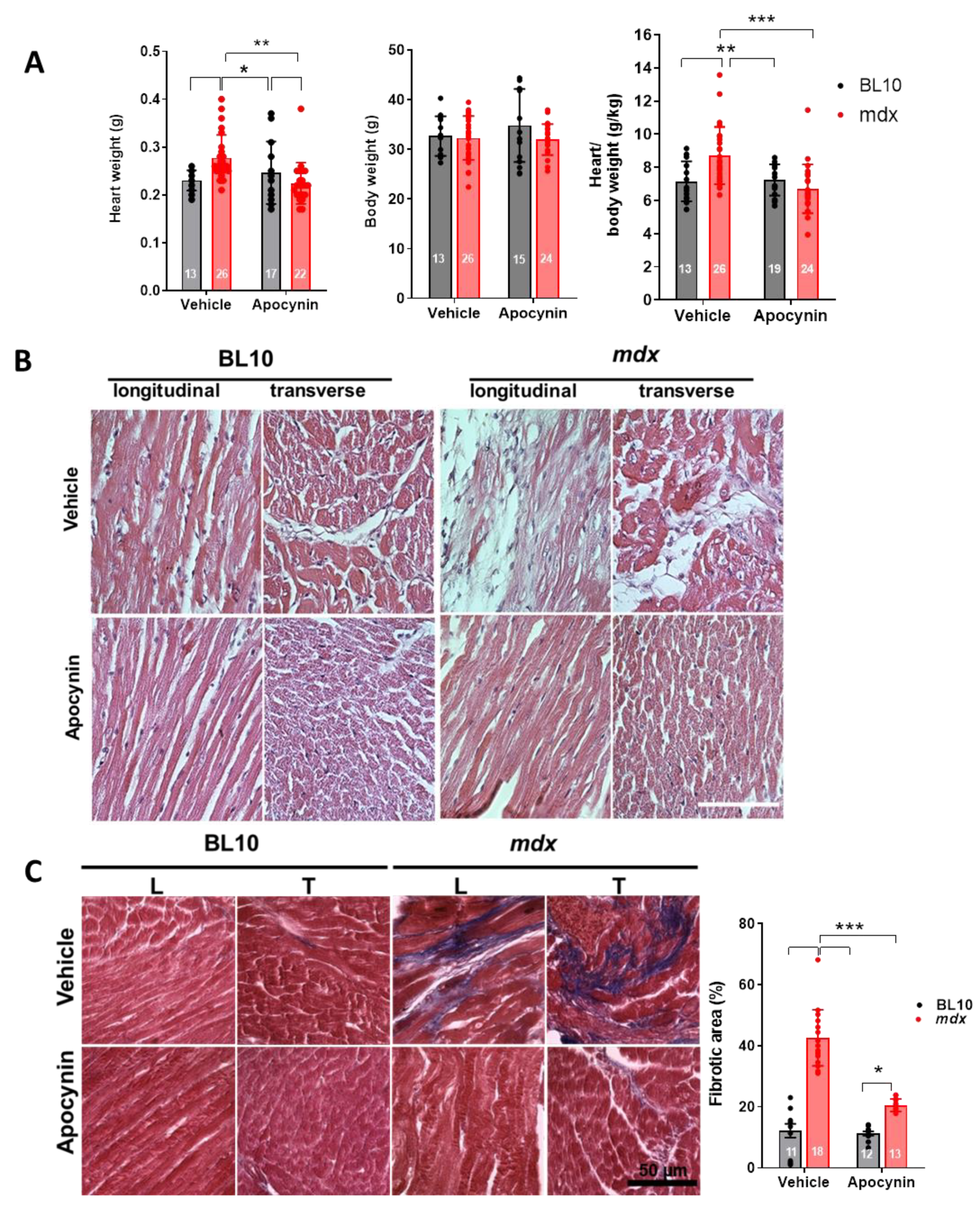
2.6. Ventricular Remodeling is Prevented by NADPH Oxidase Inhibition
3. Discussion
Limitations of the Study
4. Materials and Methods
4.1. Animals
4.2. Isolated Heart Preparation
4.3. Western Blotting
4.4. Quantification of S-Nitrosylated Proteins
4.5. Extraction of Non-Junctional and Junctional Proteins
4.6. Tissue Preparation for Histological Analysis
4.7. Immunofluorescence
4.8. Ethidium Uptake Assay
4.9. NADPH Oxidase Activity
4.10. TBARS Measurement
4.11. TUNEL Assay
4.12. Statistical Analysis
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| Cx43 | Connexin 43 |
| NOX | NADPH oxidase |
References
- Koenig, M.; Hoffman, E.P.; Bertelson, C.J.; Monaco, A.P.; Feener, C.; Kunkel, L.M. Complete cloning of the Duchenne muscular dystrophy (DMD) cDNA and preliminary genomic organization of the DMD gene in normal and affected individuals. Cell 1987, 50, 509–517. [Google Scholar] [CrossRef]
- Hoffman, E.P.; Brown, R.H., Jr.; Kunkel, L.M. Dystrophin: The protein product of the Duchenne muscular dystrophy locus. Cell 1987, 51, 919–928. [Google Scholar] [CrossRef]
- McNally, E.M.; Kaltman, J.R.; Benson, D.W.; Canter, C.E.; Cripe, L.H.; Duan, D.; Finder, J.D.; Groh, W.J.; Hoffman, E.P.; Judge, D.P.; et al. Contemporary cardiac issues in Duchenne muscular dystrophy. Working group of the national heart, lung, and blood institute in collaboration with parent project muscular dystrophy. Circulation 2015, 131, 1590–1598. [Google Scholar] [CrossRef] [Green Version]
- Guiraud, S.; Aartsma-Rus, A.; Vieira, N.M.; Davies, K.E.; van Ommen, G.J.; Kunkel, L.M. The pathogenesis and therapy of muscular dystrophies. Annu. Rev. Genom. Hum. Genet. 2015, 16, 281–308. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McNally, E.M. New approaches in the therapy of cardiomyopathy in muscular dystrophy. Annu. Rev. Med. 2007, 58, 75–88. [Google Scholar] [CrossRef]
- El-Aloul, B.; Altamirano-Diaz, L.; Zapata-Aldana, E.; Rodrigues, R.; Malvankar-Mehta, M.S.; Nguyen, C.T.; Campbell, C. Pharmacological therapy for the prevention and management of cardiomyopathy in Duchenne muscular dystrophy: A systematic review. Neuromuscul. Disord. 2017, 27, 4–14. [Google Scholar] [CrossRef]
- Falzarano, M.S.; Scotton, C.; Passarelli, C.; Ferlini, A. Duchenne muscular dystrophy: From diagnosis to therapy. Molecules 2015, 20, 18168–18184. [Google Scholar] [CrossRef] [Green Version]
- Williams, I.A.; Allen, D.G. The role of reactive oxygen species in the hearts of dystrophin-deficient mdx mice. Am. J. Physiol. Heart Circ. Physiol. 2007, 293, H1969–H1977. [Google Scholar] [CrossRef] [Green Version]
- Kuroda, J.; Sadoshima, J. NADPH oxidase and cardiac failure. J. Cardiovasc. Transl. Res. 2010, 3, 314–320. [Google Scholar] [CrossRef] [Green Version]
