Iron Metabolism in Obesity and Metabolic Syndrome

 ,
,  , ,
, ,  ,
,  and
and
Abstract
:1. Childhood Obesity
2. Metabolic Syndrome
3. Obesity Comorbidities
4. Obesity, Inflammation and Oxidative Stress
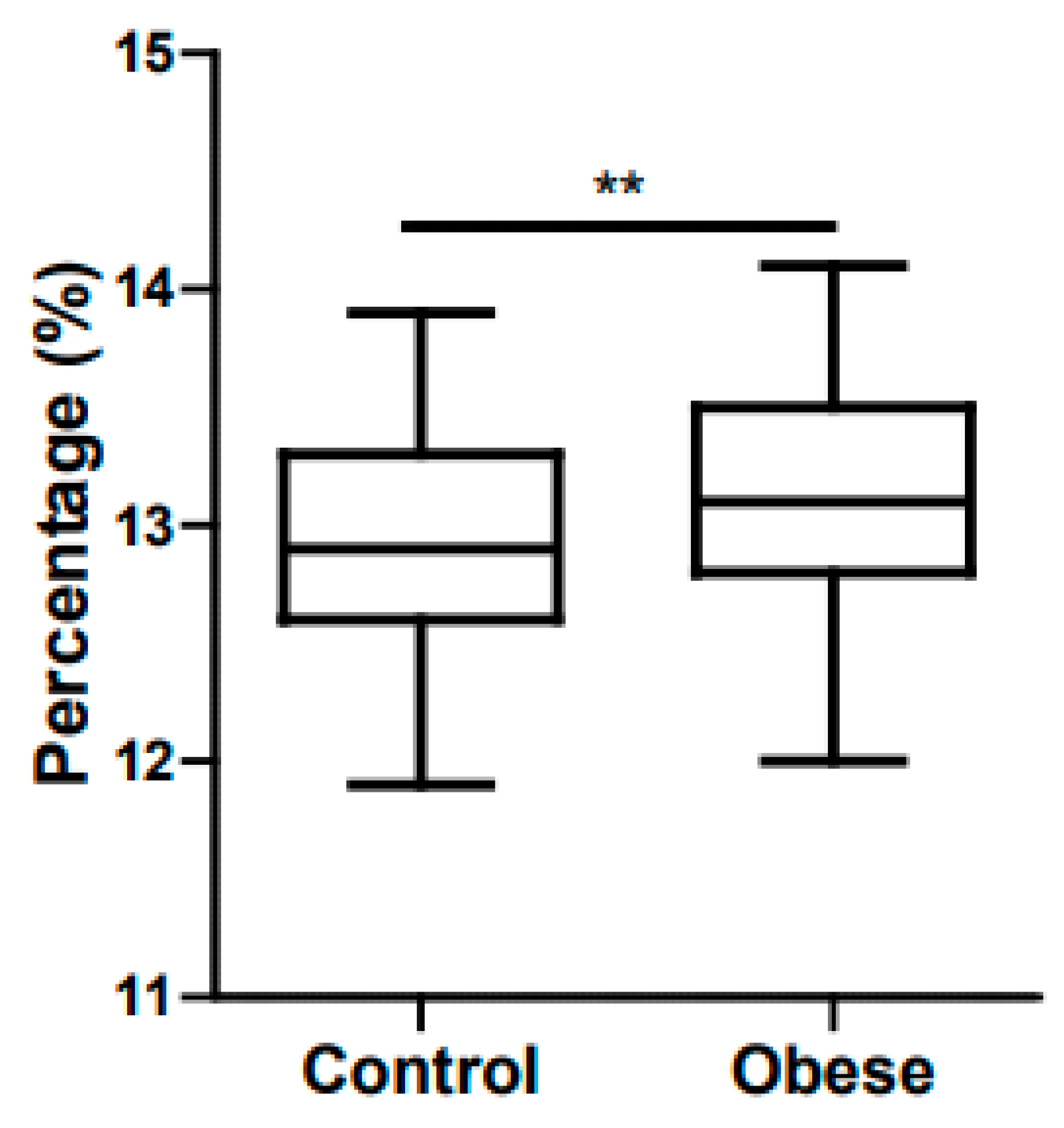
5. Childhood Obesity and Anemia
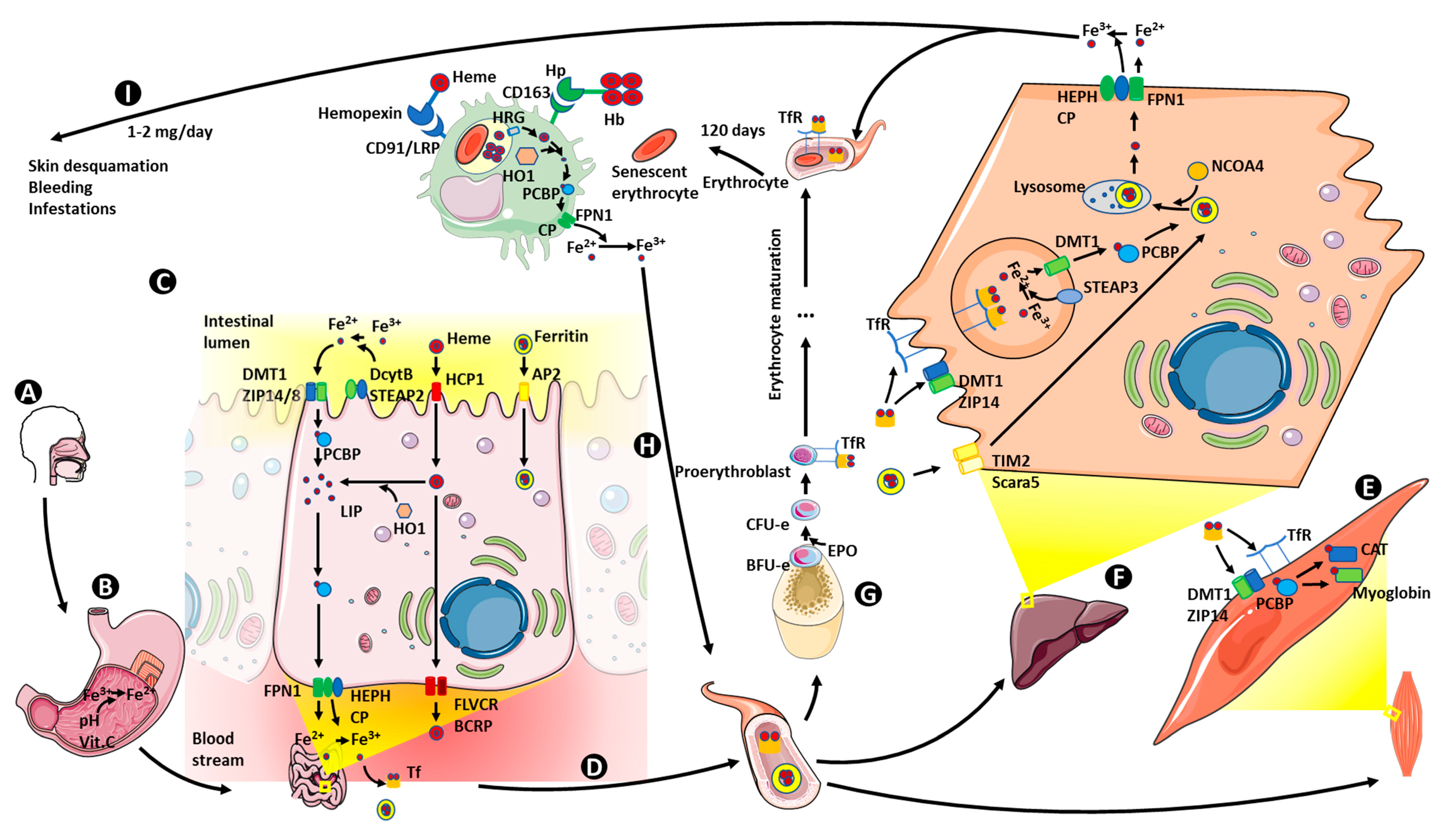
6. Iron Metabolism
6.1. Iron Absorption
6.2. Iron Storage
6.3. Iron Transport and Utilization
6.4. Iron Recycling
6.5. Iron Homeostasis Regulation
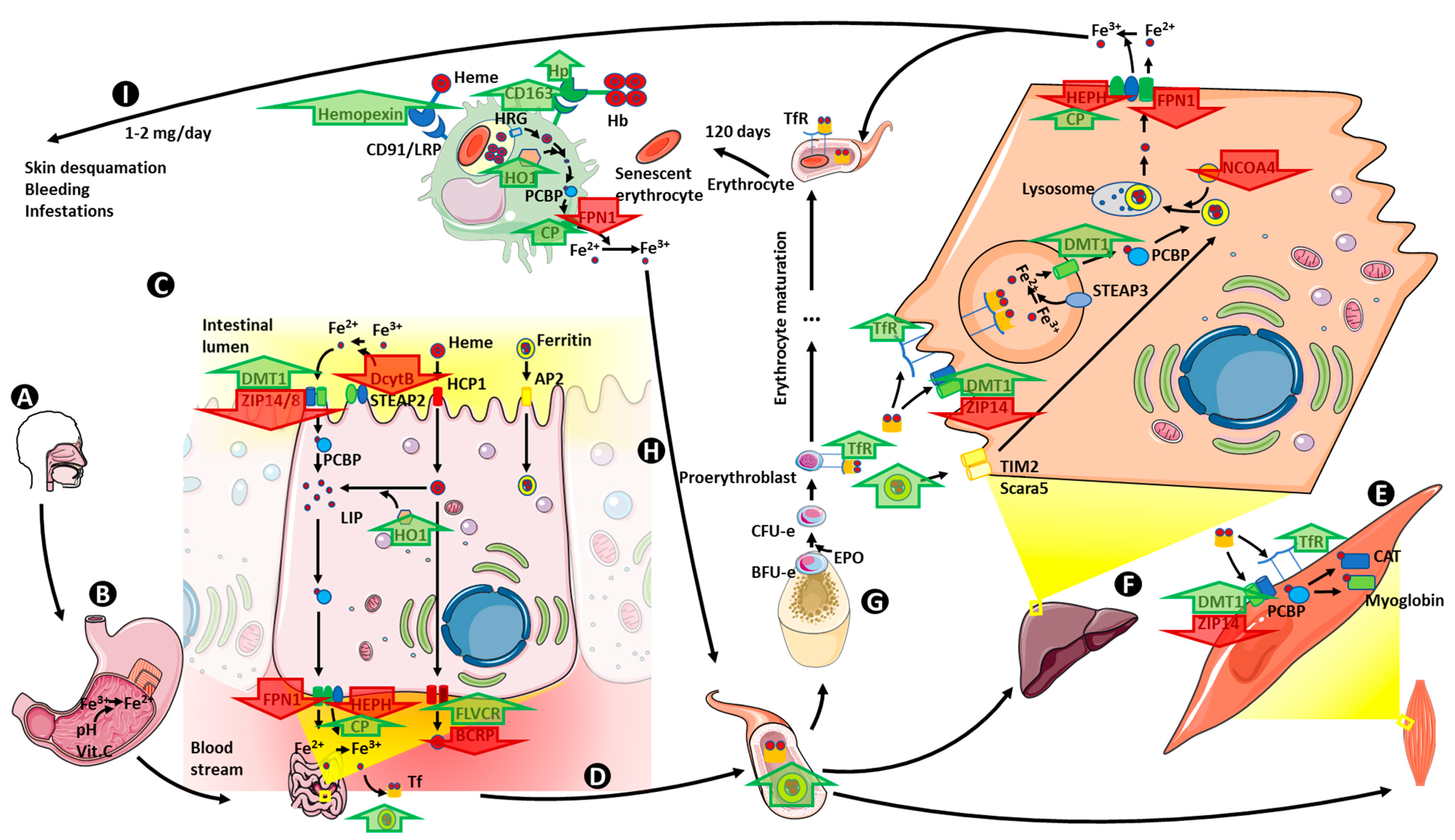
7. Iron Metabolism in Obesity
7.1. Iron Absorption
7.2. Iron Storage
7.3. Iron Transport and Utilization
7.4. Iron Recycling
7.5. Iron Homeostasis Regulation
8. Concluding Remarks
9. Future Directions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Ávila-Escalante, M.L.; Coop-Gamas, F.; Cervantes-Rodríguez, M.; Méndez-Iturbide, D.; Aranda-González, I.I. The effect of diet on oxidative stress and metabolic diseases—Clinically controlled trials. J. Food Biochem. 2020, 44, e13191. [Google Scholar] [CrossRef] [PubMed]
- World Health Organization—Obesity. Available online: https://www.who.int/health-topics/obesity#tab=tab_1 (accessed on 29 June 2020).
- World Health Organization—Childhood Obesity. Available online: https://www.who.int/dietphysicalactivity/childhood/en/ (accessed on 29 June 2020).
- Martos-Moreno, G.Á.; Gil-Campos, M.; Bueno, G.; Bahillo, P.; Bernal, S.; Feliu, A.; Lechuga-Sancho, A.M.; Palomo, E.; Ruiz, R.; Vela, A. Las alteraciones metabólicas asociadas a la obesidad están ya presentes en los primeros años de vida: Estudio colaborativo español. Nutr. Hosp. 2014, 30, 787–793. [Google Scholar] [CrossRef] [PubMed]
- Reaven, G.M. Insulin resistance and compensatory hyperinsulinemia: Role in hypertension, dyslipidemia, and coronary heart disease. Am. Heart J. 1991, 121, 1283–1288. [Google Scholar] [CrossRef]
- Bussler, S.; Penke, M.; Flemming, G.; Elhassan, Y.S.; Kratzsch, J.; Sergeyev, E.; Lipek, T.; Vogel, M.; Spielau, U.; Körner, A.; et al. Novel Insights in the Metabolic Syndrome in Childhood and Adolescence. Horm. Res. Paediatr. 2017, 88, 181–193. [Google Scholar] [CrossRef]
- Deboer, M.D. Assessing and managing the metabolic syndrome in children and adolescents. Nutrients 2019, 11, 1788. [Google Scholar] [CrossRef] [Green Version]
- Gepstein, V.; Weiss, R. Obesity as the Main Risk Factor for Metabolic Syndrome in Children. Front. Endocrinol. 2019, 10, 568. [Google Scholar] [CrossRef]
- Weihe, P.; Weihrauch-Blüher, S. Metabolic Syndrome in Children and Adolescents: Diagnostic Criteria, Therapeutic Options and Perspectives. Curr. Obes. Rep. 2019, 8, 472–479. [Google Scholar] [CrossRef]
- Luca, P.D.; Birken, C.; Grewal, P.; Dettmer, E.; Hamilton, J.K. Complex Obesity. Curr. Pediatr. Rev. 2012, 8, 179–187. [Google Scholar] [CrossRef]
- Speiser, P.W.; Rudolf, M.C.J.; Anhalt, H.; Camacho-Hubner, C.; Chiarelli, F.; Eliakim, A.; Freemark, M.; Gruters, A.; Hershkovitz, E.; Iughetti, L.; et al. Childhood Obesity. J. Clin. Endocrinol. Metab. 2005, 90, 1871–1887. [Google Scholar] [CrossRef]
- Han, J.C.; Lawlor, D.A.; Kimm, S.Y. Childhood obesity. Lancet 2010, 375, 1737–1748. [Google Scholar] [CrossRef]
- Juhola, J.; Magnussen, C.G.; Viikari, J.S.A.; Kähönen, M.; Hutri-Kähönen, N.; Jula, A.; Lehtimäki, T.; Åkerblom, H.K.; Pietikäinen, M.; Laitinen, T.; et al. Tracking of Serum Lipid Levels, Blood Pressure, and Body Mass Index from Childhood to Adulthood: The Cardiovascular Risk in Young Finns Study. J. Pediatr. 2011, 159, 584–590. [Google Scholar] [CrossRef] [PubMed]
- Pettitt, D.J.; Talton, J.; Dabelea, D.; Divers, J.; Imperatore, G.; Lawrence, J.M.; Liese, A.D.; Linder, B.; Mayer-Davis, E.J.; Pihoker, C.; et al. Prevalence of Diabetes in U.S. Youth in 2009: The SEARCH for Diabetes in Youth Study. Diabetes Care 2014, 37, 402–408. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Han, J.; Seaman, W.E.; Di, X.; Wang, W.; Willingham, M.; Torti, F.M.; Torti, S.V. Iron uptake mediated by binding of H-ferritin to the TIM-2 receptor in mouse cells. PLoS ONE 2011, 6, e23800. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nead, K.G. Overweight Children and Adolescents: A Risk Group for Iron Deficiency. Pediatrics 2004, 114, 104–108. [Google Scholar] [CrossRef] [PubMed]
