Molecular and Cellular Complexity of Glioma. Focus on Tumour Microenvironment and the Use of Molecular and Imaging Biomarkers to Overcome Treatment Resistance

 ,
,  and
and
Abstract
:1. Background
2. Glioblastoma Cell Heterogeneity, Mutations, and Models
MicroRNA and GBM Microenvironment
3. Chemoresistance and Treatments
3.1. Resistance to Temozolomide
3.2. Pharmacological Strategies to Overcome GBM Resistance
3.3. Role of miRNAs in GBM Chemoresistance
4. In Vivo Imaging for the Study of GBM Heterogeneity and Microenvironment
4.1. Cancer and Metabolism
4.2. Cancer and Hypoxia
4.3. Cancer and Inflammation
4.4. Other Targets of Tumour Microenvironment
4.5. miRNA and Imaging
5. Final Considerations and Future Direction
6. Conclusions
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| ACSS2 | Acetylcoenzime A synthetase 2 |
| ADC | Apparent diffusion coefficient |
| AMPK | 5′ AMP-Activated Protein Kinase |
| ASCT2 | Alanine, Serine, Cysteine transporter 2 |
| ASL | Arterial spin labelling |
| BBB | Blood-brain barrier |
| CAIX | Carbonic Anhydrase IX |
| [11C]AMT | alpha-[11C]methyl-L-tryptophan |
| [11C]MET | [11C]methionine |
| CNS | Central Nervous System |
| CSC/GSC | Cancer stem cell/Glioma stem cell |
| [64Cu]ATSM | [64Cu]-diacetyl-bis (N4-methylthiosemicarbazone) |
| [64Cu]RaftRGD | [64Cu]-cyclam-RAFT-c(-RGDfK-)4 |
| DCE-MRI | Dynamic contrast-enhanced Magnetic Resonance Imaging |
| DSC | Dynamic susceptibility contrast |
| DTI | Diffusion-tensor imaging |
| DWI-MRI | Diffusion-weighted Magnetic Resonance Imaging |
| ECM | Extracellular matrix |
| EGFRvIII | Epidermal Growth Factor Receptor-vIII |
| FAP | Fibroblast Activation Protein |
| [18F]BR-351 | (R)-2-(N-benzyl-4-(2-[18F]fluoroethoxy)phenylsulphonamido)-N-hydroxy-3-methylbutanamide |
| [18F]DCFPyL | 2-(3-{1-carboxy-5-[(6-[18F]fluoro-pyridine-3-carbonyl)-amino]-pentyl}-ureido)-pentanedioic acid |
| [18F]FDG | 2-deoxy-2-[18F]fluoro-D-glucose |
| [18F]DPA-714 | N,N-diethyl2-(2-(4-(2-[18F]fluoroethoxy)phenyl)-5,7-dimethylpyrazolo [1,5-a]pyrimidin-3-yl)acetamide |
| [18F]FELP | 2-[18F]-2-fluoroethyl-l-phenylalanine |
| [18F]FET | [18F]fluoroethyltyrosine |
| [18F]F-Gln | [18F]F-Glutamine |
| L-1-[18F]FETrp | L-1-[18F]fluoroethyl-tryptophan |
| [18F]GE-180 | S-N,N-diethyl-9-[2-18F-fluoroethyl]-5-methoxy-2,3,4,9-tetrahydro-1H-carbazole-4-carboxamide |
| [18F]FIMP | (S)-2-amino-3-[3-(2-18F-fluoroethoxy)-4-iodophenyl]-2-methylpropanoic acid |
| [18F]FLT | [18F]3′-deoxy-3′-fluorothymidine |
| GAMMs | Glioma associated-macrophages and microglia |
| GFAP | Glial fibrillary acidic protein |
| GLUT1 | Glucose transporter 1 |
| HIF | Hypoxia-Inducible Factor |
| HK | Hexokinase |
| IDH-1 | Isocitrate Dehydrogenase-1 |
| IDO1 | Indoleamine 2,3-dioxygenase 1 |
| [68Ga]PSMA | [68Ga]PSMA-HBED-CC |
| GBM | Glioblastoma Multiforme |
| JAK 1/2 | Janus Kinases 1/2 |
| LAT1/2 | L-type amino acid transporters 1 and 2 |
| MCT | Monocarboxylate transporter |
| MGMT | O6-methylguanine-DNA-methyl transferase |
| miRNA | microRNA |
| mTORC1 | mammalian target of rapamycin complex 1 |
| MMP | Matrix-metallo proteinase |
| MRI | Magnetic Resonance Imaging |
| MSC-IFN-β | Mesenchymal Stem/Stromal Cells-derived Interferon-β |
| mtComplex I | Mitochondrial Complex I |
| NMRS | Nuclear Magnetic Resonance Spectroscopy |
| p53 | Tumor Protein 53 |
| PD-1 | Programmed Cell Death-1 |
| PD-L1 | Programmed Cell Death-Ligand-1 |
| PARP | Poly ADP Ribose Polymerase |
| PET/CT | Positron Emission Tomography/Computed Tomography |
| PSMA | Prostate-specific membrane antigen |
| Rb | Retinoblastoma |
| RT | Radio therapy |
| SUV | Standardized Uptake Value |
| TIGIT | T-Cell Immunoreceptor With Ig and ITIM Domains |
| TIM-3 | T-Cell Immunoglobulin Mucin-3 Receptor |
| TME | Tumor micro-environment |
| TMZ | Temozolomide |
| TSPO | 18 KDa translocator protein |
| VEGF | Vascular Endothelial Growth Factor |
| WHO | World Health Organization |
| [89Zr]DFO-LEM2/15 | [89Zr]labelled-MT1-MMP-specific mAb (LEM2/15) |
References
- Wood, M.D.; Halfpenny, A.M.; Moore, S.R. Applications of molecular neuro-oncology—A review of diffuse glioma integrated diagnosis and emerging molecular entities. Diagn. Pathol. 2019, 14, 29. [Google Scholar] [CrossRef] [Green Version]
- Tamimi, A.F.; Juweid, M. Epidemiology and Outcome of Glioblastoma. In Glioblastoma; De Vleeschouwer, S., Ed.; Codon Publications: Brisbane, Australia, 2017; ISBN 978-0-9944381-2-6. [Google Scholar]
- Stupp, R.; Mason, W.P.; van den Bent, M.J.; Weller, M.; Fisher, B.; Taphoorn, M.J.B.; Belanger, K.; Brandes, A.A.; Marosi, C.; Bogdahn, U.; et al. Radiotherapy plus Concomitant and Adjuvant Temozolomide for Glioblastoma. N. Engl. J. Med. 2005, 352, 987–996. [Google Scholar] [CrossRef]
- Kazda, T.; Dziacky, A.; Burkon, P.; Pospisil, P.; Slavik, M.; Rehak, Z.; Jancalek, R.; Slampa, P.; Slaby, O.; Lakomy, R. Radiotherapy of glioblastoma 15 years after the landmark Stupp’s trial: More controversies than standards? Radiol. Oncol. 2018, 52, 121–128. [Google Scholar] [CrossRef] [Green Version]
- Carter, T.C.; Medina-Flores, R.; Lawler, B.E. Glioblastoma Treatment with Temozolomide and Bevacizumab and Overall Survival in a Rural Tertiary Healthcare Practice. BioMed Res. Int. 2018, 2018, 6204676. [Google Scholar] [CrossRef] [Green Version]
- Sorribes, I.C.; Handelman, S.K.; Jain, H.V. Mitigating temozolomide resistance in glioblastoma via DNA damage-repair inhibition. J. R. Soc. Interface 2020, 17, 20190722. [Google Scholar] [CrossRef] [Green Version]
- Bergmann, N.; Delbridge, C.; Gempt, J.; Feuchtinger, A.; Walch, A.; Schirmer, L.; Bunk, W.; Aschenbrenner, T.; Liesche-Starnecker, F.; Schlegel, J. The Intratumoral Heterogeneity Reflects the Intertumoral Subtypes of Glioblastoma Multiforme: A Regional Immunohistochemistry Analysis. Front. Oncol. 2020, 10. [Google Scholar] [CrossRef]
- Matarredona, E.R.; Pastor, A.M. Extracellular Vesicle-Mediated Communication between the Glioblastoma and Its Microenvironment. Cells 2020, 9, 96. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jeanmougin, M.; Håvik, A.B.; Cekaite, L.; Brandal, P.; Sveen, A.; Meling, T.R.; Ågesen, T.H.; Scheie, D.; Heim, S.; Lothe, R.A.; et al. Improved prognostication of glioblastoma beyond molecular subtyping by transcriptional profiling of the tumor microenvironment. Mol. Oncol. 2020, 14, 1016–1027. [Google Scholar] [CrossRef] [PubMed]
- Cheng, J.; Meng, J.; Zhu, L.; Peng, Y. Exosomal noncoding RNAs in Glioma: Biological functions and potential clinical applications. Mol. Cancer 2020, 19, 66. [Google Scholar] [CrossRef] [PubMed]
- Pottoo, F.H.; Javed, N.; Rahman, J.; Abu-Izneid, T.; Khan, F.A. Targeted delivery of miRNA based therapeuticals in the clinical management of Glioblastoma Multiforme. Semin. Cancer Biol. 2020. [Google Scholar] [CrossRef] [PubMed]
- Wright, K.; de Silva, K.; Purdie, A.C.; Plain, K.M. Comparison of methods for miRNA isolation and quantification from ovine plasma. Sci. Rep. 2020, 10, 825. [Google Scholar] [CrossRef] [PubMed]
- Kenny, A.; Jiménez-Mateos, E.M.; Zea-Sevilla, M.A.; Rábano, A.; Gili-Manzanaro, P.; Prehn, J.H.M.; Henshall, D.C.; Ávila, J.; Engel, T.; Hernández, F. Proteins and microRNAs are differentially expressed in tear fluid from patients with Alzheimer’s disease. Sci. Rep. 2019, 9, 15437. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, S.-W.; Ali, N.; Zhong, L.; Shi, J. MicroRNAs as biomarkers for human glioblastoma: Progress and potential. Acta Pharmacol. Sin. 2018, 39, 1405–1413. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sana, J.; Busek, P.; Fadrus, P.; Besse, A.; Radova, L.; Vecera, M.; Reguli, S.; Stollinova Sromova, L.; Hilser, M.; Lipina, R.; et al. Identification of microRNAs differentially expressed in glioblastoma stem-like cells and their association with patient survival. Sci. Rep. 2018, 8, 2836. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hayes, J.; Thygesen, H.; Tumilson, C.; Droop, A.; Boissinot, M.; Hughes, T.A.; Westhead, D.; Alder, J.E.; Shaw, L.; Short, S.C.; et al. Prediction of clinical outcome in glioblastoma using a biologically relevant nine-microRNA signature. Mol. Oncol. 2015, 9, 704–714. [Google Scholar] [CrossRef] [Green Version]
- Petrescu, G.E.D.; Sabo, A.A.; Torsin, L.I.; Calin, G.A.; Dragomir, M.P. MicroRNA based theranostics for brain cancer: Basic principles. J. Exp. Clin. Cancer Res. 2019, 38, 231. [Google Scholar] [CrossRef] [Green Version]
- Miyai, M.; Tomita, H.; Soeda, A.; Yano, H.; Iwama, T.; Hara, A. Current trends in mouse models of glioblastoma. J. Neurooncol. 2017, 135, 423–432. [Google Scholar] [CrossRef]
- Ramasawmy, R.; Johnson, S.P.; Roberts, T.A.; Stuckey, D.J.; David, A.L.; Pedley, R.B.; Lythgoe, M.F.; Siow, B.; Walker-Samuel, S. Monitoring the Growth of an Orthotopic Tumour Xenograft Model: Multi-Modal Imaging Assessment with Benchtop MRI (1T), High-Field MRI (9.4T), Ultrasound and Bioluminescence. PLoS ONE 2016, 11. [Google Scholar] [CrossRef]
- Thust, S.C.; Heiland, S.; Falini, A.; Jäger, H.R.; Waldman, A.D.; Sundgren, P.C.; Godi, C.; Katsaros, V.K.; Ramos, A.; Bargallo, N.; et al. Glioma imaging in Europe: A survey of 220 centres and recommendations for best clinical practice. Eur. Radiol. 2018, 28, 3306–3317. [Google Scholar] [CrossRef] [Green Version]
- la Fougère, C.; Suchorska, B.; Bartenstein, P.; Kreth, F.-W.; Tonn, J.-C. Molecular imaging of gliomas with PET: Opportunities and limitations. Neuro-oncology 2011, 13, 806–819. [Google Scholar] [CrossRef]