- Murdoch, C.E.; Zhang, M.; Cave, A.C.; Shah, A.M. NADPH oxidase-dependent redox signalling in cardiac hypertrophy, remodelling and failure. Cardiovasc. Res. 2006, 71, 208–215. [Google Scholar] [CrossRef] [Green Version]
- Gonzalez, D.R.; Treuer, A.V.; Lamirault, G.; Mayo, V.; Cao, Y.; Dulce, R.A.; Hare, J.M. NADPH oxidase-2 inhibition restores contractility and intracellular calcium handling and reduces arrhythmogenicity in dystrophic cardiomyopathy. Am. J. Physiol. Heart Circ. Physiol. 2014, 307, 710–721. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kyrychenko, S.; Polakova, E.; Kang, C.; Pocsai, K.; Ullrich, N.D.; Niggli, E.; Shirokova, N. Hierarchical accumulation of RyR post-translational modifications drives disease progression in dystrophic cardiomyopathy. Cardiovasc. Res. 2013, 97, 666–675. [Google Scholar] [CrossRef] [Green Version]
- Prosser, B.L.; Ward, C.W.; Lederer, W.J. X-ROS signaling: Rapid mechano-chemo transduction in heart. Science 2011, 333, 1440–1445. [Google Scholar] [CrossRef] [Green Version]
- Kuroda, J.; Ago, T.; Matsushima, S.; Zhai, P.; Schneider, M.D.; Sadoshima, J. NADPH oxidase 4 (Nox4) is a major source of oxidative stress in the failing heart. Proc. Natl. Acad. Sci. USA 2010, 107, 15565–15570. [Google Scholar] [CrossRef] [Green Version]
- Smyth, J.W.; Hong, T.T.; Gao, D.; Vogan, J.M.; Jensen, B.C.; Fong, T.S.; Simpson, P.C.; Stainier, D.Y.; Chi, N.C.; Shaw, R.M. Limited forward trafficking of connexin 43 reduces cell-cell coupling in stressed human and mouse myocardium. J. Clin. Investig. 2010, 120, 266–279. [Google Scholar] [CrossRef] [Green Version]
- Tomaselli, G.F. Oxidant stress derails the cardiac connexon connection. J. Clin. Investig. 2010, 120, 87–89. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saez, J.C.; Berthoud, V.M.; Branes, M.C.; Martinez, A.D.; Beyer, E.C. Plasma membrane channels formed by connexins: Their regulation and functions. Physiol. Rev. 2003, 83, 1359–1400. [Google Scholar] [CrossRef] [Green Version]
- Orellana, J.A.; Diaz, E.; Schalper, K.A.; Vargas, A.A.; Bennett, M.V.; Saez, J.C. Cation permeation through connexin 43 hemichannels is cooperative, competitive and saturable with parameters depending on the permeant species. Biochem. Biophys. Res. Commun. 2011, 409, 603–609. [Google Scholar] [CrossRef] [Green Version]
- Shintani-Ishida, K.; Uemura, K.; Yoshida, K. Hemichannels in cardiomyocytes open transiently during ischemia and contribute to reperfusion injury following brief ischemia. Am. J. Physiol. Heart Circ. Physiol. 2007, 293, 1714–1720. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- D’Hondt, C.; Iyyathurai, J.; Vinken, M.; Rogiers, V.; Leybaert, L.; Himpens, B.; Bultynck, G. Regulation of connexin- and pannexin-based channels by post-translational modifications. Biol. Cell 2013, 105, 373–398. [Google Scholar] [CrossRef] [Green Version]