- Gurnani, M.; Birken, C.; Hamilton, J.K. Childhood Obesity: Causes, Consequences, and Management. Pediatr. Clin. 2015, 62, 821–840. [Google Scholar] [CrossRef]
- Juonala, M.; Viikari, J.S.A.; Rönnemaa, T.; Helenius, H.; Taittonen, L.; Raitakari, O.T. Elevated Blood Pressure in Adolescent Boys Predicts Endothelial Dysfunction. Hypertension 2006, 48, 424–430. [Google Scholar] [CrossRef]
- Park, H.-Y.; Kwon, H.M.; Lim, H.J.; Hong, B.K.; Lee, J.Y.; Park, B.E.; Jang, Y.S.; Cho, S.Y.; Kim, H.-S. Potential role of leptin in angiogenesis: Leptin induces endothelial cell proliferation and expression of matrix metalloproteinases in vivo and in vitro. Exp. Mol. Med. 2001, 33, 95–102. [Google Scholar] [CrossRef]
- Landgraf, K.; Friebe, D.; Ullrich, T.; Kratzsch, J.; Dittrich, K.; Herberth, G.; Adams, V.; Kiess, W.; Erbs, S.; Körner, A. Chemerin as a Mediator between Obesity and Vascular Inflammation in Children. J. Clin. Endocrinol. Metab. 2012, 97, E556–E564. [Google Scholar] [CrossRef] [Green Version]
- Bruyndonckx, L.; Hoymans, V.Y.; Lemmens, K.; Ramet, J.; Vrints, C.J. Childhood obesity—Related endothelial dysfunction: An update on pathophysiological mechanisms and diagnostic advancements. Pediatr. Res. 2016, 79, 831–837. [Google Scholar] [CrossRef] [Green Version]
- Crowley, D.I.; Khoury, P.R.; Urbina, E.M.; Ippisch, H.M.; Kimball, T.R. Cardiovascular Impact of the Pediatric Obesity Epidemic: Higher Left Ventricular Mass is Related to Higher Body Mass Index. J. Pediatr. 2011, 158, 709–714. [Google Scholar] [CrossRef]
- Yu, J.J.; Yeom, H.H.; Chung, S.; Park, Y.; Lee, D.H. Left atrial diameters in overweight children with normal blood pressure. J. Pediatr. 2006, 148, 321–325. [Google Scholar] [CrossRef] [PubMed]
- Atabek, M.E.; Akyüz, E.; Eklioğlu, B.S.; Çimen, D. The Relationship between Metabolic Syndrome and Left Ventricular Mass Index in Obese Children. J. Clin. Res. Pediatr. Endocrinol. 2011, 3, 132–138. [Google Scholar] [CrossRef] [PubMed]
- Cote, A.T.; Harris, K.C.; Panagiotopoulos, C.; Sandor, G.G.S.; Devlin, A.M. Childhood Obesity and Cardiovascular Dysfunction. J. Am. Coll. Cardiol. 2013, 62, 1309–1319. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Friedemann, C.; Heneghan, C.; Mahtani, K.; Thompson, M.; Perera, R.; Ward, A.M. Cardiovascular disease risk in healthy children and its association with body mass index: Systematic review and meta-analysis. BMJ 2012, 345, e4759. [Google Scholar] [CrossRef] [Green Version]
- De Pergola, G.; De Mitrio, V.; Giorgino, F.; Sciaraffia, M.; Minenna, A.; Di Bari, L.; Pannacciulli, N.; Giorgino, R. Increase in both pro-thrombotic and anti-thrombotic factors in obese premenopausal women: Relationship with body fat distribution. Int. J. Obes. 1997, 21, 527–535. [Google Scholar] [CrossRef] [Green Version]
- Cigolini, M.; Targher, G.; Andreis, I.A.B.; Tonoli, M.; Agostino, G.; De Sandre, G. Visceral Fat Accumulation and Its Relation to Plasma Hemostatic Factors in Healthy Men. Arterioscler. Thromb. Vasc. Biol. 1996, 16, 368–374. [Google Scholar] [CrossRef]
- Mavri, A.; Alessi, M.C.; Geel-Georgelin, O.; Fina, F.; Sentocnik, J.T.; Bastelica, D.; Stegnar, M.; Juhan-Vague, I. Subcutaneous abdominal, but not femoral fat expression of plasminogen activator inhibitor-1 (PAI-1) is related to plasma PAI-1 levels and insulin resistance and decreases after weight loss. Diabetologia 2001, 44, 2025–2031. [Google Scholar] [CrossRef] [Green Version]
- Lowe, G.; Rumley, A.; Woodward, M.; Reid, E.; Rumley, J. Activated Protein C Resistance and the FV:R506Q Mutation in a Random Population Sample. Thromb. Haemost. 1999, 81, 914–918. [Google Scholar] [CrossRef]
- Kruszynska, Y.T.; Yu, J.G.; Olefsky, J.M.; Sobel, B.E. Effects of troglitazone on blood concentrations of plasminogen activator inhibitor 1 in patients with type 2 diabetes and in lean and obese normal subjects. Diabetes 2000, 49, 633–639. [Google Scholar] [CrossRef] [Green Version]
- Westerbacka, J.; Yki-Järvinen, H.; Turpeinen, A.; Rissanen, A.; Vehkavaara, S.; Syrjälä, M.; Lassila, R. Inhibition of Platelet-Collagen Interaction. Arterioscler. Thromb. Vasc. Biol. 2002, 22, 167–172. [Google Scholar] [CrossRef] [Green Version]