- Parsons, D.W.; Jones, S.; Zhang, X.; Lin, J.C.-H.; Leary, R.J.; Angenendt, P.; Mankoo, P.; Carter, H.; Siu, I.-M.; Gallia, G.L.; et al. An Integrated Genomic Analysis of Human Glioblastoma Multiforme. Science 2008, 321, 1807–1812. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Navin, N.; Kendall, J.; Troge, J.; Andrews, P.; Rodgers, L.; McIndoo, J.; Cook, K.; Stepansky, A.; Levy, D.; Esposito, D.; et al. Tumour evolution inferred by single-cell sequencing. Nature 2011, 472, 90–94. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gerlinger, M.; Rowan, A.J.; Horswell, S.; Math, M.; Larkin, J.; Endesfelder, D.; Gronroos, E.; Martinez, P.; Matthews, N.; Stewart, A.; et al. Intratumor Heterogeneity and Branched Evolution Revealed by Multiregion Sequencing. N. Engl. J. Med. 2012, 366, 883–892. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bedard, P.L.; Hansen, A.R.; Ratain, M.J.; Siu, L.L. Tumour heterogeneity in the clinic. Nature 2013, 501, 355–364. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Patel, A.P.; Tirosh, I.; Trombetta, J.J.; Shalek, A.K.; Gillespie, S.M.; Wakimoto, H.; Cahill, D.P.; Nahed, B.V.; Curry, W.T.; Martuza, R.L.; et al. Single-cell RNA-seq highlights intratumoral heterogeneity in primary glioblastoma. Science 2014, 344, 1396–1401. [Google Scholar] [CrossRef] [Green Version]
- Wang, J.; Cazzato, E.; Ladewig, E.; Frattini, V.; Rosenbloom, D.I.S.; Zairis, S.; Abate, F.; Liu, Z.; Elliott, O.; Shin, Y.-J.; et al. Clonal evolution of glioblastoma under therapy. Nat. Genet. 2016, 48, 768–776. [Google Scholar] [CrossRef] [Green Version]
- Sottoriva, A.; Spiteri, I.; Piccirillo, S.G.M.; Touloumis, A.; Collins, V.P.; Marioni, J.C.; Curtis, C.; Watts, C.; Tavaré, S. Intratumor heterogeneity in human glioblastoma reflects cancer evolutionary dynamics. Proc. Natl. Acad. Sci. USA 2013, 110, 4009–4014. [Google Scholar] [CrossRef] [Green Version]
- Wang, X.; Prager, B.C.; Wu, Q.; Kim, L.J.Y.; Gimple, R.C.; Shi, Y.; Yang, K.; Morton, A.R.; Zhou, W.; Zhu, Z.; et al. Reciprocal Signaling between Glioblastoma Stem Cells and Differentiated Tumor Cells Promotes Malignant Progression. Cell Stem Cell 2018, 22, 514–528.e5. [Google Scholar] [CrossRef] [Green Version]
- Calabrese, C.; Poppleton, H.; Kocak, M.; Hogg, T.L.; Fuller, C.; Hamner, B.; Oh, E.Y.; Gaber, M.W.; Finklestein, D.; Allen, M.; et al. A Perivascular Niche for Brain Tumor Stem Cells. Cancer Cell 2007, 11, 69–82. [Google Scholar] [CrossRef] [Green Version]
- Seidel, S.; Garvalov, B.K.; Wirta, V.; von Stechow, L.; Schänzer, A.; Meletis, K.; Wolter, M.; Sommerlad, D.; Henze, A.-T.; Nistér, M.; et al. A hypoxic niche regulates glioblastoma stem cells through hypoxia inducible factor 2α. Brain 2010, 133, 983–995. [Google Scholar] [CrossRef] [Green Version]
- Li, Z.; Bao, S.; Wu, Q.; Wang, H.; Eyler, C.; Sathornsumetee, S.; Shi, Q.; Cao, Y.; Lathia, J.; McLendon, R.E.; et al. Hypoxia-Inducible Factors Regulate Tumorigenic Capacity of Glioma Stem Cells. Cancer Cell 2009, 15, 501–513. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Najafi, M.; Farhood, B.; Mortezaee, K.; Kharazinejad, E.; Majidpoor, J.; Ahadi, R. Hypoxia in solid tumors: A key promoter of cancer stem cell (CSC) resistance. J. Cancer Res. Clin. Oncol. 2020, 146, 19–31. [Google Scholar] [CrossRef] [PubMed]
- Yan, K.; Wu, Q.; Yan, D.H.; Lee, C.H.; Rahim, N.; Tritschler, I.; DeVecchio, J.; Kalady, M.F.; Hjelmeland, A.B.; Rich, J.N. Glioma cancer stem cells secrete Gremlin1 to promote their maintenance within the tumor hierarchy. Genes Dev. 2014. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, H.; Lathia, J.D.; Wu, Q.; Wang, J.; Li, Z.; Heddleston, J.M.; Eyler, C.E.; Elderbroom, J.; Gallagher, J.; Schuschu, J.; et al. Targeting Interleukin 6 Signaling Suppresses Glioma Stem Cell Survival and Tumor Growth. Stem Cells 2009, 27, 2393–2404. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Charles, N.; Ozawa, T.; Squatrito, M.; Bleau, A.-M.; Brennan, C.W.; Hambardzumyan, D.; Holland, E.C. Perivascular Nitric Oxide Activates Notch Signaling and Promotes Stem-like Character in PDGF-Induced Glioma Cells. Cell Stem Cell 2010, 6, 141–152. [Google Scholar] [CrossRef] [Green Version]
- Zhu, T.; Costello, M.A.; Talsma, C.E.; Flack, C.G.; Crowley, J.G.; Hamm, L.L.; He, X.; Umper, S.L.H.-J.; Heth, J.A.; Muraszko, K.M.; et al. Endothelial cells create a stem cell niche in glioblastoma by providing Notch ligands that nurture self-renewal of cancer stem-like cells. Cancer Res. 2011. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lathia, J.D.; Heddleston, J.M.; Venere, M.; Rich, J.N. Deadly Teamwork: Neural Cancer Stem Cells and the Tumor Microenvironment. Cell Stem Cell 2011, 8, 482–485. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bao, S.; Wu, Q.; Sathornsumetee, S.; Hao, Y.; Li, Z.; Hjelmeland, A.B.; Shi, Q.; McLendon, R.E.; Bigner, D.D.; Rich, J.N. Stem Cell-like Glioma Cells Promote Tumor Angiogenesis through Vascular Endothelial Growth Factor. Cancer Res. 2006, 66, 7843–7848. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cheng, L.; Huang, Z.; Zhou, W.; Wu, Q.; Donnola, S.; Liu, J.K.; Fang, X.; Sloan, A.E.; Mao, Y.; Lathia, J.D.; et al. Glioblastoma Stem Cells Generate Vascular Pericytes to Support Vessel Function and Tumor Growth. Cell 2013, 153, 139–152. [Google Scholar] [CrossRef] [Green Version]
- Dirkse, A.; Golebiewska, A.; Buder, T.; Nazarov, P.V.; Muller, A.; Poovathingal, S.; Brons, N.H.C.; Leite, S.; Sauvageot, N.; Sarkisjan, D.; et al. Stem cell-associated heterogeneity in Glioblastoma results from intrinsic tumor plasticity shaped by the microenvironment. Nat. Commun. 2019, 10, 1787. [Google Scholar] [CrossRef]
- Bar-Ephraim, Y.E.; Kretzschmar, K.; Clevers, H. Organoids in immunological research. Nat. Rev. Immunol. 2020, 20, 279–293. [Google Scholar] [CrossRef] [PubMed]
- Darmanis, S.; Sloan, S.A.; Croote, D.; Mignardi, M.; Chernikova, S.; Samghababi, P.; Zhang, Y.; Neff, N.; Kowarsky, M.; Caneda, C.; et al. Single-Cell RNA-Seq Analysis of Infiltrating Neoplastic Cells at the Migrating Front of Human Glioblastoma. Cell Rep. 2017, 21, 1399–1410. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Neftel, C.; Laffy, J.; Filbin, M.G.; Hara, T.; Shore, M.E.; Rahme, G.J.; Richman, A.R.; Silverbush, D.; Shaw, M.L.; Hebert, C.M.; et al. An Integrative Model of Cellular States, Plasticity, and Genetics for Glioblastoma. Cell 2019, 178, 835–849.e21. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Babikir, H.; Muller, S.; Yagnik, G.; Shamardani, K.; Catalan, F.; Kohanbash, G.; Alvarado, B.; Lullo, E.D.; Kriegstein, A.; et al. The phenotypes of proliferating glioblastoma cells reside on a single axis of variation. Cancer Discov. 2019. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Behnan, J.; Stangeland, B.; Hosainey, S.A.M.; Joel, M.; Olsen, T.K.; Micci, F.; Glover, J.C.; Isakson, P.; Brinchmann, J.E. Differential propagation of stroma and cancer stem cells dictates tumorigenesis and multipotency. Oncogene 2017, 36, 570–584. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xiao, W.; Sohrabi, A.; Seidlits, S.K. Integrating the glioblastoma microenvironment into engineered experimental models. Future Sci. OA 2017, 3, FSO189. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bhaduri, A.; Di Lullo, E.; Jung, D.; Müller, S.; Crouch, E.E.; Espinosa, C.S.; Ozawa, T.; Alvarado, B.; Spatazza, J.; Cadwell, C.R.; et al. Outer Radial Glia-like Cancer Stem Cells Contribute to Heterogeneity of Glioblastoma. Cell Stem Cell 2020, 26, 48–63. [Google Scholar] [CrossRef]
- Pollen, A.A.; Nowakowski, T.J.; Chen, J.; Retallack, H.; Sandoval-Espinosa, C.; Nicholas, C.R.; Shuga, J.; Liu, S.J.; Oldham, M.C.; Diaz, A.; et al. Molecular Identity of Human Outer Radial Glia during Cortical Development. Cell 2015, 163, 55–67. [Google Scholar] [CrossRef] [Green Version]
- Ostrem, B.; Di Lullo, E.; Kriegstein, A. oRGs and mitotic somal translocation—A role in development and disease. Curr. Opin. Neurobiol. 2017, 42, 61–67. [Google Scholar] [CrossRef] [Green Version]
- Cancer Genome Atlas Research Network. Comprehensive genomic characterization defines human glioblastoma genes and core pathways. Nature 2008, 455, 1061–1068. [Google Scholar] [CrossRef]
- Madhavan, S.; Zenklusen, J.C.; Kotliarov, Y.; Sahni, H.; Fine, H.A.; Buetow, K. Rembrandt: Helping Personalized Medicine Become a Reality through Integrative Translational Research. Mol. Cancer Res. 2009. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Puchalski, R.B.; Shah, N.; Miller, J.; Dalley, R.; Nomura, S.R.; Yoon, J.-G.; Smith, K.A.; Lankerovich, M.; Bertagnolli, D.; Bickley, K.; et al. An anatomic transcriptional atlas of human glioblastoma. Science 2018, 360, 660–663. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hegi, M.E.; Diserens, A.-C.; Gorlia, T.; Hamou, M.-F.; de Tribolet, N.; Weller, M.; Kros, J.M.; Hainfellner, J.A.; Mason, W.; Mariani, L.; et al. MGMT Gene Silencing and Benefit from Temozolomide in Glioblastoma. N. Engl. J. Med. 2005, 352, 997–1003. [Google Scholar] [CrossRef] [Green Version]
- Shah, N.; Schroeder, B.; Cobbs, C. MGMT methylation in glioblastoma: Tale of the tail. Neuro-oncology 2015, 17, 167–168. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wick, W.; Weller, M.; van den Bent, M.; Sanson, M.; Weiler, M.; von Deimling, A.; Plass, C.; Hegi, M.; Platten, M.; Reifenberger, G. MGMT testing—The challenges for biomarker-based glioma treatment. Nat. Rev. Neurol. 2014, 10, 372–385. [Google Scholar] [CrossRef] [Green Version]
- Robertson, F.L.; Marqués-Torrejón, M.-A.; Morrison, G.M.; Pollard, S.M. Experimental models and tools to tackle glioblastoma. Dis. Models Mech. 2019, 12. [Google Scholar] [CrossRef] [Green Version]
- Hackam, D.G.; Redelmeier, D.A. Translation of research evidence from animals to humans. JAMA 2006, 296, 1731–1732. [Google Scholar] [CrossRef]
- Shanks, N.; Greek, R.; Greek, J. Are animal models predictive for humans? Philos. Ethics Humanit. Med. 2009, 4, 2. [Google Scholar] [CrossRef] [Green Version]
- van der Worp, H.B.; Howells, D.W.; Sena, E.S.; Porritt, M.J.; Rewell, S.; O’Collins, V.; Macleod, M.R. Can animal models of disease reliably inform human studies? PLoS Med. 2010, 7, e1000245. [Google Scholar] [CrossRef] [Green Version]