- Wang, N.; De Bock, M.; Antoons, G.; Gadicherla, A.K.; Bol, M.; Decrock, E.; Evans, W.H.; Sipido, K.R.; Bukauskas, F.F.; Leybaert, L. Connexin mimetic peptides inhibit Cx43 hemichannel opening triggered by voltage and intracellular Ca2+ elevation. Basic Res. Cardiol. 2012, 107, 304. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ismail, H.M.; Scapozza, L.; Ruegg, U.T.; Dorchies, O.M. Diapocynin, a dimer of the NADPH oxidase inhibitor apocynin, reduces ROS production and prevents force loss in eccentrically contracting dystrophic muscle. PLoS ONE 2014, 9, e110708. [Google Scholar] [CrossRef] [PubMed]
- John, S.A.; Kondo, R.; Wang, S.Y.; Goldhaber, J.I.; Weiss, J.N. Connexin-43 hemichannels opened by metabolic inhibition. J. Biol. Chem. 1999, 274, 236–240. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hesketh, G.G.; Shah, M.H.; Halperin, V.L.; Cooke, C.A.; Akar, F.G.; Yen, T.E.; Kass, D.A.; Machamer, C.E.; Van Eyk, J.E.; Tomaselli, G.F. Ultrastructure and regulation of lateralized connexin43 in the failing heart. Circ. Res. 2010, 106, 1153–1163. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Retamal, M.A.; Cortes, C.J.; Reuss, L.; Bennett, M.V.; Saez, J.C. S-nitrosylation and permeation through connexin 43 hemichannels in astrocytes: Induction by oxidant stress and reversal by reducing agents. Proc. Natl. Acad. Sci. USA 2006, 103, 4475–4480. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maejima, Y.; Kuroda, J.; Matsushima, S.; Ago, T.; Sadoshima, J. Regulation of myocardial growth and death by NADPH oxidase. J. Mol. Cell Cardiol. 2011, 50, 408–416. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, N.; De Vuyst, E.; Ponsaerts, R.; Boengler, K.; Palacios-Prado, N.; Wauman, J.; Lai, C.P.; De Bock, M.; Decrock, E.; Bol, M.; et al. Selective inhibition of Cx43 hemichannels by Gap19 and its impact on myocardial ischemia/reperfusion injury. Basic Res. Cardiol. 2013, 108, 309. [Google Scholar] [CrossRef] [Green Version]
- Chung, H.S.; Kim, G.E.; Holewinski, R.J.; Venkatraman, V.; Zhu, G.; Bedja, D.; Kass, D.A.; Van Eyk, J.E. Transient receptor potential channel 6 regulates abnormal cardiac S-nitrosylation in Duchenne muscular dystrophy. Proc. Natl. Acad. Sci. USA 2017, 114, e10763–e10771. [Google Scholar] [CrossRef] [Green Version]
- Straub, A.C.; Billaud, M.; Johnstone, S.R.; Best, A.K.; Yemen, S.; Dwyer, S.T.; Looft-Wilson, R.; Lysiak, J.J.; Gaston, B.; Palmer, L.; et al. Compartmentalized connexin 43 s-nitrosylation/denitrosylation regulates heterocellular communication in the vessel wall. Arter. Thromb. Vasc. Biol. 2011, 31, 399–407. [Google Scholar] [CrossRef] [Green Version]
- Lillo, M.A.; Himelman, E.; Shirokova, N.; Xie, L.H.; Fraidenraich, D.; Contreras, J.E. S-nitrosylation of connexin43 hemichannels elicits cardiac stress-induced arrhythmias in Duchenne muscular dystrophy mice. JCI Insight 2019, 4, 24. [Google Scholar] [CrossRef]