- De Pergola, G.; Pannacciulli, N. Coagulation and fibrinolysis abnormalities in obesity. J. Endocrinol. Investig. 2002, 25, 899–904. [Google Scholar] [CrossRef] [PubMed]
- Rogero, M.; Calder, P. Obesity, Inflammation, Toll-Like Receptor 4 and Fatty Acids. Nutrients 2018, 10, 432. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Luo, X.; Li, Y.; Yang, P.; Chen, Y.; Wei, L.; Yu, T.; Xia, J.; Ruan, X.Z.; Zhao, L.; Chen, Y. Obesity induces preadipocyte CD36 expression promoting inflammation via the disruption of lysosomal calcium homeostasis and lysosome function. EBioMedicine 2020, 56, 102797. [Google Scholar] [CrossRef] [PubMed]
- Pessentheiner, A.R.; Ducasa, G.M.; Gordts, P.L.S.M. Proteoglycans in Obesity-Associated Metabolic Dysfunction and Meta-Inflammation. Front. Immunol. 2020, 11, 769. [Google Scholar] [CrossRef]
- Kataru, R.P.; Park, H.J.; Baik, J.E.; Li, C.; Shin, J.; Mehrara, B.J. Regulation of Lymphatic Function in Obesity. Front. Physiol. 2020, 11, 459. [Google Scholar] [CrossRef]
- Benova, A.; Tencerova, M. Obesity-Induced Changes in Bone Marrow Homeostasis. Front. Endocrinol. 2020, 11, 294. [Google Scholar] [CrossRef]
- Ellulu, M.S.; Patimah, I.; Khaza’ai, H.; Rahmat, A.; Abed, Y. Obesity and inflammation: The linking mechanism and the complications. Arch. Med. Sci. 2017, 13, 851–863. [Google Scholar] [CrossRef]
- Zorena, K.; Jachimowicz-Duda, O.; Ślęzak, D.; Robakowska, M.; Mrugacz, M. Adipokines and Obesity. Potential Link to Metabolic Disorders and Chronic Complications. Int. J. Mol. Sci. 2020, 21, 3570. [Google Scholar] [CrossRef]
- Rivera, P.; Martos-Moreno, G.Á.; Barrios, V.; Suárez, J.; Pavón, F.J.; Chowen, J.A.; de Fonseca, F.R.; Argente, J. A novel approach to childhood obesity: Circulating chemokines and growth factors as biomarkers of insulin resistance. Pediatr. Obes. 2019, 14, e12473. [Google Scholar] [CrossRef]
- Hagman, E.; Besor, O.; Hershkop, K.; Santoro, N.; Pierpont, B.; Mata, M.; Caprio, S.; Weiss, R. Relation of the degree of obesity in childhood to adipose tissue insulin resistance. Acta Diabetol. 2019, 56, 219–226. [Google Scholar] [CrossRef] [Green Version]
- Mărginean, C.O.; Meliţ, L.E.; Huțanu, A.; Ghiga, D.V.; Săsăran, M.O. The adipokines and inflammatory status in the era of pediatric obesity. Cytokine 2020, 126, 154925. [Google Scholar] [CrossRef] [PubMed]
- Alissa, E.M.; Sutaih, R.H.; Kamfar, H.Z.; Alagha, A.E.; Marzouki, Z.M. Serum progranulin levels in relation to insulin resistance in childhood obesity. J. Pediatr. Endocrinol. Metab. 2017, 30, 1251–1256. [Google Scholar] [CrossRef] [PubMed]
- Mărginean, C.O.; Meliţ, L.E.; Ghiga, D.V.; Mărginean, M.O. Early Inflammatory Status Related to Pediatric Obesity. Front. Pediatr. 2019, 7, 241. [Google Scholar] [CrossRef] [PubMed]
- Hussain, T.; Tan, B.; Yin, Y.; Blachier, F.; Tossou, M.C.B.; Rahu, N. Oxidative Stress and Inflammation: What Polyphenols Can Do for Us? Oxidative Med. Cell. Longev. 2016, 2016, 1–9. [Google Scholar] [CrossRef] [Green Version]
- Biswas, S.K. Does the Interdependence between Oxidative Stress and Inflammation Explain the Antioxidant Paradox? Oxidative Med. Cell. Longev. 2016, 2016, 17–19. [Google Scholar] [CrossRef] [Green Version]
- Avelar, T.M.T.; Storch, A.S.; Castro, L.A.; Azevedo, G.V.M.M.; Ferraz, L.; Lopes, P.F. Oxidative stress in the pathophysiology of metabolic syndrome: Which mechanisms are involved? J. Bras. Patol. Med. Lab. 2015, 51, 231–239. [Google Scholar] [CrossRef]
- Lechuga-Sancho, A.M.; Gallego-Andujar, D.; Ruiz-Ocaña, P.; Visiedo, F.M.; Saez-Benito, A.; Schwarz, M.; Segundo, C.; Mateos, R.M. Obesity induced alterations in redox homeostasis and oxidative stress are present from an early age. PLoS ONE 2018, 13, e191547. [Google Scholar] [CrossRef]
- Kilic, E.; Özer, Ö.F.; Toprak, A.E.; Erman, H.; Torun, E.; Ayhan, S.K.; Caglar, H.G.; Selek, S.; Kocyigit, A. Oxidative Stress Status in Childhood Obesity: A Potential Risk Predictor. Med. Sci. Monit. 2016, 22, 3673–3679. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Correia-Costa, L.; Sousa, T.; Morato, M.; Cosme, D.; Afonso, J.; Areias, J.C.; Schaefer, F.; Guerra, A.; Afonso, A.C.; Azevedo, A.; et al. Oxidative stress and nitric oxide are increased in obese children and correlate with cardiometabolic risk and renal function. Br. J. Nutr. 2016, 116, 805–815. [Google Scholar] [CrossRef] [Green Version]