- Ben-David, U.; Ha, G.; Tseng, Y.-Y.; Greenwald, N.F.; Oh, C.; Shih, J.; McFarland, J.M.; Wong, B.; Boehm, J.S.; Beroukhim, R.; et al. Patient-derived xenografts undergo mouse-specific tumor evolution. Nat. Genet. 2017, 49, 1567–1575. [Google Scholar] [CrossRef] [Green Version]
- Hubert, C.G.; Rivera, M.; Spangler, L.C.; Wu, Q.; Mack, S.C.; Prager, B.C.; Couce, M.; McLendon, R.E.; Sloan, A.E.; Rich, J.N. A Three-Dimensional Organoid Culture System Derived from Human Glioblastomas Recapitulates the Hypoxic Gradients and Cancer Stem Cell Heterogeneity of Tumors Found In Vivo. Cancer Res. 2016, 76, 2465–2477. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ogawa, J.; Pao, G.M.; Shokhirev, M.N.; Verma, I.M. Glioblastoma Model Using Human Cerebral Organoids. Cell Rep. 2018, 23, 1220–1229. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Linkous, A.; Balamatsias, D.; Snuderl, M.; Edwards, L.; Miyaguchi, K.; Milner, T.; Reich, B.; Cohen-Gould, L.; Storaska, A.; Nakayama, Y.; et al. Modeling Patient-Derived Glioblastoma with Cerebral Organoids. Cell Rep. 2019, 26, 3203–3211. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lancaster, M.A.; Knoblich, J.A. Generation of cerebral organoids from human pluripotent stem cells. Nat. Protoc. 2014, 9, 2329–2340. [Google Scholar] [CrossRef] [Green Version]
- Mansour, A.A.; Gonçalves, J.T.; Bloyd, C.W.; Li, H.; Fernandes, S.; Quang, D.; Johnston, S.; Parylak, S.L.; Jin, X.; Gage, F.H. An in vivo model of functional and vascularized human brain organoids. Nat. Biotechnol. 2018, 36, 432–441. [Google Scholar] [CrossRef]
- Jacob, F.; Salinas, R.D.; Zhang, D.Y.; Nguyen, P.T.T.; Schnoll, J.G.; Wong, S.Z.H.; Thokala, R.; Sheikh, S.; Saxena, D.; Prokop, S.; et al. A Patient-Derived Glioblastoma Organoid Model and Biobank Recapitulates Inter- and Intra-tumoral Heterogeneity. Cell 2020, 180, 188–204.e22. [Google Scholar] [CrossRef]
- Ling, H.; Fabbri, M.; Calin, G.A. MicroRNAs and other non-coding RNAs as targets for anticancer drug development. Nat. Rev. Drug Discov. 2013, 12, 847–865. [Google Scholar] [CrossRef] [Green Version]
- Catalanotto, C.; Cogoni, C.; Zardo, G. MicroRNA in Control of Gene Expression: An Overview of Nuclear Functions. Int. J. Mol. Sci. 2016, 17, 1712. [Google Scholar] [CrossRef] [Green Version]
- Forero, D.A.; González-Giraldo, Y.; Castro-Vega, L.J.; Barreto, G.E. qPCR-based methods for expression analysis of miRNAs. BioTechniques 2019, 67, 192–199. [Google Scholar] [CrossRef] [Green Version]
- Goldman, J.G.; Andrews, H.; Amara, A.; Naito, A.; Alcalay, R.N.; Shaw, L.M.; Taylor, P.; Xie, T.; Tuite, P.; Henchcliffe, C.; et al. Cerebrospinal fluid, plasma, and saliva in the BioFIND study: Relationships among biomarkers and Parkinson’s disease Features. Mov. Disord. 2018, 33, 282–288. [Google Scholar] [CrossRef] [Green Version]
- Urbanek, M.O.; Nawrocka, A.U.; Krzyzosiak, W.J. Small RNA Detection by in Situ Hybridization Methods. Int. J. Mol. Sci. 2015, 16, 13259–13286. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eyileten, C.; Wicik, Z.; De Rosa, S.; Mirowska-Guzel, D.; Soplinska, A.; Indolfi, C.; Jastrzebska-Kurkowska, I.; Czlonkowska, A.; Postula, M. MicroRNAs as Diagnostic and Prognostic Biomarkers in Ischemic Stroke—A Comprehensive Review and Bioinformatic Analysis. Cells 2018, 7, 249. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Halle, B.; Marcusson, E.G.; Aaberg-Jessen, C.; Jensen, S.S.; Meyer, M.; Schulz, M.K.; Andersen, C.; Kristensen, B.W. Convection-enhanced delivery of an anti-miR is well-tolerated, preserves anti-miR stability and causes efficient target de-repression: A proof of concept. J. Neuro-Oncol. 2016, 126, 47–55. [Google Scholar] [CrossRef] [PubMed]
- Shaji, S.K.; Sunilkumar, D.; Mahalakshmi, N.V.; Kumar, G.B.; Nair, B.G. Analysis of microarray data for identification of key microRNA signatures in glioblastoma multiforme. Oncol. Lett. 2019, 18, 1938–1948. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Møller, H.G.; Rasmussen, A.P.; Andersen, H.H.; Johnsen, K.B.; Henriksen, M.; Duroux, M. A Systematic Review of MicroRNA in Glioblastoma Multiforme: Micro-modulators in the Mesenchymal Mode of Migration and Invasion. Mol. Neurobiol. 2013, 47, 131–144. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shea, A.; Harish, V.; Afzal, Z.; Chijioke, J.; Kedir, H.; Dusmatova, S.; Roy, A.; Ramalinga, M.; Harris, B.; Blancato, J.; et al. MicroRNAs in glioblastoma multiforme pathogenesis and therapeutics. Cancer Med. 2016, 5, 1917–1946. [Google Scholar] [CrossRef]
- Malzkorn, B.; Wolter, M.; Liesenberg, F.; Grzendowski, M.; Stühler, K.; Meyer, H.E.; Reifenberger, G. Identification and Functional Characterization of microRNAs Involved in the Malignant Progression of Gliomas. Brain Pathol. 2010, 20, 539–550. [Google Scholar] [CrossRef]
- Wang, B.-C.; Ma, J. Role of MicroRNAs in Malignant Glioma. Chin. Med. J. (Engl.) 2015, 128, 1238–1244. [Google Scholar] [CrossRef]
- Condrat, C.E.; Thompson, D.C.; Barbu, M.G.; Bugnar, O.L.; Boboc, A.; Cretoiu, D.; Suciu, N.; Cretoiu, S.M.; Voinea, S.C. miRNAs as Biomarkers in Disease: Latest Findings Regarding Their Role in Diagnosis and Prognosis. Cells 2020, 9, 276. [Google Scholar] [CrossRef] [Green Version]
- Hübner, M.; Moellhoff, N.; Effinger, D.; Hinske, C.L.; Hirschberger, S.; Wu, T.; Müller, M.B.; Strauß, G.; Kreth, F.-W.; Kreth, S. MicroRNA-93 acts as an “anti-inflammatory tumor suppressor” in glioblastoma. Neuro-Oncol. Adv. 2020, 2. [Google Scholar] [CrossRef]
- Buruiană, A.; Florian, Ș.I.; Florian, A.I.; Timiș, T.-L.; Mihu, C.M.; Miclăuș, M.; Oșan, S.; Hrapșa, I.; Cataniciu, R.C.; Farcaș, M.; et al. The Roles of miRNA in Glioblastoma Tumor Cell Communication: Diplomatic and Aggressive Negotiations. Int. J. Mol. Sci. 2020, 21, 1950. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Beyer, S.; Fleming, J.; Meng, W.; Singh, R.; Haque, S.J.; Chakravarti, A. The Role of miRNAs in Angiogenesis, Invasion and Metabolism and Their Therapeutic Implications in Gliomas. Cancers 2017, 9, 85. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, X.; Wang, W. Tumor suppressor microRNA-613 inhibits glioma cell proliferation, invasion and angiogenesis by targeting vascular endothelial growth factor A. Mol. Med. Rep. 2017, 16, 6729–6735. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Z.-F.; Liao, F.; Wu, H.; Dai, J. Glioma stem cells-derived exosomal miR-26a promotes angiogenesis of microvessel endothelial cells in glioma. J. Exp. Clin. Cancer Res. 2019, 38, 201. [Google Scholar] [CrossRef] [Green Version]
- Chen, X.; Yang, F.; Zhang, T.; Wang, W.; Xi, W.; Li, Y.; Zhang, D.; Huo, Y.; Zhang, J.; Yang, A.; et al. MiR-9 promotes tumorigenesis and angiogenesis and is activated by MYC and OCT4 in human glioma. J. Exp. Clin. Cancer Res. 2019, 38, 99. [Google Scholar] [CrossRef] [Green Version]
- Svensson, A.; Özen, I.; Genové, G.; Paul, G.; Bengzon, J. Endogenous Brain Pericytes Are Widely Activated and Contribute to Mouse Glioma Microvasculature. PLoS ONE 2015, 10, e0123553. [Google Scholar] [CrossRef] [Green Version]
- Jin, Y.; An, X.; Ye, Z.; Cully, B.; Wu, J.; Li, J. RGS5, a Hypoxia-inducible Apoptotic Stimulator in Endothelial Cells. J. Biol. Chem. 2009, 284, 23436–23443. [Google Scholar] [CrossRef] [Green Version]
- Matias, D.; Balça-Silva, J.; da Graça, G.C.; Wanjiru, C.M.; Macharia, L.W.; Nascimento, C.P.; Roque, N.R.; Coelho-Aguiar, J.M.; Pereira, C.M.; Dos Santos, M.F.; et al. Microglia/Astrocytes-Glioblastoma Crosstalk: Crucial Molecular Mechanisms and Microenvironmental Factors. Front. Cell. Neurosci. 2018, 12. [Google Scholar] [CrossRef] [Green Version]
- Groblewska, M.; Litman-Zawadzka, A.; Mroczko, B. The Role of Selected Chemokines and Their Receptors in the Development of Gliomas. Int. J. Mol. Sci. 2020, 21, 3704. [Google Scholar] [CrossRef]
- Morimura, T.; Neuchrist, C.; Kitz, K.; Budka, H.; Scheiner, O.; Kraft, D.; Lassmann, H. Monocyte subpopulations in human gliomas: Expression of Fc and complement receptors and correlation with tumor proliferation. Acta Neuropathol. 1990, 80, 287–294. [Google Scholar] [CrossRef]
- Roggendorf, W.; Strupp, S.; Paulus, W. Distribution and characterization of microglia/macrophages in human brain tumors. Acta Neuropathol. 1996, 92, 288–293. [Google Scholar] [CrossRef] [PubMed]
- Graeber, M.B.; Scheithauer, B.W.; Kreutzberg, G.W. Microglia in brain tumors. Glia 2002, 40, 252–259. [Google Scholar] [CrossRef] [PubMed]
- Wei, J.; Gabrusiewicz, K.; Heimberger, A. The Controversial Role of Microglia in Malignant Gliomas. Clin. Dev. Immunol. 2013, 2013. [Google Scholar] [CrossRef] [PubMed]
- Tili, E.; Michaille, J.-J.; Wernicke, D.; Alder, H.; Costinean, S.; Volinia, S.; Croce, C.M. Mutator activity induced by microRNA-155 (miR-155) links inflammation and cancer. Proc. Natl. Acad. Sci. USA 2011, 108, 4908–4913. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Prabowo, A.S.; van Scheppingen, J.; Iyer, A.M.; Anink, J.J.; Spliet, W.G.M.; van Rijen, P.C.; Meeteren, A.Y.N.S.; Aronica, E. Differential expression and clinical significance of three inflammation-related microRNAs in gangliogliomas. J. Neuroinflamm. 2015, 12. [Google Scholar] [CrossRef] [Green Version]
- Iyer, A.; Zurolo, E.; Prabowo, A.; Fluiter, K.; Spliet, W.G.M.; van Rijen, P.C.; Gorter, J.A.; Aronica, E. MicroRNA-146a: A Key Regulator of Astrocyte-Mediated Inflammatory Response. PLoS ONE 2012, 7. [Google Scholar] [CrossRef] [Green Version]
- Li, Y.; Wang, Y.; Yu, L.; Sun, C.; Cheng, D.; Yu, S.; Wang, Q.; Yan, Y.; Kang, C.; Jin, S.; et al. miR-146b-5p inhibits glioma migration and invasion by targeting MMP16. Cancer Lett. 2013, 339, 260–269. [Google Scholar] [CrossRef]
- Würdinger, T.; Tannous, B.A.; Saydam, O.; Skog, J.; Grau, S.; Soutschek, J.; Weissleder, R.; Breakefield, X.O.; Krichevsky, A.M. miR-296 Regulates Growth Factor Receptor Overexpression in Angiogenic Endothelial Cells. Cancer Cell 2008, 14, 382–393. [Google Scholar] [CrossRef] [Green Version]