- Vielma, A.Z.; Leon, L.; Fernandez, I.C.; Gonzalez, D.R.; Boric, M.P. Nitric oxide synthase 1 modulates basal and beta-adrenergic-stimulated contractility by rapid and reversible redox-dependent S-Nitrosylation of the heart. PLoS ONE 2016, 11, e0160813. [Google Scholar] [CrossRef] [Green Version]
- Kyrychenko, S.; Kyrychenko, V.; Badr, M.A.; Ikeda, Y.; Sadoshima, J.; Shirokova, N. Pivotal role of miR-448 in the development of ROS-induced cardiomyopathy. Cardiovasc. Res. 2015, 108, 324–334. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kitajima, N.; Numaga-Tomita, T.; Watanabe, M.; Kuroda, T.; Nishimura, A.; Miyano, K.; Yasuda, S.; Kuwahara, K.; Sato, Y.; Ide, T.; et al. TRPC3 positively regulates reactive oxygen species driving maladaptive cardiac remodeling. Sci. Rep. 2016, 6, 37001. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, F.; Sugishita, K.; Su, Z.; Ueda, I.; Barry, W.H. Activation of connexin-43 hemichannels can elevate [Ca(2+)]i and [Na(+)]i in rabbit ventricular myocytes during metabolic inhibition. J. Mol. Cell. Cardiol. 2001, 33, 2145–2155. [Google Scholar] [CrossRef]
- Kondo, R.P.; Wang, S.Y.; John, S.A.; Weiss, J.N.; Goldhaber, J.I. Metabolic inhibition activates a non-selective current through connexin hemichannels in isolated ventricular myocytes. J. Mol. Cell. Cardiol. 2000, 32, 1859–1872. [Google Scholar] [CrossRef]
- Gonzalez, J.P.; Ramachandran, J.; Xie, L.H.; Contreras, J.E.; Fraidenraich, D. Selective connexin43 Inhibition prevents isoproterenol-induced arrhythmias and lethality in muscular dystrophy mice. Sci. Rep. 2015, 5, 13490. [Google Scholar] [CrossRef]
- Severs, N.J.; Bruce, A.F.; Dupont, E.; Rothery, S. Remodelling of gap junctions and connexin expression in diseased myocardium. Cardiovasc. Res. 2008, 80, 9–19. [Google Scholar] [CrossRef] [Green Version]
- Colussi, C.; Rosati, J.; Straino, S.; Spallotta, F.; Berni, R.; Stilli, D.; Rossi, S.; Musso, E.; Macchi, E.; Mai, A.; et al. Nepsilon-lysine acetylation determines dissociation from GAP junctions and lateralization of connexin 43 in normal and dystrophic heart. Proc. Natl. Acad. Sci. USA 2011, 108, 2795–2800. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fauconnier, J.; Thireau, J.; Reiken, S.; Cassan, C.; Richard, S.; Matecki, S.; Marks, A.R.; Lacampagne, A. Leaky RyR2 trigger ventricular arrhythmias in Duchenne muscular dystrophy. Proc. Natl. Acad. Sci. USA 2010, 107, 1559–1564. [Google Scholar] [CrossRef] [Green Version]