- Leo, F.; Rossodivita, A.; Segni, C.; Raimondo, S.; Canichella, S.; Silvestrini, A.; Miggiano, G.; Meucci, E.; Mancini, A. Frailty of Obese Children: Evaluation of Plasma Antioxidant Capacity in Pediatric Obesity. Exp. Clin. Endocrinol. Diabetes 2016, 124, 481–486. [Google Scholar] [CrossRef]
- Stenzel, A.; Carvalho, R.; Jesus, P.; Bull, A.; Pereira, S.; Saboya, C.; Ramalho, A. Serum Antioxidant Associations with Metabolic Characteristics in Metabolically Healthy and Unhealthy Adolescents with Severe Obesity: An Observational Study. Nutrients 2018, 10, 150. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wenzel, B.; Stults, H.; Mayer, J. Hypoferræmia in Obese Adolescents. Lancet 1962, 280, 327–328. [Google Scholar] [CrossRef]
- Menzie, C.M.; Yanoff, L.B.; Denkinger, B.I.; McHugh, T.; Sebring, N.G.; Calis, K.A.; Yanovski, J.A. Obesity-Related Hypoferremia Is Not Explained by Differences in Reported Intake of Heme and Nonheme Iron or Intake of Dietary Factors that Can Affect Iron Absorption. J. Am. Diet. Assoc. 2008, 108, 145–148. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Coimbra, S.; Catarino, C.; Santos-Silva, A. The role of adipocytes in the modulation of iron metabolism in obesity. Obes. Rev. 2013, 14, 771–779. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, F.M.; Weschenfelder, J.; Sander, C.; Minkwitz, J.; Thormann, J.; Chittka, T.; Mergl, R.; Kirkby, K.C.; Faßhauer, M.; Stumvoll, M.; et al. Inflammatory Cytokines in General and Central Obesity and Modulating Effects of Physical Activity. PLoS ONE 2015, 10, e0121971. [Google Scholar] [CrossRef] [PubMed]
- Ferrucci, L.; Guralnik, J.M.; Woodman, R.C.; Bandinelli, S.; Lauretani, F.; Corsi, A.M.; Chaves, P.H.M.; Ershler, W.B.; Longo, D.L. Proinflammatory state and circulating erythropoietin in persons with and without anemia. Am. J. Med. 2005, 118, 1288.e11–1288.e19. [Google Scholar] [CrossRef]
- Zhao, L.; Zhang, X.; Shen, Y.; Fang, X.; Wang, Y.; Wang, F. Obesity and iron deficiency: A quantitative meta-analysis. Obes. Rev. 2015, 16, 1081–1093. [Google Scholar] [CrossRef]
- Manios, Y.; Moschonis, G.; Chrousos, G.P.; Lionis, C.; Mougios, V.; Kantilafti, M.; Tzotzola, V.; Skenderi, K.P.; Petridou, A.; Tsalis, G.; et al. The double burden of obesity and iron deficiency on children and adolescents in Greece: The Healthy Growth Study. J. Hum. Nutr. Diet. 2013, 26, 470–478. [Google Scholar] [CrossRef]
- Del Giudice, E.M.; Santoro, N.; Amato, A.; Brienza, C.; Calabrò, P.; Wiegerinck, E.T.; Cirillo, G.; Tartaglione, N.; Grandone, A.; Swinkels, D.W.; et al. Hepcidin in obese children as a potential mediator of the association between obesity and iron deficiency. J. Clin. Endocrinol. Metab. 2009, 94, 5102–5107. [Google Scholar] [CrossRef] [Green Version]
- Sal, E.; Yenicesu, I.; Celik, N.; Pasaoglu, H.; Celik, B.; Pasaoglu, O.T.; Kaya, Z.; Kocak, U.; Camurdan, O.; Bideci, A.; et al. Relationship between obesity and iron deficiency anemia: Is there a role of hepcidin? Hematology 2018, 23, 542–548. [Google Scholar] [CrossRef] [Green Version]
- Aigner, E.; Feldman, A.; Datz, C. Obesity as an emerging risk factor for iron deficiency. Nutrients 2014, 6, 3587–3600. [Google Scholar] [CrossRef] [PubMed]
- Bertinato, J.; Aroche, C.; Plouffe, L.J.; Lee, M.; Murtaza, Z.; Kenney, L.; Lavergne, C.; Aziz, A. Diet-induced obese rats have higher iron requirements and are more vulnerable to iron deficiency. Eur. J. Nutr. 2014, 53, 885–895. [Google Scholar] [CrossRef] [PubMed]
- Fujita, B.; Strodthoff, D.; Fritzenwanger, M.; Pfeil, A.; Ferrari, M.; Goebel, B.; Figulla, H.R.; Gerdes, N.; Jung, C. Altered red blood cell distribution width in overweight adolescents and its association with markers of inflammation. Pediatr. Obes. 2013, 8, 385–391. [Google Scholar] [CrossRef] [PubMed]
- Doğan, G.; Andiran, N.; Çelik, N.; Uysal, S. Iron parameters, pro-hepcidin and soluble transferrin receptor levels in obese children. Minerva Pediatr. 2016, 72, 175–181. [Google Scholar] [CrossRef] [PubMed]
- World Health Organization—IDA. Available online: https://www.who.int/nutrition/topics/ida/en/ (accessed on 29 June 2020).