- Karsy, M.; Arslan, E.; Moy, F. Current Progress on Understanding MicroRNAs in Glioblastoma Multiforme. Genes Cancer 2012, 3, 3–15. [Google Scholar] [CrossRef]
- Nikaki, A.; Piperi, C.; Papavassiliou, A.G. Role of microRNAs in gliomagenesis: Targeting miRNAs in glioblastoma multiforme therapy. Expert Opin. Investig. Drugs 2012, 21, 1475–1488. [Google Scholar] [CrossRef]
- Silber, J.; James, C.D.; Hodgson, J.G. microRNAs in Gliomas: Small Regulators of a Big Problem. Neuromol. Med. 2009, 11, 208–222. [Google Scholar] [CrossRef]
- Chen, F.; Chen, L.; He, H.; Huang, W.; Zhang, R.; Li, P.; Meng, Y.; Jiang, X. Up-regulation of microRNA-16 in Glioblastoma Inhibits the Function of Endothelial Cells and Tumor Angiogenesis by Targeting Bmi-1. Anti-Cancer Agents Med. Chem. 2016, 16, 609–620. [Google Scholar] [CrossRef]
- Lucero, R.; Zappulli, V.; Sammarco, A.; Murillo, O.D.; Cheah, P.S.; Srinivasan, S.; Tai, E.; Ting, D.T.; Wei, Z.; Roth, M.E.; et al. Glioma-Derived miRNA-Containing Extracellular Vesicles Induce Angiogenesis by Reprogramming Brain Endothelial Cells. Cell Rep. 2020, 30, 2065–2074.e4. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sakata, J.; Sasayama, T.; Tanaka, K.; Nagashima, H.; Nakada, M.; Tanaka, H.; Hashimoto, N.; Kagawa, N.; Kinoshita, M.; Nakamizo, S.; et al. MicroRNA regulating stanniocalcin-1 is a metastasis and dissemination promoting factor in glioblastoma. J. Neuro-Oncol. 2019, 142, 241–251. [Google Scholar] [CrossRef] [PubMed]
- Dudvarski Stanković, N.; Bicker, F.; Keller, S.; Jones, D.T.; Harter, P.N.; Kienzle, A.; Gillmann, C.; Arnold, P.; Golebiewska, A.; Keunen, O.; et al. EGFL7 enhances surface expression of integrin α5β1 to promote angiogenesis in malignant brain tumors. EMBO Mol. Med. 2018, 10. [Google Scholar] [CrossRef] [PubMed]
- Zhang, G.; Chen, L.; Khan, A.A.; Li, B.; Gu, B.; Lin, F.; Su, X.; Yan, J. miRNA-124-3p/neuropilin-1(NRP-1) axis plays an important role in mediating glioblastoma growth and angiogenesis. Int. J. Cancer 2018, 143, 635–644. [Google Scholar] [CrossRef] [Green Version]
- Zeng, A.; Yin, J.; Li, Y.; Li, R.; Wang, Z.; Zhou, X.; Jin, X.; Shen, F.; Yan, W.; You, Y. miR-129-5p targets Wnt5a to block PKC/ERK/NF-κB and JNK pathways in glioblastoma. Cell Death Dis. 2018, 9, 1–16. [Google Scholar] [CrossRef] [Green Version]
- Li, W.; Liu, Y.; Yang, W.; Han, X.; Li, S.; Liu, H.; Gerweck, L.E.; Fukumura, D.; Loeffler, J.S.; Yang, B.B.; et al. MicroRNA-378 enhances radiation response in ectopic and orthotopic implantation models of glioblastoma. J. Neuro-Oncol. 2018, 136, 63–71. [Google Scholar] [CrossRef] [Green Version]
- Xu, X.; Zhang, F.; Chen, X.; Ying, Q. MicroRNA-518b functions as a tumor suppressor in glioblastoma by targeting PDGFRB. Mol. Med. Rep. 2017, 16, 5326–5332. [Google Scholar] [CrossRef]
- Chung, H.J.; Choi, Y.E.; Kim, E.S.; Han, Y.-H.; Park, M.-J.; Bae, I.H. miR-29b attenuates tumorigenicity and stemness maintenance in human glioblastoma multiforme by directly targeting BCL2L2. Oncotarget 2015, 6, 18429–18444. [Google Scholar] [CrossRef]
- Zhi, T.; Jiang, K.; Xu, X.; Yu, T.; Wu, W.; Nie, E.; Zhou, X.; Jin, X.; Zhang, J.; Wang, Y.; et al. MicroRNA-520d-5p inhibits human glioma cell proliferation and induces cell cycle arrest by directly targeting PTTG1. Am. J. Transl. Res. 2017, 9, 4872–4887. [Google Scholar] [PubMed]
- Wong, H.-K.A.; Fatimy, R.E.; Onodera, C.; Wei, Z.; Yi, M.; Mohan, A.; Gowrisankaran, S.; Karmali, P.; Marcusson, E.; Wakimoto, H.; et al. The Cancer Genome Atlas Analysis Predicts MicroRNA for Targeting Cancer Growth and Vascularization in Glioblastoma. Mol. Ther. 2015, 23, 1234–1247. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rabé, M.; Dumont, S.; Álvarez-Arenas, A.; Janati, H.; Belmonte-Beitia, J.; Calvo, G.F.; Thibault-Carpentier, C.; Séry, Q.; Chauvin, C.; Joalland, N.; et al. Identification of a transient state during the acquisition of temozolomide resistance in glioblastoma. Cell Death Dis. 2020, 11, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Jiapaer, S.; Furuta, T.; Tanaka, S.; Kitabayashi, T.; Nakada, M. Potential Strategies Overcoming the Temozolomide Resistance for Glioblastoma. Neurol. Med. Chir. (Tokyo) 2018, 58, 405–421. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aasland, D.; Götzinger, L.; Hauck, L.; Berte, N.; Meyer, J.; Effenberger, M.; Schneider, S.; Reuber, E.E.; Roos, W.P.; Tomicic, M.T.; et al. Temozolomide induces senescence and repression of DNA repair pathways in glioblastoma cells via activation of ATR-CHK1, p21, and NF-kB. Cancer Res. 2018. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kanazawa, T.; Minami, Y.; Jinzaki, M.; Toda, M.; Yoshida, K.; Sasaki, H. Predictive markers for MGMT promoter methylation in glioblastomas. Neurosurg. Rev. 2019, 42, 867–876. [Google Scholar] [CrossRef]
- Fan, C.-H.; Liu, W.-L.; Cao, H.; Wen, C.; Chen, L.; Jiang, G. O6-methylguanine DNA methyltransferase as a promising target for the treatment of temozolomide-resistant gliomas. Cell Death Dis. 2013, 4, e876. [Google Scholar] [CrossRef] [Green Version]
- Jalili, C.; Rashidi, I.; Pazhouhi, M. Novel approaches to reduce temozolomide resistance in glioblastoma multiforme: A review of the literature. World Cancer Res. J. 2019, 6. [Google Scholar] [CrossRef]
- Higuchi, F.; Nagashima, H.; Ning, J.; Koerner, M.V.A.; Wakimoto, H.; Cahill, D.P. Restoration of Temozolomide Sensitivity by Poly(ADP-Ribose) Polymerase inhibitors in Mismatch Repair Deficient Glioblastoma is Independent of Base Excision Repair. Clin. Cancer Res. 2020. [Google Scholar] [CrossRef]
- Gupta, S.K.; Smith, E.J.; Mladek, A.C.; Tian, S.; Decker, P.A.; Kizilbash, S.H.; Kitange, G.J.; Sarkaria, J.N. PARP Inhibitors for Sensitization of Alkylation Chemotherapy in Glioblastoma: Impact of Blood-Brain Barrier and Molecular Heterogeneity. Front. Oncol. 2019, 8. [Google Scholar] [CrossRef] [Green Version]
- Chen, X.; Zhang, M.; Gan, H.; Wang, H.; Lee, J.-H.; Fang, D.; Kitange, G.J.; He, L.; Hu, Z.; Parney, I.F.; et al. A novel enhancer regulates MGMT expression and promotes temozolomide resistance in glioblastoma. Nat. Commun. 2018, 9, 2949. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Strobel, H.; Baisch, T.; Fitzel, R.; Schilberg, K.; Siegelin, M.D.; Karpel-Massler, G.; Debatin, K.-M.; Westhoff, M.-A. Temozolomide and Other Alkylating Agents in Glioblastoma Therapy. Biomedicines 2019, 7, 69. [Google Scholar] [CrossRef] [Green Version]
- Belter, A.; Barciszewski, J.; Barciszewska, A.-M. Revealing the epigenetic effect of temozolomide on glioblastoma cell lines in therapeutic conditions. PLoS ONE 2020, 15. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lin, C.-J.; Lee, C.-C.; Shih, Y.-L.; Lin, C.-H.; Wang, S.-H.; Chen, T.-H.; Shih, C.-M. Inhibition of Mitochondria- and Endoplasmic Reticulum Stress-Mediated Autophagy Augments Temozolomide-Induced Apoptosis in Glioma Cells. PLoS ONE 2012, 7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- He, Y.; Su, J.; Lan, B.; Gao, Y.; Zhao, J. Targeting off-target effects: Endoplasmic reticulum stress and autophagy as effective strategies to enhance temozolomide treatment. Onco Targets Ther. 2019, 12, 1857–1865. [Google Scholar] [CrossRef] [Green Version]
- Yi, G.; Huang, G.; Guo, M.; Zhang, X.; Wang, H.; Deng, S.; Li, Y.; Xiang, W.; Chen, Z.; Pan, J.; et al. Acquired temozolomide resistance in MGMT-deficient glioblastoma cells is associated with regulation of DNA repair by DHC2. Brain 2019, 142, 2352–2366. [Google Scholar] [CrossRef]
- Sun, D.-P.; Lee, Y.-W.; Chen, J.-T.; Lin, Y.-W.; Chen, R.-M. The Bradykinin-BDKRB1 Axis Regulates Aquaporin 4 Gene Expression and Consequential Migration and Invasion of Malignant Glioblastoma Cells via a Ca2+-MEK1-ERK1/2-NF-κB Mechanism. Cancers 2020, 12, 667. [Google Scholar] [CrossRef] [Green Version]
- Kim, M.; Umemura, Y.; Leung, D. Bevacizumab and Glioblastoma: Past, Present, and Future Directions. Cancer J. 2018, 24, 180–186. [Google Scholar] [CrossRef]
- Wick, W.; Gorlia, T.; Bendszus, M.; Taphoorn, M.; Sahm, F.; Harting, I.; Brandes, A.A.; Taal, W.; Domont, J.; Idbaih, A.; et al. Lomustine and Bevacizumab in Progressive Glioblastoma. N. Engl. J. Med. 2017, 377, 1954–1963. [Google Scholar] [CrossRef]
- Herrlinger, U.; Tzaridis, T.; Mack, F.; Steinbach, J.; Schlegel, U.; Sabel, M.; Hau, P.; Kortman, R.D.; Krex, D.; Grauer, O.; et al. Phase III Trial of CCNU/Temozolomide (TMZ) Combination Therapy vs. Standard TMZ Therapy for Newly Diagnosed MGMT-methylated Glioblastoma Patients. Neuro-oncology 2017, 19, vi13–vi14. [Google Scholar] [CrossRef]
- Herrlinger, U.; Tzaridis, T.; Mack, F.; Steinbach, J.P.; Schlegel, U.; Sabel, M.; Hau, P.; Kortmann, R.-D.; Krex, D.; Grauer, O.; et al. Lomustine-temozolomide combination therapy versus standard temozolomide therapy in patients with newly diagnosed glioblastoma with methylated MGMT promoter (CeTeG/NOA-09): A randomised, open-label, phase 3 trial. Lancet 2019, 393, 678–688. [Google Scholar] [CrossRef]
- Lombardi, G.; Salvo, G.L.D.; Brandes, A.A.; Eoli, M.; Rudà, R.; Faedi, M.; Lolli, I.; Pace, A.; Daniele, B.; Pasqualetti, F.; et al. Regorafenib compared with lomustine in patients with relapsed glioblastoma (REGOMA): A multicentre, open-label, randomised, controlled, phase 2 trial. Lancet Oncol. 2019, 20, 110–119. [Google Scholar] [CrossRef]
- An, Z.; Aksoy, O.; Zheng, T.; Fan, Q.-W.; Weiss, W.A. Epidermal growth factor receptor (EGFR) and EGFRvIII in glioblastoma (GBM): Signaling pathways and targeted therapies. Oncogene 2018, 37, 1561–1575. [Google Scholar] [CrossRef] [PubMed]
- Nimotuzumab Plus Radiotherapy with Concomitant and Adjuvant Temozolomide for Cerebral Glioblastoma—Full Text View—ClinicalTrials.gov. Available online: https://clinicaltrials.gov/ct2/show/NCT03388372 (accessed on 11 July 2020).