- Sanchez, G.; Araneda, F.; Pena, J.P.; Finkelstein, J.P.; Riquelme, J.A.; Montecinos, L.; Barrientos, G.; Llanos, P.; Pedrozo, Z.; Said, M.; et al. High-fat-diet-induced obesity produces spontaneous ventricular arrhythmias and increases the activity of ryanodine receptors in mice. Int. J. Mol. Sci. 2018, 19, 533. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Valdes, A.; Treuer, A.V.; Barrios, G.; Ponce, N.; Fuentealba, R.; Dulce, R.A.; Gonzalez, D.R. NOX inhibition improves beta-adrenergic stimulated contractility and intracellular calcium handling in the aged rat heart. Int. J. Mol. Sci. 2018, 19, 2404. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Khairallah, R.J.; Shi, G.; Sbrana, F.; Prosser, B.L.; Borroto, C.; Mazaitis, M.J.; Hoffman, E.P.; Mahurkar, A.; Sachs, F.; Sun, Y.; et al. Microtubules underlie dysfunction in duchenne muscular dystrophy. Sci. Signal. 2012, 5, ra56. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Prins, K.W.; Asp, M.L.; Zhang, H.; Wang, W.; Metzger, J.M. Microtubule-mediated misregulation of junctophilin-2 underlies t-tubule disruptions and calcium mishandling in mdx mice. JACC Basic Transl. Sci. 2016, 1, 122–130. [Google Scholar] [CrossRef] [Green Version]
- Shaw, R.M.; Fay, A.J.; Puthenveedu, M.A.; von Zastrow, M.; Jan, Y.N.; Jan, L.Y. Microtubule plus-end-tracking proteins target gap junctions directly from the cell interior to adherens junctions. Cell 2007, 128, 547–560. [Google Scholar] [CrossRef] [Green Version]
- Himelman, E.; Lillo, M.A.; Nouet, J.; Gonzalez, J.P.; Zhao, Q.; Xie, L.H.; Li, H.; Liu, T.; Wehrens, X.H.; Lampe, P.D.; et al. Prevention of connexin-43 remodeling protects against Duchenne muscular dystrophy cardiomyopathy. J. Clin. Investig. 2020, 130, 1713–1727. [Google Scholar] [CrossRef] [PubMed]
- Willebrords, J.; Maes, M.; Crespo Yanguas, S.; Vinken, M. Inhibitors of connexin and pannexin channels as potential therapeutics. Pharmacol. Ther. 2017, 180, 144–160. [Google Scholar] [CrossRef] [Green Version]
- Cea, L.A.; Cisterna, B.A.; Puebla, C.; Frank, M.; Figueroa, X.F.; Cardozo, C.; Willecke, K.; Latorre, R.; Saez, J.C. De novo expression of connexin hemichannels in denervated fast skeletal muscles leads to atrophy. Proc. Natl. Acad. Sci. USA 2013, 110, 16229–16234. [Google Scholar] [CrossRef] [Green Version]
- Molica, F.; Stierlin, F.B.; Fontana, P.; Kwak, B.R. Pannexin- and Connexin-Mediated Intercellular Communication in Platelet Function. Int. J. Mol. Sci. 2017, 18, 850. [Google Scholar] [CrossRef]
- Molica, F.; Figueroa, X.F.; Kwak, B.R.; Isakson, B.E.; Gibbins, J.M. Connexins and pannexins in vascular function and disease. Int. J. Mol. Sci. 2018, 19, 1663. [Google Scholar] [CrossRef] [Green Version]
- Gaete, P.S.; Lillo, M.A.; Puebla, M.; Poblete, I.; Figueroa, X.F. CGRP signalling inhibits NO production through pannexin-1 channel activation in endothelial cells. Sci. Rep. 2019, 9, 7932. [Google Scholar] [CrossRef] [Green Version]