- Abbaspour, N.; Hurrell, R.; Kelishadi, R. Review on iron and its importance for human health. J. Res. Med. Sci. 2014, 19, 164–174. [Google Scholar]
- Yiannikourides, A.; Latunde-Dada, G. A Short Review of Iron Metabolism and Pathophysiology of Iron Disorders. Medicines 2019, 6, 85. [Google Scholar] [CrossRef] [Green Version]
- Hower, V.; Mendes, P.; Torti, F.M.; Laubenbacher, R.; Akman, S.; Shulaev, V.; Torti, S.V. A general map of iron metabolism and tissue-specific subnetworks. Mol. Biosyst. 2009, 5, 422. [Google Scholar] [CrossRef] [Green Version]
- Laftah, A.H.; Latunde-Dada, G.O.; Fakih, S.; Hider, R.C.; Simpson, R.J.; McKie, A.T. Haem and folate transport by proton-coupled folate transporter/haem carrier protein 1 (SLC46A1). Br. J. Nutr. 2008, 101, 1150–1156. [Google Scholar] [CrossRef] [Green Version]
- Soares, M.P.; Bach, F.H. Heme oxygenase-1: From biology to therapeutic potential. Trends Mol. Med. 2009, 15, 50–58. [Google Scholar] [CrossRef]
- Araujo, J.A.; Zhang, M.; Yin, F. Heme Oxygenase-1, Oxidation, Inflammation, and Atherosclerosis. Front. Pharmacol. 2012, 3, 119. [Google Scholar] [CrossRef] [Green Version]
- Krishnamurthy, P.; Xie, T.; Schuetz, J. The role of transporters in cellular heme and porphyrin homeostasis. Pharmacol. Ther. 2007, 114, 345–358. [Google Scholar] [CrossRef] [PubMed]
- Gulec, S.; Anderson, G.J.; Collins, J.F. Mechanistic and regulatory aspects of intestinal iron absorption. Am. J. Physiol. Gastrointest. Liver Physiol. 2014, 307, G397–G409. [Google Scholar] [CrossRef] [Green Version]
- Silva, B.; Faustino, P. An overview of molecular basis of iron metabolism regulation and the associated pathologies. Biochim. Biophys. Acta BBA Mol. Basis Dis. 2015, 1852, 1347–1359. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bogdan, A.R.; Miyazawa, M.; Hashimoto, K.; Tsuji, Y. Regulators of Iron Homeostasis: New Players in Metabolism, Cell Death, and Disease. Trends Biochem. Sci. 2016, 41, 274–286. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- San Martin, C.D.; Garri, C.; Pizarro, F.; Walter, T.; Theil, E.C.; Núñez, M.T. Caco-2 Intestinal Epithelial Cells Absorb Soybean Ferritin by μ2 (AP2)-Dependent Endocytosis. J. Nutr. 2008, 138, 659–666. [Google Scholar] [CrossRef]
- Li, J.Y.; Paragas, N.; Ned, R.M.; Qiu, A.; Viltard, M.; Leete, T.; Drexler, I.R.; Chen, X.; Sanna-Cherchi, S.; Mohammed, F.; et al. Scara5 Is a Ferritin Receptor Mediating Non-Transferrin Iron Delivery. Dev. Cell 2009, 16, 35–46. [Google Scholar] [CrossRef] [Green Version]
- Drakesmith, H.; Nemeth, E.; Ganz, T. Ironing out Ferroportin. Cell Metab. 2015, 22, 777–787. [Google Scholar] [CrossRef] [Green Version]
- Wang, F.; Paradkar, P.N.; Custodio, A.O.; Ward, D.M.; Fleming, M.D.; Campagna, D.; Roberts, K.A.; Boyartchuk, V.; Dietrich, W.F.; Kaplan, J.; et al. Genetic variation in Mon1a affects protein trafficking and modifies macrophage iron loading in mice. Nat. Genet. 2007, 39, 1025–1032. [Google Scholar] [CrossRef]
- Petrak, J.; Vyoral, D. Hephaestin—A ferroxidase of cellular iron export. Int. J. Biochem. Cell Biol. 2005, 37, 1173–1178. [Google Scholar] [CrossRef]
- Xu, E.; Chen, M.; Zheng, J.; Maimaitiming, Z.; Zhong, T.; Chen, H. Deletion of hephaestin and ceruloplasmin induces a serious systemic iron deficiency and disrupts iron homeostasis. Biochem. Biophys. Res. Commun. 2018, 503, 1905–1910. [Google Scholar] [CrossRef]
- Gkouvatsos, K.; Papanikolaou, G.; Pantopoulos, K. Regulation of iron transport and the role of transferrin. Biochim. Biophys. Acta BBA Gen. Subj. 2012, 1820, 188–202. [Google Scholar] [CrossRef] [PubMed]
- Mancias, J.D.; Wang, X.; Gygi, S.P.; Harper, J.W.; Kimmelman, A.C. Quantitative proteomics identifies NCOA4 as the cargo receptor mediating ferritinophagy. Nature 2014, 509, 105–109. [Google Scholar] [CrossRef] [PubMed]
- Kawabata, H. Transferrin and transferrin receptors update. Free Radic. Biol. Med. 2019, 133, 46–54. [Google Scholar] [CrossRef] [PubMed]
- Rosa, L.; Cutone, A.; Lepanto, M.S.; Paesano, R.; Valenti, P. Lactoferrin: A natural glycoprotein involved in iron and inflammatory homeostasis. Int. J. Mol. Sci. 2017, 18, 1985. [Google Scholar] [CrossRef]
- Devireddy, L.R.; Gazin, C.; Zhu, X.; Green, M.R. A Cell-Surface Receptor for Lipocalin 24p3 Selectively Mediates Apoptosis and Iron Uptake. Cell 2005, 123, 1293–1305. [Google Scholar] [CrossRef] [Green Version]
- Xiao, X.; Yeoh, B.S.; Vijay-Kumar, M. Lipocalin 2: An Emerging Player in Iron Homeostasis and Inflammation. Annu. Rev. Nutr. 2017, 37, 103–130. [Google Scholar] [CrossRef]
- Yoon, T.; Cowan, J.A. Frataxin-mediated iron delivery to ferrochelatase in the final step of heme biosynthesis. J. Biol. Chem. 2004, 279, 25943–25946. [Google Scholar] [CrossRef] [Green Version]
- Paul, B.T.; Manz, D.H.; Torti, F.M.; Torti, S.V. Mitochondria and Iron: Current questions. Expert Rev. Hematol. 2017, 10, 65–79. [Google Scholar] [CrossRef] [Green Version]
- Ward, D.M.; Cloonan, S.M. Mitochondrial Iron in Human Health and Disease. Annu. Rev. Physiol. 2019, 81, 453–482. [Google Scholar] [CrossRef]
- Du, J.; Zhou, Y.; Li, Y.; Xia, J.; Chen, Y.; Chen, S.; Wang, X.; Sun, W.; Wang, T.; Ren, X.; et al. Identification of Frataxin as a regulator of ferroptosis. Redox Biol. 2020, 32, 101483. [Google Scholar] [CrossRef]
- Waldvogel-Abramowski, S.; Waeber, G.; Gassner, C.; Buser, A.; Frey, B.M.; Favrat, B.; Tissot, J.-D. Physiology of Iron Metabolism. Transfus. Med. Hemother. 2014, 41, 213–221. [Google Scholar] [CrossRef] [PubMed]
- Beaumont, C.; Delaby, C. Recycling Iron in Normal and Pathological States. Semin. Hematol. 2009, 46, 328–338. [Google Scholar] [CrossRef] [PubMed]
- Sukhbaatar, N.; Weichhart, T. Iron Regulation: Macrophages in Control. Pharmaceuticals 2018, 11, 137. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lutz, H.U.; Bogdanova, A. Mechanisms tagging senescent red blood cells for clearance in healthy humans. Front. Physiol. 2013, 4, 387. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mena, N.P.; Esparza, A.; Tapia, V.; Valdés, P.; Núñez, M.T. Hepcidin inhibits apical iron uptake in intestinal cells. Am. J. Physiol. Gastrointest. Liver Physiol. 2007, 294, G192–G198. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ambachew, S.; Biadgo, B. Hepcidin in Iron Homeostasis: Diagnostic and Therapeutic Implications in Type 2 Diabetes Mellitus Patients. Acta Haematol. 2017, 138, 183–193. [Google Scholar] [CrossRef] [PubMed]
- Babitt, J.L.; Lin, H.Y. Molecular Mechanisms of Hepcidin Regulation: Implications for the Anemia of CKD. Am. J. Kidney Dis. 2010, 55, 726–741. [Google Scholar] [CrossRef] [Green Version]
- Ganz, T.; Nemeth, E. Hepcidin and iron homeostasis. Biochim. Biophys. Acta BBA Mol. Cell Res. 2012, 1823, 1434–1443. [Google Scholar] [CrossRef] [Green Version]
- Coffey, R.; Ganz, T. Erythroferrone: An Erythroid Regulator of Hepcidin and Iron Metabolism. HemaSphere 2018, 2, e35. [Google Scholar] [CrossRef]
- Camaschella, C.; Nai, A.; Silvestri, L. Iron metabolism and iron disorders revisited in the hepcidin era. Haematologica 2020, 105, 260–272. [Google Scholar] [CrossRef] [Green Version]
- Wrighting, D.M.; Andrews, N.C. Interleukin-6 induces hepcidin expression through STAT3. Blood 2006, 108, 3204–3209. [Google Scholar] [CrossRef] [PubMed]