- Du, X.-J.; Li, X.-M.; Cai, L.-B.; Sun, J.-C.; Wang, S.-Y.; Wang, X.-C.; Pang, X.-L.; Deng, M.-L.; Chen, F.-F.; Wang, Z.-Q.; et al. Efficacy and safety of nimotuzumab in addition to radiotherapy and temozolomide for cerebral glioblastoma: A phase II multicenter clinical trial. J. Cancer 2019, 10, 3214–3223. [Google Scholar] [CrossRef] [Green Version]
- Yuan, A.L.; Ricks, C.B.; Bohm, A.K.; Lun, X.; Maxwell, L.; Safdar, S.; Bukhari, S.; Gerber, A.; Sayeed, W.; Bering, E.A.; et al. ABT-888 restores sensitivity in temozolomide resistant glioma cells and xenografts. PLoS ONE 2018, 13, e0202860. [Google Scholar] [CrossRef] [Green Version]
- Lemasson, B.; Wang, H.; Galbán, S.; Li, Y.; Zhu, Y.; Heist, K.A.; Tsein, C.; Chenevert, T.L.; Rehemtulla, A.; Galbán, C.J.; et al. Evaluation of Concurrent Radiation, Temozolomide and ABT-888 Treatment Followed by Maintenance Therapy with Temozolomide and ABT-888 in a Genetically Engineered Glioblastoma Mouse Model. Neoplasia 2016, 18, 82–89. [Google Scholar] [CrossRef] [Green Version]
- Gupta, S.K.; Kizilbash, S.H.; Carlson, B.L.; Mladek, A.C.; Boakye-Agyeman, F.; Bakken, K.K.; Pokorny, J.L.; Schroeder, M.A.; Decker, P.A.; Cen, L.; et al. Delineation of MGMT Hypermethylation as a Biomarker for Veliparib-Mediated Temozolomide-Sensitizing Therapy of Glioblastoma. J. Natl. Cancer Inst. 2016, 108. [Google Scholar] [CrossRef] [Green Version]
- Khasraw, M.; McDonald, K.L.; Rosenthal, M.; Lwin, Z.; Ashley, D.M.; Wheeler, H.; Barnes, E.; Foote, M.C.; Koh, E.-S.; Sulman, E.P.; et al. A randomized phase II trial of veliparib (V), radiotherapy (RT) and temozolomide (TMZ) in patients (pts) with unmethylated MGMT (uMGMT) glioblastoma (GBM). J. Clin. Oncol. 2019, 37, 2011. [Google Scholar] [CrossRef]
- National Cancer Institute (NCI). A Phase II/III Randomized Trial of Veliparib or Placebo in Combination with Adjuvant Temozolomide in Newly Diagnosed Glioblastoma with MGMT Promoter Hypermethylation. U.S. National Library of Medicine Homepage. Available online: https://clinicaltrials.gov/.
- Hanna, C.; Kurian, K.M.; Williams, K.; Watts, C.; Jackson, A.; Carruthers, R.; Strathdee, K.; Cruickshank, G.; Dunn, L.; Erridge, S.; et al. Pharmacokinetics, safety and tolerability of olaparib and temozolomide for recurrent glioblastoma: Results of the phase I OPARATIC trial. Neuro-oncology 2020. [Google Scholar] [CrossRef]
- Kiyokawa, J.; Wakimoto, H. Preclinical and Clinical Development of Oncolytic Adenovirus for The Treatment of Malignant Glioma. Oncolytic Virotherapy 2019, 8, 27–37. [Google Scholar] [CrossRef] [Green Version]
- Tejada, S.; Valle, R.D.; Gallego, J.; Alonso, M.M.; Peterkin, J. ACTR-15. A phase I study of the oncolytic virus DNX-2401 and a short course temozolomide for glioblastoma at first recurrence. Neuro-oncology 2016, 18, vi4. [Google Scholar] [CrossRef]
- Virus DNX2401 and Temozolomide in Recurrent Glioblastoma—Full Text View—ClinicalTrials.gov. Available online: https://clinicaltrials.gov/ct2/show/NCT01956734 (accessed on 11 July 2020).
- Radiation Therapy with Temozolomide and Pembrolizumab in Treating Patients with Newly Diagnosed Glioblastoma—Full Text View—ClinicalTrials.gov. Available online: https://clinicaltrials.gov/ct2/show/NCT02530502 (accessed on 11 July 2020).
- National Cancer Institute (NCI). A Randomized, Double Blind. Phase II Trial of Surgery, Radiation Therapy Plus Temozolomide and Pembrolizumab with and without HSPPC-96 in Newly Diagnosed Glioblastoma (GBM). U.S. National Library of Medicine Homepage. Available online: https://clinicaltrials.gov/.
- Park, J.-H.; Ryu, C.H.; Kim, M.J.; Jeun, S.-S. Combination Therapy for Gliomas Using Temozolomide and Interferon-Beta Secreting Human Bone Marrow Derived Mesenchymal Stem Cells. J. Korean Neurosurg. Soc. 2015, 57, 323–328. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, T.; Li, A.; Xu, Y.; Xin, Y. Momelotinib sensitizes glioblastoma cells to temozolomide by enhancement of autophagy via JAK2/STAT3 inhibition. Oncol. Rep. 2019, 41, 1883–1892. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Valtorta, S.; Dico, A.L.; Raccagni, I.; Gaglio, D.; Belloli, S.; Politi, L.S.; Martelli, C.; Diceglie, C.; Bonanomi, M.; Ercoli, G.; et al. Metformin and temozolomide, a synergic option to overcome resistance in glioblastoma multiforme models. Oncotarget 2017, 8, 113090–113104. [Google Scholar] [CrossRef] [Green Version]
- Mazurek, M.; Litak, J.; Kamieniak, P.; Kulesza, B.; Jonak, K.; Baj, J.; Grochowski, C. Metformin as Potential Therapy for High-Grade Glioma. Cancers 2020, 12, 210. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Temozolomide, Memantine Hydrochloride, Mefloquine, and Metformin Hydrochloride in Treating Patients with Glioblastoma Multiforme After Radiation Therapy—Full Text View—ClinicalTrials.gov. Available online: https://clinicaltrials.gov/ct2/show/NCT01430351 (accessed on 11 July 2020).
- Shenouda, G. Metformin and Neo-adjuvant Temozolomide and Hypofractionated Accelerated Limited-margin Radiotherapy Followed by Adjuvant Temozolomide in Patients with Glioblastoma Multiforme (M-HARTT STUDY). U.S. National Library of Medicine Homepage. Available online: https://clinicaltrials.gov/.
- Study on Low Dose Temozolomide Plus Metformin or Placebo in Patient with Recurrent or Refractory Glioblastoma—Full Text View—ClinicalTrials.gov. Available online: https://clinicaltrials.gov/ct2/show/NCT03243851 (accessed on 11 July 2020).
- Yu, Z.; Zhao, G.; Zhang, Z.; Li, Y.; Chen, Y.; Wang, N.; Zhao, Z.; Xie, G. Efficacy and safety of bevacizumab for the treatment of glioblastoma (Review). Exp. Ther. Med. 2016, 11, 371–380. [Google Scholar] [CrossRef] [Green Version]
- Garcia, J.; Hurwitz, H.I.; Sandler, A.B.; Miles, D.; Coleman, R.L.; Deurloo, R.; Chinot, O.L. Bevacizumab (Avastin®) in cancer treatment: A review of 15 years of clinical experience and future outlook. Cancer Treat. Rev. 2020, 86. [Google Scholar] [CrossRef]
- Bevacizumab and Temozolomide in Treating Older Patients with Newly-Diagnosed Glioblastoma Multiforme or Gliosarcoma—Full Text View—ClinicalTrials.gov. Available online: https://clinicaltrials.gov/ct2/show/NCT01149850 (accessed on 11 July 2020).
- Grupo Español de Investigación en Neurooncología a Phase II Open Label. Randomised Multicentric Study in Patients with Unresectable Glioblastoma Using Neo-adjuvant Treatment with Two Cycles of Temozolomide Previous Temozolomide Plus Radiation Therapy and Adjuvant Temozolomide vs. Neo-adjuvant Treatment with Two Cycles of Temozolomide Plus Bevacizumab Previous Temozolomide, Bevacizumab and Radiation Therapy and Adjuvant Temozolomide. U.S. National Library of Medicine Homepage. Available online: https://clinicaltrials.gov/.
- Robins, H.I.; Zhang, P.; Gilbert, M.R.; Chakravarti, A.; de Groot, J.F.; Grimm, S.A.; Wang, F.; Lieberman, F.S.; Krauze, A.; Trotti, A.M.; et al. A randomized phase I/II study of ABT-888 in combination with temozolomide in recurrent temozolomide resistant glioblastoma: An NRG oncology RTOG group study. J. Neurooncol. 2016, 126, 309–316. [Google Scholar] [CrossRef] [Green Version]
- Cancer Research UK A Cancer Research UK Phase I Trial of Olaparib (AZD2281), an Oral PARP Inhibitor, in Combination with Extended Low-Dose Oral Temozolomide in Patients with Relapsed Glioblastoma. U.S. National Library of Medicine Homepage. Available online: https://clinicaltrials.gov.
- Reardon, D.A.; Brandes, A.A.; Omuro, A.; Mulholland, P.; Lim, M.; Wick, A.; Baehring, J.; Ahluwalia, M.S.; Roth, P.; Bähr, O.; et al. Effect of Nivolumab vs. Bevacizumab in Patients with Recurrent Glioblastoma: The CheckMate 143 Phase 3 Randomized Clinical Trial. JAMA Oncol. 2020. [Google Scholar] [CrossRef]
- Bristol-Myers Squibb A Randomized Phase 3 Open Label. Study of Nivolumab Versus Bevacizumab and Multiple Phase 1 Safety Cohorts of Nivolumab or Nivolumab in Combination with Ipilimumab Across Different Lines of Glioblastoma. U.S. National Library of Medicine Homepage. Available online: https://clinicaltrials.gov/.
- National Cancer Institute (NCI). A Phase II Trial of Nivolumab for IDH-mutated Gliomas with Hypermutator Phenotype. U.S. National Library of Medicine Homepage. Available online: https://clinicaltrials.gov/.
- Machida, Y.; Nakagawa, M.; Matsunaga, H.; Yamaguchi, M.; Ogawara, Y.; Shima, Y.; Yamagata, K.; Katsumoto, T.; Hattori, A.; Itoh, M.; et al. A Potent Blood–Brain Barrier-Permeable Mutant IDH1 Inhibitor Suppresses the Growth of Glioblastoma with IDH1 Mutation in a Patient-Derived Orthotopic Xenograft Model. Mol. Cancer Ther. 2019. [Google Scholar] [CrossRef]
- Study of DS-1001b in Patients with Gene IDH1-Mutated Gliomas—Full Text View—ClinicalTrials.gov. Available online: https://clinicaltrials.gov/ct2/show/NCT03030066 (accessed on 11 July 2020).