- Begandt, D.; Good, M.E.; Keller, A.S.; DeLalio, L.J.; Rowley, C.; Isakson, B.E.; Figueroa, X.F. Pannexin channel and connexin hemichannel expression in vascular function and inflammation. BMC Cell Biol. 2017, 18, 1–16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mayhan, W.G.; Arrick, D.M.; Sharpe, G.M.; Patel, K.P.; Sun, H. Inhibition of NAD(P)H oxidase alleviates impaired NOS-dependent responses of pial arterioles in type 1 diabetes mellitus. Microcirculation 2006, 13, 567–575. [Google Scholar] [CrossRef] [PubMed]
- Paliege, A.; Pasumarthy, A.; Mizel, D.; Yang, T.; Schnermann, J.; Bachmann, S. Effect of apocynin treatment on renal expression of COX-2, NOS1, and renin in Wistar-Kyoto and spontaneously hypertensive rats. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2006, 290, 694–700. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bruce, A.F.; Rothery, S.; Dupont, E.; Severs, N.J. Gap junction remodelling in human heart failure is associated with increased interaction of connexin43 with ZO-1. Cardiovasc. Res. 2008, 77, 757–765. [Google Scholar] [CrossRef] [Green Version]
- Bolte, S.; Cordelieres, F.P. A guided tour into subcellular colocalization analysis in light microscopy. J. Microsc. 2006, 224, 213–232. [Google Scholar] [CrossRef]
- van Steensel, B.; van Binnendijk, E.P.; Hornsby, C.D.; van der Voort, H.T.; Krozowski, Z.S.; de Kloet, E.R.; van Driel, R. Partial colocalization of glucocorticoid and mineralocorticoid receptors in discrete compartments in nuclei of rat hippocampus neurons. J. Cell Sci. 1996, 109, 787–792. [Google Scholar]
- Ramanathan, L.; Das, N.P.; Li, Q.T. Studies on lipid oxidation in fish phospholipid liposomes. Biol. Trace Elem. Res. 1994, 40, 59–70. [Google Scholar] [CrossRef]










© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Vielma, A.Z.; Boric, M.P.; Gonzalez, D.R. Apocynin Treatment Prevents Cardiac Connexin 43 Hemichannels Hyperactivity by Reducing Nitroso-Redox Stress in Mdx Mice. Int. J. Mol. Sci. 2020, 21, 5415. https://doi.org/10.3390/ijms21155415
Vielma AZ, Boric MP, Gonzalez DR. Apocynin Treatment Prevents Cardiac Connexin 43 Hemichannels Hyperactivity by Reducing Nitroso-Redox Stress in Mdx Mice. International Journal of Molecular Sciences. 2020; 21(15):5415. https://doi.org/10.3390/ijms21155415
Chicago/Turabian StyleVielma, Alejandra Z., Mauricio P. Boric, and Daniel R. Gonzalez. 2020. "Apocynin Treatment Prevents Cardiac Connexin 43 Hemichannels Hyperactivity by Reducing Nitroso-Redox Stress in Mdx Mice" International Journal of Molecular Sciences 21, no. 15: 5415. https://doi.org/10.3390/ijms21155415
APA StyleVielma, A. Z., Boric, M. P., & Gonzalez, D. R. (2020). Apocynin Treatment Prevents Cardiac Connexin 43 Hemichannels Hyperactivity by Reducing Nitroso-Redox Stress in Mdx Mice. International Journal of Molecular Sciences, 21(15), 5415. https://doi.org/10.3390/ijms21155415