- Zhang, D.L.; Ghosh, M.C.; Rouault, T.A. The physiological functions of iron regulatory proteins in iron homeostasis—An update. Front. Pharmacol. 2014, 5, 124. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Renassia, C.; Peyssonnaux, C. New insights into the links between hypoxia and iron homeostasis. Curr. Opin. Hematol. 2019, 26, 125–130. [Google Scholar] [CrossRef] [PubMed]
- Castoldi, M.; Spasic, M.V.; Altamura, S.; Elmén, J.; Lindow, M.; Kiss, J.; Stolte, J.; Sparla, R.; D’Alessandro, L.A.; Klingmüller, U.; et al. The liver-specific microRNA miR-122 controls systemic iron homeostasis in mice. J. Clin. Investig. 2011, 121, 1386–1396. [Google Scholar] [CrossRef] [PubMed]
- Jais, A.; Einwallner, E.; Sharif, O.; Gossens, K.; Lu, T.T.-H.; Soyal, S.M.; Medgyesi, D.; Neureiter, D.; Paier-Pourani, J.; Dalgaard, K.; et al. Heme Oxygenase-1 Drives Metaflammation and Insulin Resistance in Mouse and Man. Cell 2014, 158, 25–40. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carmona-Montesinos, E.; Velazquez-Perez, R.; Aguirre, E.P.; Rivas-Arancibia, S. Obesity, Oxidative Stress, and Their Effect on Serum Heme Oxygenase-1 Concentrations and Insulin in Children Aged 3 to 5 Years in a Pediatric Hospital of the Ministry of Health CDMX. Child. Obes. 2016, 12, 474–481. [Google Scholar] [CrossRef]
- Tu, T.H.; Joe, Y.; Choi, H.-S.; Chung, H.T.; Yu, R. Induction of Heme Oxygenase-1 with Hemin Reduces Obesity-Induced Adipose Tissue Inflammation via Adipose Macrophage Phenotype Switching. Mediat. Inflamm. 2014, 2014, 1–10. [Google Scholar] [CrossRef]
- Peterson, S.J.; Rubinstein, R.; Faroqui, M.; Raza, A.; Boumaza, I.; Zhang, Y.; Stec, D.; Abraham, N.G. Positive Effects of Heme Oxygenase Upregulation on Adiposity and Vascular Dysfunction: Gene Targeting vs. Pharmacologic Therapy. Int. J. Mol. Sci. 2019, 20, 2514. [Google Scholar] [CrossRef] [Green Version]
- Mishra, J.; Simonsen, R.; Kumar, N. Intestinal breast cancer resistance protein (BCRP) requires Janus kinase 3 activity for drug efflux and barrier functions in obesity. J. Biol. Chem. 2019, 294, 18337–18348. [Google Scholar] [CrossRef]
- Moreno-Navarrete, J.M.; Rodríguez, A.; Ortega, F.J.; Becerril, S.; Sabater-Masdeu, M.; Latorre, J.; Ricart, W.; Frühbeck, G.; Fernández-Real, J.M. Increased adipose tissue heme levels and exportation are associated with altered systemic glucose metabolism. Sci. Rep. 2017, 7, 1–9. [Google Scholar] [CrossRef] [Green Version]
- Sonnweber, T.; Ress, C.; Nairz, M.; Theurl, I.; Schroll, A.; Murphy, A.T.; Wroblewski, V.; Witcher, D.R.; Moser, P.; Ebenbichler, C.F.; et al. High-fat diet causes iron deficiency via hepcidin-independent reduction of duodenal iron absorption. J. Nutr. Biochem. 2012, 23, 1600–1608. [Google Scholar] [CrossRef] [PubMed]
- Briffa, J.F.; Grinfeld, E.; Jenkin, K.A.; Mathai, M.L.; Poronnik, P.; Mcainch, A.J.; Hryciw, D.H.; McAinch, A. Diet induced obesity in rats reduces NHE3 and Na+/K+-ATPase expression in the kidney. Clin. Exp. Pharmacol. Physiol. 2015, 42, 1118–1126. [Google Scholar] [CrossRef] [PubMed]
- Noh, H.; Paik, H.Y.; Kim, J.; Chung, J. The Alteration of Zinc Transporter Gene Expression Is Associated with Inflammatory Markers in Obese Women. Biol. Trace Elem. Res. 2014, 158, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Cozzolino, S.M.F.; Marreiro, D.D.N.; Fisberg, M. Zinc Nutritional Status in Obese Children and Adolescents. Biol. Trace Elem. Res. 2002, 86, 107–122. [Google Scholar] [CrossRef]
- Cruz, K.J.C.; de Oliveira, A.R.S.; Morais, J.B.S.; Severo, J.S.; Mendes, P.M.V.; de Sousa Melo, S.R.; de Sousa, G.S.; do Marreiro, D.N. Zinc and Insulin Resistance: Biochemical and Molecular Aspects. Biol. Trace Elem. Res. 2018, 186, 407–412. [Google Scholar] [CrossRef]
- Ikeda, Y.; Watanabe, H.; Shiuchi, T.; Hamano, H.; Horinouchi, Y.; Imanishi, M.; Goda, M.; Zamami, Y.; Takechi, K.; Izawa-Ishizawa, Y.; et al. Deletion of H-ferritin in macrophages alleviates obesity and diabetes induced by high-fat diet in mice. Diabetologia 2020, 63, 1588–1602. [Google Scholar] [CrossRef]
- Suárez-Ortegón, M.F.; Echeverri, I.; Prats-Puig, A.; Bassols, J.; Carreras-Badosa, G.; López-Bermejo, A.; Fernández-Real, J.M. Iron Status and Metabolically Unhealthy Obesity in Prepubertal Children. Obesity 2019, 27, 636–644. [Google Scholar] [CrossRef]
- Sorokman, T.V.; Sokolnyk, S.V.; Popelyuk, A.-M.V.; Sokolnyk, S.O.; Makarova, O.V.; Bezruk, V.V. Indices of metal proteins (transferrin ceruloplasmin) in overweight and obese children. Arch. Balk. Med. Union 2019, 54, 300–306. [Google Scholar] [CrossRef]
- Kim, O.Y.; Shin, M.J.; Moon, J.; Chung, J.H. Plasma ceruloplasmin as a biomarker for obesity: A proteomic approach. Clin. Biochem. 2011, 44, 351–356. [Google Scholar] [CrossRef]
- Arner, E.; Forrest, A.R.R.; Ehrlund, A.; Mejhert, N.; Itoh, M.; Kawaji, H.; Lassmann, T.; Laurencikiene, J.; Rydén, M.; Arner, P.; et al. Ceruloplasmin is a novel adipokine which is overexpressed in adipose tissue of obese subjects and in obesity-associated cancer cells. PLoS ONE 2014, 9, e80274. [Google Scholar] [CrossRef]
- Jiang, C.; Zhang, S.; Li, D.; Chen, L.; Zhao, Y.; Mei, G.; Liu, J.; Tang, Y.; Gao, C.; Yao, P. Impaired ferritinophagy flux induced by high fat diet mediates hepatic insulin resistance via endoplasmic reticulum stress. Food Chem. Toxicol. 2020, 140, 111329. [Google Scholar] [CrossRef] [PubMed]
- Freixenet, N.; Remacha, Á.; Berlanga, E.; Caixàs, A.; Giménez-Palop, O.; Blanco-Vaca, F.; Bach, V.; Baiget, M.; Sánchez, Y.; Félez, J.; et al. Serum soluble transferrin receptor concentrations are increased in central obesity. Results from a screening programme for hereditary hemochromatosis in men with hyperferritinemia. Clin. Chim. Acta 2009, 400, 111–116. [Google Scholar] [CrossRef] [PubMed]
- Fernández-Cao, J.C.; Arija, V.; Aranda, N.; Basora, J.; Diez-Espino, J.; Estruch, R.; Fitó, M.; Corella, D.; Salas-Salvadó, J. Soluble transferrin receptor and risk of type 2 diabetes in the obese and nonobese. Eur. J. Clin. Investig. 2017, 47, 221–230. [Google Scholar] [CrossRef] [PubMed]
- Moreno-Navarrete, J.M.; Ortega, F.J.; Bassols, J.; Castro, A.; Ricart, W.; Fernández-Real, J.M. Association of Circulating Lactoferrin Concentration and 2 Nonsynonymous LTF Gene Polymorphisms with Dyslipidemia in Men Depends on Glucose-Tolerance Status. Clin. Chem. 2008, 54, 301–309. [Google Scholar] [CrossRef] [Green Version]
- Moreno-Navarrete, J.M.; Ortega, F.J.; Bassols, J.; Ricart, W.; Fernández-Real, J.M. Decreased circulating lactoferrin in insulin resistance and altered glucose tolerance as a possible marker of neutrophil dysfunction in type 2 diabetes. J. Clin. Endocrinol. Metab. 2009, 94, 4036–4044. [Google Scholar] [CrossRef] [Green Version]
- Catalán, V.; Gómez-Ambrosi, J.; Rodríguez, A.; Ramírez, B.; Silva, C.; Rotellar, F.; Gil, M.J.; Cienfuegos, J.A.; Salvador, J.; Frühbeck, G. Increased adipose tissue expression of lipocalin-2 in obesity is related to inflammation and matrix metalloproteinase-2 and metalloproteinase-9 activities in humans. J. Mol. Med. 2009, 87, 803–813. [Google Scholar] [CrossRef]
- Ishii, A.; Katsuura, G.; Imamaki, H.; Kimura, H.; Mori, K.P.; Kuwabara, T.; Kasahara, M.; Yokoi, H.; Ohinata, K.; Kawanishi, T.; et al. Obesity-promoting and anti-thermogenic effects of neutrophil gelatinase-associated lipocalin in mice. Sci. Rep. 2017, 7, 1–11. [Google Scholar] [CrossRef] [Green Version]