- Natsume, A.; Wakabayashi, T.; Miyakita, Y.; Narita, Y.; Mineharu, Y.; Arakawa, Y.; Yamasaki, F.; Sugiyama, K.; Hata, N.; Muragaki, Y.; et al. Phase I study of a brain penetrant mutant IDH1 inhibitor DS-1001b in patients with recurrent or progressive IDH1 mutant gliomas. J. Clin. Oncol. 2019, 37, 2004. [Google Scholar] [CrossRef]
- Reardon, D.A.; De Groot, J.F.; Colman, H.; Jordan, J.T.; Daras, M.; Clarke, J.L.; Nghiemphu, P.L.; Gaffey, S.C.; Peters, K.B. Safety of pembrolizumab in combination with bevacizumab in recurrent glioblastoma (rGBM). J. Clin. Oncol. 2016, 34, 2010. [Google Scholar] [CrossRef]
- MD, D.R. Phase II Study of Pembrolizumab (MK-3475) With and Without Bevacizumab for Recurrent Glioblastoma. U.S. National Library of Medicine Homepage. Available online: https://clinicaltrials.gov/.
- Kim, J.E.; Patel, M.A.; Mangraviti, A.; Kim, E.S.; Theodros, D.; Velarde, E.; Liu, A.; Sankey, E.W.; Tam, A.; Xu, H.; et al. Combination Therapy with Anti-PD-1, Anti-TIM-3, and Focal Radiation Results in Regression of Murine Gliomas. Clin. Cancer Res. 2016. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hung, A.L.; Maxwell, R.; Theodros, D.; Belcaid, Z.; Mathios, D.; Luksik, A.S.; Kim, E.; Wu, A.; Xia, Y.; Garzon-Muvdi, T.; et al. TIGIT and PD-1 dual checkpoint blockade enhances antitumor immunity and survival in GBM. Oncoimmunology 2018, 7, e1466769. [Google Scholar] [CrossRef]
- Hanif, F.; Muzaffar, K.; Perveen, K.; Malhi, S.M.; Simjee, S.U. Glioblastoma Multiforme: A Review of its Epidemiology and Pathogenesis through Clinical Presentation and Treatment. Asian Pac. J. Cancer Prev. 2017, 18, 3–9. [Google Scholar] [CrossRef]
- Yin, J.; Zeng, A.; Zhang, Z.; Shi, Z.; Yan, W.; You, Y. Exosomal transfer of miR-1238 contributes to temozolomide-resistance in glioblastoma. EBioMedicine 2019, 42, 238–251. [Google Scholar] [CrossRef] [Green Version]
- Zeng, A.; Wei, Z.; Yan, W.; Yin, J.; Huang, X.; Zhou, X.; Li, R.; Shen, F.; Wu, W.; Wang, X.; et al. Exosomal transfer of miR-151a enhances chemosensitivity to temozolomide in drug-resistant glioblastoma. Cancer Lett. 2018, 436, 10–21. [Google Scholar] [CrossRef]
- Figueroa, J.; Phillips, L.M.; Shahar, T.; Hossain, A.; Gumin, J.; Kim, H.; Bean, A.J.; Calin, G.A.; Fueyo, J.; Walters, E.T.; et al. Exosomes from Glioma-Associated Mesenchymal Stem Cells Increase the Tumorigenicity of Glioma Stem-like Cells via Transfer of miR-1587. Cancer Res. 2017, 77, 5808–5819. [Google Scholar] [CrossRef] [Green Version]
- Seymour, T.; Nowak, A.; Kakulas, F. Targeting Aggressive Cancer Stem Cells in Glioblastoma. Front. Oncol. 2015, 5. [Google Scholar] [CrossRef]
- Yang, L.; Li, N.; Yan, Z.; Li, C.; Zhao, Z. MiR-29a-Mediated CD133 Expression Contributes to Cisplatin Resistance in CD133+ Glioblastoma Stem Cells. J. Mol. Neurosci. 2018, 66, 369–377. [Google Scholar] [CrossRef]
- Xiao, S.; Yang, Z.; Qiu, X.; Lv, R.; Liu, J.; Wu, M.; Liao, Y.; Liu, Q. miR-29c contribute to glioma cells temozolomide sensitivity by targeting O 6 -methylguanine-DNA methyltransferases indirectly. Oncotarget 2016, 7, 50229–50238. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, S.; Zeng, A.; Hu, Q.; Yan, W.; Liu, Y.; You, Y. miR-423-5p contributes to a malignant phenotype and temozolomide chemoresistance in glioblastomas. Neuro-oncology 2017, 19, 55–65. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yan, W.; Liu, Y.; Yang, P.; Wang, Z.; You, Y.; Jiang, T. MicroRNA profiling of Chinese primary glioblastoma reveals a temozolomide-chemoresistant subtype. Oncotarget 2015, 6, 11676–11682. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Luo, W.; Yan, D.; Song, Z.; Zhu, X.; Liu, X.; Li, X.; Zhao, S. miR-126-3p sensitizes glioblastoma cells to temozolomide by inactivating Wnt/β-catenin signaling via targeting SOX2. Life Sci. 2019, 226, 98–106. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Xu, J.; Li, H.; Sun, C.; Yu, L.; Li, Y.; Shi, C.; Zhou, X.; Bian, X.; Ping, Y.; et al. miR-146b-5p functions as a tumor suppressor by targeting TRAF6 and predicts the prognosis of human gliomas. Oncotarget 2015, 6, 29129–29142. [Google Scholar] [CrossRef] [Green Version]
- Lin, Y.; Qiu, Y.; Xu, C.; Liu, Q.; Peng, B.; Kaufmann, G.F.; Chen, X.; Lan, B.; Wei, C.; Lu, D.; et al. Functional Role of Asparaginyl Endopeptidase Ubiquitination by TRAF6 in Tumor Invasion and Metastasis. J. Natl. Cancer Inst. 2014, 106. [Google Scholar] [CrossRef] [Green Version]
- Sun, H.; Li, X.; Fan, L.; Wu, G.; Li, M.; Fang, J. TRAF6 is upregulated in colon cancer and promotes proliferation of colon cancer cells. Int. J. Biochem. Cell Biol. 2014, 53, 195–201. [Google Scholar] [CrossRef]
- Yan, Y.; Xu, Z.; Dai, S.; Qian, L.; Sun, L.; Gong, Z. Targeting autophagy to sensitive glioma to temozolomide treatment. J. Exp. Clin. Cancer Res. 2016, 35, 23. [Google Scholar] [CrossRef] [Green Version]
- Huang, B.-S.; Luo, Q.-Z.; Han, Y.; Huang, D.; Tang, Q.-P.; Wu, L.-X. MiR-223/PAX6 Axis Regulates Glioblastoma Stem Cell Proliferation and the Chemo Resistance to TMZ via Regulating PI3K/Akt Pathway. J. Cell. Biochem. 2017, 118, 3452–3461. [Google Scholar] [CrossRef]
- Cheng, Q.; Ma, X.; Cao, H.; Chen, Z.; Wan, X.; Chen, R.; Peng, R.; Huang, J.; Jiang, B. Role of miR-223/paired box 6 signaling in temozolomide chemoresistance in glioblastoma multiforme cells. Mol. Med. Rep. 2017, 15, 597–604. [Google Scholar] [CrossRef]
- Munoz, J.L.; Walker, N.D.; Mareedu, S.; Pamarthi, S.H.; Sinha, G.; Greco, S.J.; Rameshwar, P. Cycling Quiescence in Temozolomide Resistant Glioblastoma Cells Is Partly Explained by microRNA-93 and -193-Mediated Decrease of Cyclin, D. Front. Pharmacol. 2019, 10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jia, B.; Liu, W.; Gu, J.; Wang, J.; Lv, W.; Zhang, W.; Hao, Q.; Pang, Z.; Mu, N.; Zhang, W.; et al. MiR-7-5p suppresses stemness and enhances temozolomide sensitivity of drug-resistant glioblastoma cells by targeting Yin Yang 1. Exp. Cell Res. 2019, 375, 73–81. [Google Scholar] [CrossRef] [PubMed]
- Sarvagalla, S.; Kolapalli, S.P.; Vallabhapurapu, S. The Two Sides of YY1 in Cancer: A Friend and a Foe. Front. Oncol. 2019, 9. [Google Scholar] [CrossRef] [Green Version]
- Li, J.; Song, J.; Guo, F. miR-186 reverses cisplatin resistance and inhibits the formation of the glioblastoma-initiating cell phenotype by degrading Yin Yang 1 in glioblastoma. Int. J. Mol. Med. 2019, 43, 517–524. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, S.; Qi, P.; Zhang, T.; Li, F.; He, X. The HIF-1α/miR-224-3p/ATG5 axis affects cell mobility and chemosensitivity by regulating hypoxia-induced protective autophagy in glioblastoma and astrocytoma. Oncol. Rep. 2019, 41, 1759–1768. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, H.; Chen, L.; Li, J.; Zhou, Q.; Huang, A.; Liu, W.; Wang, K.; Gao, L.; Qi, S.; Lu, Y. miR-519a enhances chemosensitivity and promotes autophagy in glioblastoma by targeting STAT3/Bcl2 signaling pathway. J. Hematol. Oncol. 2018, 11, 70. [Google Scholar] [CrossRef] [Green Version]
- Stojcheva, N.; Schechtmann, G.; Sass, S.; Roth, P.; Florea, A.-M.; Stefanski, A.; Stühler, K.; Wolter, M.; Müller, N.S.; Theis, F.J.; et al. MicroRNA-138 promotes acquired alkylator resistance in glioblastoma by targeting the Bcl-2-interacting mediator BIM. Oncotarget 2016, 7, 12937–12950. [Google Scholar] [CrossRef] [Green Version]
- Zhou, D.; Wan, Y.; Xie, D.; Wang, Y.; Wei, J.; Yan, Q.; Lu, P.; Mo, L.; Xie, J.; Yang, S.; et al. DNMT1 mediates chemosensitivity by reducing methylation of miRNA-20a promoter in glioma cells. Exp. Mol. Med. 2015, 47, e182. [Google Scholar] [CrossRef] [Green Version]
- Vietheer, J.-M.; Rieger, J.; Wagner, M.; Senft, C.; Tichy, J.; Foerch, C. Serum concentrations of glial fibrillary acidic protein (GFAP) do not indicate tumor recurrence in patients with glioblastoma. J. Neurooncol. 2017, 135, 193–199. [Google Scholar] [CrossRef]
- Fortier, A.-M.; Asselin, E.; Cadrin, M. Keratin 8 and 18 loss in epithelial cancer cells increases collective cell migration and cisplatin sensitivity through claudin1 up-regulation. J. Biol. Chem. 2013, 288, 11555–11571. [Google Scholar] [CrossRef] [Green Version]
- Chattopadhyay, R.; Das, S.; Maiti, A.K.; Boldogh, I.; Xie, J.; Hazra, T.K.; Kohno, K.; Mitra, S.; Bhakat, K.K. Regulatory role of human AP-endonuclease (APE1/Ref-1) in YB-1-mediated activation of the multidrug resistance gene MDR1. Mol. Cell. Biol. 2008, 28, 7066–7080. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fantini, D.; Vascotto, C.; Deganuto, M.; Bivi, N.; Gustincich, S.; Marcon, G.; Quadrifoglio, F.; Damante, G.; Bhakat, K.K.; Mitra, S.; et al. APE1/Ref-1 regulates PTEN expression mediated by Egr-1. Free Radic. Res. 2008, 42, 20–29. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chun-Zhi, Z.; Lei, H.; An-Ling, Z.; Yan-Chao, F.; Xiao, Y.; Guang-Xiu, W.; Zhi-Fan, J.; Pei-Yu, P.; Qing-Yu, Z.; Chun-Sheng, K. MicroRNA-221 and microRNA-222 regulate gastric carcinoma cell proliferation and radioresistance by targeting PTEN. BMC Cancer 2010, 10, 367. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, J.-J.; Lin, J.; Yang, H.; Kong, W.; He, L.; Ma, X.; Coppola, D.; Cheng, J.Q. MicroRNA-221/222 negatively regulates estrogen receptor α and is associated with tamoxifen resistance in breast cancer. J. Biol. Chem. 2016, 291, 22859. [Google Scholar] [CrossRef] [Green Version]