- Kanaka-Gantenbein, C.; Margeli, A.; Pervanidou, P.; Sakka, S.; Mastorakos, G.; Chrousos, G.P.; Papassotiriou, I. Retinol-binding protein 4 and lipocalin-2 in childhood and adolescent obesity: When children are not just “small adults”. Clin. Chem. 2008, 54, 1176–1182. [Google Scholar] [CrossRef]
- Bartolome, F.; Antequera, D.; de la Cueva, M.; Rubio-Fernandez, M.; Castro, N.; Pascual, C.; Camins, A.; Carro, E. Endothelial-specific deficiency of megalin in the brain protects mice against high-fat diet challenge. J. Neuroinflamm. 2020, 17, 22. [Google Scholar] [CrossRef] [Green Version]
- Pomplun, D.; Voigt, A.; Schulz, T.J.; Thierbach, R.; Pfeiffer, A.F.; Ristow, M. Reduced expression of mitochondrial frataxin in mice exacerbates diet-induced obesity. Proc. Natl. Acad. Sci. USA 2007, 104, 6377–6381. [Google Scholar] [CrossRef] [Green Version]
- Chiellini, C.; Santini, F.; Marsili, A.; Berti, P.; Bertacca, A.; Pelosini, C.; Scartabelli, G.; Pardini, E.; López-Soriano, J.; Centoni, R.; et al. Serum Haptoglobin: A Novel Marker of Adiposity in Humans. J. Clin. Endocrinol. Metab. 2004, 89, 2678–2683. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Pergola, G.; Di Roma, P.; Paoli, G.; Guida, P.; Pannacciulli, N.; Giorgino, R. Haptoglobin serum levels are independently associated with insulinemia in overweight and obese women. J. Endocrinol. Investig. 2007, 30, 399–403. [Google Scholar] [CrossRef] [PubMed]
- Chiellini, C.; Bertacca, A.; Novelli, S.E.; Görgün, C.Z.; Ciccarone, A.; Giordano, A.; Xu, H.; Soukas, A.; Costa, M.; Gandini, D.; et al. Obesity modulates the expression of haptoglobin in the white adipose tissue via TNFα. J. Cell. Physiol. 2002, 190, 251–258. [Google Scholar] [CrossRef] [PubMed]
- Vázquez-Moreno, M.; Locia-Morales, D.; Perez-Herrera, A.; Gomez-Diaz, R.A.; Gonzalez-Dzib, R.; Valdez-González, A.L.; Flores-Alfaro, E.; Corona-Salazar, P.; Suarez-Sanchez, F.; Gomez-Zamudio, J.; et al. Causal Association of Haptoglobin With Obesity in Mexican Children: A Mendelian Randomization Study. J. Clin. Endocrinol. Metab. 2020, 105, dgaa213. [Google Scholar] [CrossRef]
- Aguiar, L.; Marinho, C.; Martins, R.; Alho, I.; Ferreira, J.; Levy, P.; Faustino, P.; Bicho, M.; Inacio, A. Interaction between HFE and haptoglobin polymorphisms and its relation with plasma glutathione levels in obese children. Cell. Mol. Biol. 2019, 65, 69–74. [Google Scholar] [CrossRef]
- Fjeldborg, K.; Pedersen, S.B.; Møller, H.J.; Christiansen, T.; Bennetzen, M.; Richelsen, B. Human Adipose Tissue Macrophages Are Enhanced but Changed to an Anti-Inflammatory Profile in Obesity. J. Immunol. Res. 2014, 2014, 309548. [Google Scholar] [CrossRef]
- Kazankov, K.; Møller, H.J.; Lange, A.; Birkebaek, N.H.; Holland-Fischer, P.; Solvig, J.; Hørlyck, A.; Kristensen, K.; Rittig, S.; Handberg, A.; et al. The macrophage activation marker sCD163 is associated with changes in NAFLD and metabolic profile during lifestyle intervention in obese children. Pediatr. Obes. 2015, 10, 226–233. [Google Scholar] [CrossRef]
- Franklin, J.L.; Bennett, W.L.; Messina, J.L. Insulin attenuates TNFα-induced hemopexin mRNA: An anti-inflammatory action of insulin in rat H4IIE hepatoma cells. Biochem. Biophys. Rep. 2017, 9, 211–216. [Google Scholar] [CrossRef]
- Lawson, H.A.; Zayed, M.; Wayhart, J.P.; Fabbrini, E.; Love-Gregory, L.; Klein, S.; Semenkovich, C.F. Physiologic and genetic evidence links hemopexin to triglycerides in mice and humans. Int. J. Obes. 2017, 41, 631–638. [Google Scholar] [CrossRef] [Green Version]
- Lee, S. The genetic and epigenetic association of LDL Receptor Related Protein 1B (LRP1B) gene with childhood obesity. Sci. Rep. 2019, 9, 1815. [Google Scholar] [CrossRef] [Green Version]
- Altunkaynak, B.Z.; Ozbek, E.; Altunkaynak, M.E. A stereological and histological analysis of spleen on obese female rats, fed with high fat diet. Saudi Med. J. 2007, 28, 353. [Google Scholar] [PubMed]
- Rebours, V.; Garteiser, P.; Ribeiro-Parenti, L.; Cavin, J.B.; Doblas, S.; Pagé, G.; Bado, A.; Couvineau, A.; Ruszniewski, P.; Paradis, V.; et al. Obesity-induced pancreatopathy in rats is reversible after bariatric surgery. Sci. Rep. 2018, 8, 1–11. [Google Scholar] [CrossRef] [PubMed]
- He, Y.; Ren, L.; Zhang, Q.; Zhang, M.; Shi, J.; Hu, W.; Peng, H. Deficient serum furin predicts risk of abdominal obesity: Findings from a prospective cohort of Chinese adults. Postgrad. Med. J. 2020. [Google Scholar] [CrossRef] [PubMed]
- Gajewska, J.; Ambroszkiewicz, J.; Klemarczyk, W.; Głąb-Jabłońska, E.; Weker, H.; Chełchowska, M. Ferroportin-hepcidin axis in prepubertal obese children with sufficient daily iron intake. Int. J. Environ. Res. Public Health 2018, 15, 2156. [Google Scholar] [CrossRef] [Green Version]
- Park, C.Y.; Chung, J.; Koo, K.-O.; Kim, M.S.; Han, S.N. Hepatic iron storage is related to body adiposity and hepatic inflammation. Nutr. Metab. 2017, 14, 1–9. [Google Scholar] [CrossRef] [Green Version]
- Luciani, N.; Brasse-Lagnel, C.; Poli, M.; Anty, R.; Lesueur, C.; Cormont, M.; Laquerriere, A.; Folope, V.; Lemarchand-Brustel, Y.; Gugenheim, J.; et al. Hemojuvelin: A new link between obesity and iron homeostasis. Obesity 2011, 19, 1545–1551. [Google Scholar] [CrossRef]
- Blázquez-Medela, A.M.; Jumabay, M.; Boström, K.I. Beyond the bone: Bone morphogenetic protein signaling in adipose tissue. Obes. Rev. 2019, 20, 648–658. [Google Scholar] [CrossRef] [Green Version]
- Böttcher, Y.; Unbehauen, H.; Klöting, N.; Ruschke, K.; Körner, A.; Schleinitz, D.; Tönjes, A.; Enigk, B.; Wolf, S.; Dietrich, K.; et al. Adipose tissue expression and genetic variants of the bone morphogenetic protein receptor 1A gene (BMPR1A) are associated with human obesity. Diabetes 2009, 58, 2119–2128. [Google Scholar] [CrossRef] [Green Version]
- Folgueras, A.R.; Freitas-Rodríguez, S.; Ramsay, A.J.; Garabaya, C.; Rodríguez, F.; Velasco, G.; López-Otín, C. Matriptase-2 deficiency protects from obesity by modulating iron homeostasis. Nat. Commun. 2018, 9, 1–12. [Google Scholar] [CrossRef]
- Seong, H.-A.; Manoharan, R.; Ha, H. Smad proteins differentially regulate obesity-induced glucose and lipid abnormalities and inflammation via class-specific control of AMPK-related kinase MPK38/MELK activity. Cell Death Dis. 2018, 9, 1–13. [Google Scholar] [CrossRef] [Green Version]
- Katz, O.; Stuible, M.; Golishevski, N.; Lifshitz, L.; Tremblay, M.L.; Gassmann, M.; Mittelman, M.; Neumann, D. Erythropoietin treatment leads to reduced blood glucose levels and body mass: Insights from murine models. J. Endocrinol. 2010, 205, 87–95. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Teng, R.; Gavrilova, O.; Suzuki, N.; Chanturiya, T.; Schimel, D.; Hugendubler, L.; Mammen, S.; Yver, D.R.; Cushman, S.W.; Mueller, E.; et al. Disrupted erythropoietin signalling promotes obesity and alters hypothalamus proopiomelanocortin production. Nat. Commun. 2011, 2, 1–12. [Google Scholar] [CrossRef] [Green Version]
- Liu, Y.; Luo, B.; Shi, R.; Wang, J.; Liu, Z.; Liu, W.; Wang, S.; Zhang, Z. Nonerythropoietic Erythropoietin-Derived Peptide Suppresses Adipogenesis, Inflammation, Obesity and Insulin Resistance. Sci. Rep. 2015, 5, 15134. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, Y.; Xu, D.; Yin, C.; Wang, S.; Wang, M.; Xiao, Y. IL-10/STAT3 is reduced in childhood obesity with hypertriglyceridemia and is related to triglyceride level in diet-induced obese rats. BMC Endocr. Disord. 2018, 18, 1–9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wunderlich, C.M.; Hövelmeyer, N.; Wunderlich, F.T. Mechanisms of chronic JAK-STAT3-SOCS3 signaling in obesity. Jak-stat 2013, 2, e23878. [Google Scholar] [CrossRef]
- Lee, Y.S.; Riopel, M.; Cabrales, P.; Bandyopadhyay, G.K. Hepatocyte-specific HIF-1α ablation improves obesity-induced glucose intolerance by reducing first-pass GLP-1 degradation. Sci. Adv. 2019, 5, eaaw4176. [Google Scholar] [CrossRef] [Green Version]
- Jun, J.C.; Devera, R.; Unnikrishnan, D.; Shin, M.-K.; Bevans-Fonti, S.; Yao, Q.; Rathore, A.; Younas, H.; Halberg, N.; Scherer, P.E.; et al. Adipose HIF-1α causes obesity by suppressing brown adipose tissue thermogenesis. J. Mol. Med. 2017, 95, 287–297. [Google Scholar] [CrossRef] [Green Version]
- Saito, H.; Tanaka, T.; Sugahara, M.; Tanaka, S.; Fukui, K.; Wakashima, T.; Nangaku, M. Inhibition of prolyl hydroxylase domain (PHD) by JTZ-951 reduces obesity-related diseases in the liver, white adipose tissue, and kidney in mice with a high-fat diet. Lab. Investig. 2019, 99, 1217–1232. [Google Scholar] [CrossRef]
- Lee, Y.S.; Kim, J.; Osborne, O.; Oh, D.Y.; Sasik, R.; Schenk, S.; Chen, A.; Chung, H.; Murphy, A.; Watkins, S.M.; et al. Increased Adipocyte O2 Consumption Triggers HIF-1α, Causing Inflammation and Insulin Resistance in Obesity. Cell 2014, 157, 1339–1352. [Google Scholar] [CrossRef] [Green Version]
- López, P.; Castro, A.; Flórez, M.; Miranda, K.; Aranda, P.; Sánchez-González, C.; Llopis, J.; Arredondo, M. miR-155 and miR-122 expression of spermatozoa in obese subjects. Front. Genet. 2018, 9, 175. [Google Scholar] [CrossRef] [Green Version]




{kind=link}
{kind=link}
{kind=link}
| Iron Metabolism Level | Protein | Experimental Procedure | Result | Reference |
|---|---|---|---|---|
| Iron absorption | HO-1 | Mice Ho-1 depletion | IR and inflammation | [108] |
| Quantification in children | Higher levels in obesity | [109] | ||
| Induction in HFD-mice and cell cultures | anti-inflammatory phenotype, insulin sensitivity, repressed adipogenesis | [110,111] | ||
| BCRP | Obese humans | Intestinal dysfunction of the transporter | [112] | |
| FLVCR1 | Quantification in HFD mice | mRNA levels positive relation with fasting glucose and negative with insulin resistance | [113] | |
| Dcytb | Quantification in HFD mice | Lower mRNA levels | [114] | |
| Hephaestatin | Quantification in HFD mice | Lower mRNA levels | [114] | |
| TfR1 | Quantification in HFD mice | Higher mRNA and protein levels | [114] | |
| DMT1 | Quantification in HFD mice | Higher mRNA and protein levels | [114] | |
| NHE3 | Quantification in HFD mice | Reduced renal expression | [115] | |
| Zinc transporters | Obese women | Reduced mRNA levels | [116] | |
| Iron storage | Ferritin | H-Ferritin deletion in HFD mice | Anti-obesogenic state | [119] |
| Obese children | Relation with metabolically unhealthy obesity | [120] | ||
| Iron transport and utilization | FPN1 | Quantification in HFD mice | Decreased levels of FPN1 | [144,145] |
| Quantification in HFD mice | Decreased levels of FPN1 in obese children | [147] | ||
| Hephaestatin | Quantification in HFD mice | Lower mRNA levels | [114] | |
| CP | Obese adults and children | Higher circulating CP levels | [121,122,123] | |
| NCOA4 | Quantification in HFD mice | Impaired expression | [124] | |
| sTfR | Obese humans | Related to BMI in hyperferritinemia | [125] | |
| Lactoferrin | Obese humans | Inversely related to BMI and obesity | [127,128] | |
| LCN2 | Obese humans | Increased levels | [129] | |
| LCN2 knockout mice | Obesogenic and anti-thermogenic activity | [130] | ||
| Obese children | Decreased levels | [131] | ||
| Megalin | Endothelial megalin-deficient mice | Protective role against HFD-induced obesity | [132] | |
| Frataxin | Frataxin deletion in mice | Impaired oxidative metabolism and higher predisposition to suffer from high-caloric diet-induced obesity | [133] | |
| Iron recycling | Haptoglobin | Obese women | Relation with BMI, HOMA-IR, fasting insulin or blood glucose blood levels | [134,135] |
| Obese adults and children | Positive association with obesity and allele related to haptoglobin levels | [137] | ||
| Obese children | Allele related to oxidative stress in obesity | [138] | ||
| CD163 | Obese humans | Elevated expression and relation with HOMA-IR | [139] | |
| Obese children submitted to improved lifestyle | Changes in CD163 associated with better insulin sensitivity | [140] | ||
| Hemopexin | Quantification in cell cultures | Higher mRNA levels in inflammatory states and relation of hemopexin and adipogenesis | [141,142] | |
| Quantification in humans | Variations according to metabolic disease status and triglyceride levels | [142] | ||
| LRP | Obese children | SNPs and epigenetic modifications related to BMI | [143] | |
| Iron homeostasis regulation | Furin | Obese humans | Furin deficiency related to obesity risk | [146] |
| Hepcidin | Obese children | Increased levels | [147] | |
| HJV | Obese humans | Increased mRNA levels in adipose tissue | [149] | |
| BMP 2 | Obese humans | Higher levels | [150] | |
| BMP 4 | BMP4 deletion in mice | Obesogenic effects | [150] | |
| BMP 7 | HFD mice treated with BMP 7 | Anti-obesogenic and anti-inflammatory effects | [150] | |
| BMPR1A | Obese humans | Increased mRNA levels and SNPs involved | [151] | |
| Matriptase-2 | Deficient matriptase-2 mice | Protection against obesity and its complications | [152] | |
| SMAD proteins | HFD mice overexpressing SMAD isoforms | SMAD 2, 3 and 4 improve obesity-related metabolic parameters and inflammation. SMAD7 has detrimental effects by regulating MPK38 activity | [153] | |
| EPO | Mice | Anti-obesogenic effects | [154,155,156] | |
| STAT3 | Obese children and HFD mice | Lower mRNA levels | [157] | |
| HIF1 | Mice | Obesogenic effects | [159,160] | |
| HIF1 | Mice | Anti-obesogenic effects | [161] | |
| HIF2 | Mice | Anti-obesogenic effects | [162] | |
| miRNA122 | Obese humans | Increased levels | [163] |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
González-Domínguez, Á.; Visiedo-García, F.M.; Domínguez-Riscart, J.; González-Domínguez, R.; Mateos, R.M.; Lechuga-Sancho, A.M. Iron Metabolism in Obesity and Metabolic Syndrome. Int. J. Mol. Sci. 2020, 21, 5529. https://doi.org/10.3390/ijms21155529
González-Domínguez Á, Visiedo-García FM, Domínguez-Riscart J, González-Domínguez R, Mateos RM, Lechuga-Sancho AM. Iron Metabolism in Obesity and Metabolic Syndrome. International Journal of Molecular Sciences. 2020; 21(15):5529. https://doi.org/10.3390/ijms21155529
Chicago/Turabian StyleGonzález-Domínguez, Álvaro, Francisco M. Visiedo-García, Jesús Domínguez-Riscart, Raúl González-Domínguez, Rosa M. Mateos, and Alfonso María Lechuga-Sancho. 2020. "Iron Metabolism in Obesity and Metabolic Syndrome" International Journal of Molecular Sciences 21, no. 15: 5529. https://doi.org/10.3390/ijms21155529
APA StyleGonzález-Domínguez, Á., Visiedo-García, F. M., Domínguez-Riscart, J., González-Domínguez, R., Mateos, R. M., & Lechuga-Sancho, A. M. (2020). Iron Metabolism in Obesity and Metabolic Syndrome. International Journal of Molecular Sciences, 21(15), 5529. https://doi.org/10.3390/ijms21155529