- Acunzo, M.; Visone, R.; Romano, G.; Veronese, A.; Lovat, F.; Palmieri, D.; Bottoni, A.; Garofalo, M.; Gasparini, P.; Condorelli, G.; et al. miR-130a targets MET and induces TRAIL-sensitivity in NSCLC by downregulating miR-221 and 222. Oncogene 2012, 31, 634–642. [Google Scholar] [CrossRef] [Green Version]
- Zhang, C.; Kang, C.; Wang, P.; Cao, Y.; Lv, Z.; Yu, S.; Wang, G.; Zhang, A.; Jia, Z.; Han, L.; et al. MicroRNA-221 and -222 Regulate Radiation Sensitivity by Targeting the PTEN Pathway. Int. J. Radiat. Oncol. Biol. Phys. 2011, 80, 240–248. [Google Scholar] [CrossRef]
- Miller, T.E.; Ghoshal, K.; Ramaswamy, B.; Roy, S.; Datta, J.; Shapiro, C.L.; Jacob, S.; Majumder, S. MicroRNA-221/222 Confers Tamoxifen Resistance in Breast Cancer by Targeting p27Kip1. J. Biol. Chem. 2008, 283, 29897–29903. [Google Scholar] [CrossRef] [Green Version]
- Li, R.; Gao, K.; Luo, H.; Wang, X.; Shi, Y.; Dong, Q.; Luan, W.; You, Y. Identification of intrinsic subtype-specific prognostic microRNAs in primary glioblastoma. J. Exp. Clin. Cancer Res. 2014, 33, 9. [Google Scholar] [CrossRef] [Green Version]
- Tivnan, A.; Heilinger, T.; Ramsey, J.M.; O’Connor, G.; Pokorny, J.L.; Sarkaria, J.N.; Stringer, B.W.; Day, B.W.; Boyd, A.W.; Kim, E.L.; et al. Anti-GD2-ch14.18/CHO coated nanoparticles mediate glioblastoma (GBM)-specific delivery of the aromatase inhibitor, Letrozole, reducing proliferation, migration and chemoresistance in patient-derived GBM tumor cells. Oncotarget 2017, 8, 16605–16620. [Google Scholar] [CrossRef]
- Di Leva, G.; Piovan, C.; Gasparini, P.; Ngankeu, A.; Taccioli, C.; Briskin, D.; Cheung, D.G.; Bolon, B.; Anderlucci, L.; Alder, H.; et al. Estrogen mediated-activation of miR-191/425 cluster modulates tumorigenicity of breast cancer cells depending on estrogen receptor status. PLoS Genet. 2013, 9, e1003311. [Google Scholar] [CrossRef]
- Pyko, I.V.; Nakada, M.; Sabit, H.; Teng, L.; Furuyama, N.; Hayashi, Y.; Kawakami, K.; Minamoto, T.; Fedulau, A.S.; Hamada, J. Glycogen synthase kinase 3β inhibition sensitizes human glioblastoma cells to temozolomide by affecting O6-methylguanine DNA methyltransferase promoter methylation via c-Myc signaling. Carcinogenesis 2013, 34, 2206–2217. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fu, Y.; Hu, D.; Qiu, J.; Xie, X.; Ye, F.; Lu, W.-G. Overexpression of glycogen synthase kinase-3 in ovarian carcinoma cells with acquired paclitaxel resistance. Int. J. Gynecol. Cancer 2011, 21, 439–444. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grassilli, E.; Ianzano, L.; Bonomo, S.; Missaglia, C.; Cerrito, M.G.; Giovannoni, R.; Masiero, L.; Lavitrano, M. GSK3A is redundant with GSK3B in modulating drug resistance and chemotherapy-induced necroptosis. PLoS ONE 2014, 9, e100947. [Google Scholar] [CrossRef] [PubMed]
- Tian, T.; Mingyi, M.; Qiu, X.; Qiu, Y. MicroRNA-101 reverses temozolomide resistance by inhibition of GSK3β in glioblastoma. Oncotarget 2016, 7, 79584–79595. [Google Scholar] [CrossRef] [PubMed]
- Salvatore, D.; Lo Dico, A.; Martelli, C.; Diceglie, C.; Ottobrini, L. PET biomarkers and probes for treatment response assessment in glioblastoma: A work in progress. Clin. Transl. Imaging 2019, 7, 285–294. [Google Scholar] [CrossRef]
- John, F.; Bosnyák, E.; Robinette, N.L.; Amit-Yousif, A.J.; Barger, G.R.; Shah, K.D.; Michelhaugh, S.K.; Klinger, N.V.; Mittal, S.; Juhász, C. Multimodal imaging-defined subregions in newly diagnosed glioblastoma: Impact on overall survival. Neuro-oncology 2019, 21, 264–273. [Google Scholar] [CrossRef] [Green Version]
- Verduin, M.; Compter, I.; Steijvers, D.; Postma, A.A.; Eekers, D.B.P.; Anten, M.M.; Ackermans, L.; Ter Laan, M.; Leijenaar, R.T.H.; van de Weijer, T.; et al. Noninvasive Glioblastoma Testing: Multimodal Approach to Monitoring and Predicting Treatment Response. Dis. Markers 2018, 2018, 2908609. [Google Scholar] [CrossRef] [Green Version]
- Ata, E.S.; Turgut, M.; Eraslan, C.; Dayanır, Y.Ö. Comparison between dynamic susceptibility contrast magnetic resonance imaging and arterial spin labeling techniques in distinguishing malignant from benign brain tumors. Eur. J. Radiol. 2016, 85, 1545–1553. [Google Scholar] [CrossRef]
- Pope, W.B.; Brandal, G. Conventional and advanced magnetic resonance imaging in patients with high-grade glioma. Q. J. Nucl. Med. Mol. Imaging 2018, 62, 239–253. [Google Scholar] [CrossRef]
- Zhang, H.; Ma, L.; Shu, C.; Wang, Y.; Dong, L. Diagnostic accuracy of diffusion MRI with quantitative ADC measurements in differentiating glioma recurrence from radiation necrosis. J. Neurol. Sci. 2015, 351, 65–71. [Google Scholar] [CrossRef]
- Valtorta, S.; Nicolini, G.; Tripodi, F.; Meregalli, C.; Cavaletti, G.; Avezza, F.; Crippa, L.; Bertoli, G.; Sanvito, F.; Fusi, P.; et al. A novel AMPK activator reduces glucose uptake and inhibits tumor progression in a mouse xenograft model of colorectal cancer. Investig. New Drugs 2014, 32, 1123–1133. [Google Scholar] [CrossRef] [PubMed]
- Venneti, S.; Dunphy, M.P.; Zhang, H.; Pitter, K.L.; Zanzonico, P.; Campos, C.; Carlin, S.D.; La Rocca, G.; Lyashchenko, S.; Ploessl, K.; et al. Glutamine-based PET imaging facilitates enhanced metabolic evaluation of gliomas in vivo. Sci. Transl. Med. 2015, 7, 274ra17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miner, M.W.; Liljenbäck, H.; Virta, J.; Merisaari, J.; Oikonen, V.; Westermarck, J.; Li, X.-G.; Roivainen, A. (2S, 4R)-4-[18F]Fluoroglutamine for In vivo PET Imaging of Glioma Xenografts in Mice: An Evaluation of Multiple Pharmacokinetic Models. Mol. Imaging Biol. 2020, 22, 969–978. [Google Scholar] [CrossRef] [Green Version]
- Dunphy, M.P.S.; Harding, J.J.; Venneti, S.; Zhang, H.; Burnazi, E.M.; Bromberg, J.; Omuro, A.M.; Hsieh, J.J.; Mellinghoff, I.K.; Staton, K.; et al. In Vivo PET Assay of Tumor Glutamine Flux and Metabolism: In-Human Trial of 18F-(2 S, 4 R)-4-Fluoroglutamine. Radiology 2018, 287, 667–675. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, S.; Kim, D.; Kim, S.H.; Park, M.; Chang, J.H.; Yun, M. The roles of 11C-acetate PET/CT in predicting tumor differentiation and survival in patients with cerebral glioma. Eur. J. Nucl. Med. Mol. Imaging 2018, 45, 1012–1020. [Google Scholar] [CrossRef] [PubMed]
- Koyasu, S.; Shimizu, Y.; Morinibu, A.; Saga, T.; Nakamoto, Y.; Togashi, K.; Harada, H. Increased 14C-acetate accumulation in IDH-mutated human glioblastoma: Implications for detecting IDH-mutated glioblastoma with 11C-acetate PET imaging. J. Neurooncol. 2019, 145, 441–447. [Google Scholar] [CrossRef]
- Lukas, R.V.; Juhász, C.; Wainwright, D.A.; James, C.D.; Kennedy, E.; Stupp, R.; Lesniak, M.S. Imaging tryptophan uptake with positron emission tomography in glioblastoma patients treated with indoximod. J. Neurooncol. 2019, 141, 111–120. [Google Scholar] [CrossRef]
- Guastella, A.R.; Michelhaugh, S.K.; Klinger, N.V.; Kupsky, W.J.; Polin, L.A.; Muzik, O.; Juhász, C.; Mittal, S. Tryptophan PET Imaging of the Kynurenine Pathway in Patient-Derived Xenograft Models of Glioblastoma. Mol. Imaging 2016. [Google Scholar] [CrossRef] [Green Version]
- Bosnyák, E.; Michelhaugh, S.K.; Klinger, N.V.; Kamson, D.O.; Barger, G.R.; Mittal, S.; Juhász, C. Prognostic Molecular and Imaging Biomarkers in Primary Glioblastoma. Clin. Nucl. Med. 2017, 42, 341–347. [Google Scholar] [CrossRef] [Green Version]
- John, F.; Robinette, N.L.; Amit-Yousif, A.J.; Bosnyák, E.; Barger, G.R.; Shah, K.D.; Mittal, S.; Juhász, C. Multimodal Imaging of Nonenhancing Glioblastoma Regions. Mol. Imaging 2019, 18. [Google Scholar] [CrossRef]
- Xin, Y.; Gao, X.; Liu, L.; Ge, W.-P.; Jain, M.K.; Cai, H. Evaluation of l-1-[18F] Fluoroethyl-Tryptophan for PET Imaging of Cancer. Mol. Imaging Biol. 2019, 21. [Google Scholar] [CrossRef] [PubMed]
- Henderson, F.; Brem, S.; O’Rourke, D.M.; Nasrallah, M.; Buch, V.P.; Young, A.J.; Doot, R.K.; Pantel, A.; Desai, A.; Bagley, S.J.; et al. 18F-Fluciclovine PET to distinguish treatment-related effects from disease progression in recurrent glioblastoma: PET fusion with MRI guides neurosurgical sampling. Neuro-Oncol. Pract. 2020, 7, 152–157. [Google Scholar] [CrossRef] [PubMed]
- Verhoeven, J.; Hulpia, F.; Kersemans, K.; Bolcaen, J.; De Lombaerde, S.; Goeman, J.; Descamps, B.; Hallaert, G.; Van den Broecke, C.; Deblaere, K.; et al. New fluoroethyl phenylalanine analogues as potential LAT1-targeting PET tracers for glioblastoma. Sci. Rep. 2019, 9, 2878. [Google Scholar] [CrossRef] [PubMed]
- Nozaki, S.; Nakatani, Y.; Mawatari, A.; Shibata, N.; Hume, W.E.; Hayashinaka, E.; Wada, Y.; Doi, H.; Watanabe, Y. 18 F-FIMP: A LAT1-specific PET probe for discrimination between tumor tissue and inflammation. Sci. Rep. 2019, 9, 15718. [Google Scholar] [CrossRef]
- Schwarzenberg, J.; Czernin, J.; Cloughesy, T.F.; Ellingson, B.M.; Pope, W.B.; Geist, C.; Dahlbom, M.; Silverman, D.H.S.; Satyamurthy, N.; Phelps, M.E.; et al. 3′-deoxy-3′-18F-fluorothymidine PET and MRI for early survival predictions in patients with recurrent malignant glioma treated with bevacizumab. J. Nucl. Med. 2012, 53, 29–36. [Google Scholar] [CrossRef] [Green Version]
- Viel, T.; Schelhaas, S.; Wagner, S.; Wachsmuth, L.; Schwegmann, K.; Kuhlmann, M.; Faber, C.; Kopka, K.; Schäfers, M.; Jacobs, A.H. Early assessment of the efficacy of temozolomide chemotherapy in experimental glioblastoma using [18F] FLT-PET imaging. PLoS ONE 2013, 8, e67911. [Google Scholar] [CrossRef] [Green Version]
- O’Halloran, P.J.; Viel, T.; Murray, D.W.; Wachsmuth, L.; Schwegmann, K.; Wagner, S.; Kopka, K.; Jarzabek, M.A.; Dicker, P.; Hermann, S.; et al. Mechanistic interrogation of combination bevacizumab/dual PI3K/mTOR inhibitor response in glioblastoma implementing novel MR and PET imaging biomarkers. Eur. J. Nucl. Med. Mol. Imaging 2016, 43, 1673–1683. [Google Scholar] [CrossRef]
- Lo Dico, A.; Martelli, C.; Valtorta, S.; Raccagni, I.; Diceglie, C.; Belloli, S.; Gianelli, U.; Vaira, V.; Politi, L.S.; Bosari, S.; et al. Identification of imaging biomarkers for the assessment of tumour response to different treatments in a preclinical glioma model. Eur. J. Nucl. Med. Mol. Imaging 2015, 42, 1093–1105. [Google Scholar] [CrossRef]
- Corroyer-Dulmont, A.; Pérès, E.A.; Gérault, A.N.; Savina, A.; Bouquet, F.; Divoux, D.; Toutain, J.; Ibazizène, M.; MacKenzie, E.T.; Barré, L.; et al. Multimodal imaging based on MRI and PET reveals [(18)F]FLT PET as a specific and early indicator of treatment efficacy in a preclinical model of recurrent glioblastoma. Eur. J. Nucl. Med. Mol. Imaging 2016, 43, 682–694. [Google Scholar] [CrossRef]
- Lo Dico, A.; Valtorta, S.; Martelli, C.; Belloli, S.; Gianelli, U.; Tosi, D.; Bosari, S.; Degrassi, A.; Russo, M.; Raccagni, I.; et al. Validation of an engineered cell model for in vitro and in vivo HIF-1α evaluation by different imaging modalities. Mol. Imaging Biol. 2014, 16, 210–223. [Google Scholar] [CrossRef]
- Wehrl, H.F.; Schwab, J.; Hasenbach, K.; Reischl, G.; Tabatabai, G.; Quintanilla-Martinez, L.; Jiru, F.; Chughtai, K.; Kiss, A.; Cay, F.; et al. Multimodal elucidation of choline metabolism in a murine glioma model using magnetic resonance spectroscopy and 11C-choline positron emission tomography. Cancer Res. 2013, 73, 1470–1480. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Takei, H.; Shinoda, J.; Ikuta, S.; Maruyama, T.; Muragaki, Y.; Kawasaki, T.; Ikegame, Y.; Okada, M.; Ito, T.; Asano, Y.; et al. Usefulness of positron emission tomography for differentiating gliomas according to the 2016 World Health Organization classification of tumors of the central nervous system. J. Neurosurg. 2019, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Raccagni, I.; Valtorta, S.; Moresco, R.M.; Belloli, S. Tumour hypoxia: Lessons learnt from preclinical imaging. Clin. Transl. Imaging 2017, 5, 407–425. [Google Scholar] [CrossRef]
- Gangemi, V.; Mignogna, C.; Guzzi, G.; Lavano, A.; Bongarzone, S.; Cascini, G.L.; Sabatini, U. Impact of [64Cu] [Cu(ATSM)] PET/CT in the evaluation of hypoxia in a patient with Glioblastoma: A case report. BMC Cancer 2019, 19, 1197. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Toriihara, A.; Ohtake, M.; Tateishi, K.; Hino-Shishikura, A.; Yoneyama, T.; Kitazume, Y.; Inoue, T.; Kawahara, N.; Tateishi, U. Prognostic implications of 62Cu-diacetyl-bis (N4-methylthiosemicarbazone) PET/CT in patients with glioma. Ann. Nucl. Med. 2018, 32, 264–271. [Google Scholar] [CrossRef]
- Jin, Z.-H.; Tsuji, A.B.; Degardin, M.; Sugyo, A.; Yoshii, Y.; Nagatsu, K.; Zhang, M.-R.; Fujibayashi, Y.; Dumy, P.; Boturyn, D.; et al. Uniform intratumoral distribution of radioactivity produced using two different radioagents, 64Cu-cyclam-RAFT-c(-RGDfK-)4 and 64Cu-ATSM, improves therapeutic efficacy in a small animal tumor model. EJNMMI Res. 2018, 8, 54. [Google Scholar] [CrossRef]
- Yoshii, Y.; Matsumoto, H.; Yoshimoto, M.; Zhang, M.-R.; Oe, Y.; Kurihara, H.; Narita, Y.; Jin, Z.-H.; Tsuji, A.B.; Yoshinaga, K.; et al. Multiple Administrations of 64Cu-ATSM as a Novel Therapeutic Option for Glioblastoma: A Translational Study Using Mice with Xenografts. Transl. Oncol. 2017, 11, 24–30. [Google Scholar] [CrossRef]
- Pérès, E.A.; Toutain, J.; Paty, L.-P.; Divoux, D.; Ibazizène, M.; Guillouet, S.; Barré, L.; Vidal, A.; Cherel, M.; Bourgeois, M.; et al. 64Cu-ATSM/64Cu-Cl2 and their relationship to hypoxia in glioblastoma: A preclinical study. EJNMMI Res. 2019, 9, 114. [Google Scholar] [CrossRef] [Green Version]
- Su, Z.; Roncaroli, F.; Durrenberger, P.F.; Coope, D.J.; Karabatsou, K.; Hinz, R.; Thompson, G.; Turkheimer, F.E.; Janczar, K.; Du Plessis, D.; et al. The 18-kDa mitochondrial translocator protein in human gliomas: An 11C-(R)PK11195 PET imaging and neuropathology study. J. Nucl. Med. 2015, 56, 512–517. [Google Scholar] [CrossRef] [Green Version]
- Zinnhardt, B.; Pigeon, H.; Thézé, B.; Viel, T.; Wachsmuth, L.; Fricke, I.B.; Schelhaas, S.; Honold, L.; Schwegmann, K.; Wagner, S.; et al. Combined PET Imaging of the Inflammatory Tumor Microenvironment Identifies Margins of Unique Radiotracer Uptake. Cancer Res. 2017, 77, 1831–1841. [Google Scholar] [CrossRef] [Green Version]
- Zinnhardt, B.; Müther, M.; Roll, W.; Backhaus, P.; Jeibmann, A.; Foray, C.; Barca, C.; Döring, C.; Tavitian, B.; Dollé, F.; et al. TSPO imaging-guided characterization of the immunosuppressive myeloid tumor microenvironment in patients with malignant glioma. Neuro-oncology 2020, 22, 1030–1043. [Google Scholar] [CrossRef] [PubMed]
- Pigeon, H.; Pérès, E.A.; Truillet, C.; Jego, B.; Boumezbeur, F.; Caillé, F.; Zinnhardt, B.; Jacobs, A.H.; Le Bihan, D.; Winkeler, A. TSPO-PET and diffusion-weighted MRI for imaging a mouse model of infiltrative human glioma. Neuro-oncology 2019, 21, 755–764. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Belloli, S.; Zanotti, L.; Murtaj, V.; Mazzon, C.; Di Grigoli, G.; Monterisi, C.; Masiello, V.; Iaccarino, L.; Cappelli, A.; Poliani, P.L.; et al. 18F-VC701-PET and MRI in the in vivo neuroinflammation assessment of a mouse model of multiple sclerosis. J. Neuroinflamm. 2018, 15, 33. [Google Scholar] [CrossRef] [PubMed]
- Albert, N.L.; Unterrainer, M.; Fleischmann, D.F.; Lindner, S.; Vettermann, F.; Brunegraf, A.; Vomacka, L.; Brendel, M.; Wenter, V.; Wetzel, C.; et al. TSPO PET for glioma imaging using the novel ligand 18F-GE-180: First results in patients with glioblastoma. Eur. J. Nucl. Med. Mol. Imaging 2017, 44, 2230–2238. [Google Scholar] [CrossRef] [PubMed]
- Unterrainer, M.; Fleischmann, D.F.; Vettermann, F.; Ruf, V.; Kaiser, L.; Nelwan, D.; Lindner, S.; Brendel, M.; Wenter, V.; Stöcklein, S.; et al. TSPO PET, tumour grading and molecular genetics in histologically verified glioma: A correlative 18F-GE-180 PET study. Eur. J. Nucl. Med. Mol. Imaging 2020, 47, 1368–1380. [Google Scholar] [CrossRef] [PubMed]
- Blair, A.; Zmuda, F.; Malviya, G.; Tavares, A.A.S.; Tamagnan, G.D.; Chalmers, A.J.; Dewar, D.; Pimlott, S.L.; Sutherland, A. A novel 18F-labelled high affinity agent for PET imaging of the translocator protein. Chem. Sci. 2015, 6, 4772–4777. [Google Scholar] [CrossRef] [Green Version]
- Perrone, M.; Moon, B.S.; Park, H.S.; Laquintana, V.; Jung, J.H.; Cutrignelli, A.; Lopedota, A.; Franco, M.; Kim, S.E.; Lee, B.C.; et al. A Novel PET Imaging Probe for the Detection and Monitoring of Translocator Protein 18 kDa Expression in Pathological Disorders. Sci. Rep. 2016, 6, 20422. [Google Scholar] [CrossRef]
- Sasikumar, A.; Joy, A.; Pillai, M.R.A.; Nanabala, R.; Thomas, B. 68Ga-PSMA PET/CT Imaging in Multiple Myeloma. Clin. Nucl. Med. 2017, 42, e126–e127. [Google Scholar] [CrossRef]
- Fragomeni, R.A.S.; Menke, J.R.; Holdhoff, M.; Ferrigno, C.; Laterra, J.J.; Solnes, L.B.; Javadi, M.S.; Szabo, Z.; Pomper, M.G.; Rowe, S.P. Prostate-Specific Membrane Antigen–Targeted Imaging With [18F] DCFPyL in High-Grade Gliomas. Clin. Nucl. Med. 2017, 42, e433–e435. [Google Scholar] [CrossRef] [Green Version]
- Unterrainer, M.; Niyazi, M.; Ruf, V.; Bartenstein, P.; Albert, N.L. The endothelial prostate-specific membrane antigen is highly expressed in gliosarcoma and visualized by [68Ga]-PSMA-11 PET: A theranostic outlook for brain tumor patients? Neuro-oncology 2017, 19, 1698–1699. [Google Scholar] [CrossRef]
- Matsuda, M.; Ishikawa, E.; Yamamoto, T.; Hatano, K.; Joraku, A.; Iizumi, Y.; Masuda, Y.; Nishiyama, H.; Matsumura, A. Potential use of prostate specific membrane antigen (PSMA) for detecting the tumor neovasculature of brain tumors by PET imaging with 89Zr-Df-IAB2M anti-PSMA minibody. J. Neurooncol. 2018, 138, 581–589. [Google Scholar] [CrossRef] [PubMed]
- Verma, P.; Malhotra, G.; Agrawal, R.; Sonavane, S.; Meshram, V.; Asopa, R.V. Evidence of Prostate-Specific Membrane Antigen Expression in Metastatic Differentiated Thyroid Cancer Using 68Ga-PSMA-HBED-CC PET/CT. Clin. Nucl. Med. 2018, 43, e265–e268. [Google Scholar] [CrossRef] [PubMed]
- Sasikumar, A.; Kashyap, R.; Joy, A.; Charan Patro, K.; Bhattacharya, P.; Reddy Pilaka, V.K.; Oommen, K.E.; Pillai, M.R.A. Utility of 68Ga-PSMA-11 PET/CT in Imaging of Glioma-A Pilot Study. Clin. Nucl. Med. 2018, 43, e304–e309. [Google Scholar] [CrossRef] [PubMed]
- Salas Fragomeni, R.A.; Pienta, K.J.; Pomper, M.G.; Gorin, M.A.; Rowe, S.P. Uptake of Prostate-Specific Membrane Antigen-Targeted 18F-DCFPyL in Cerebral Radionecrosis: Implications for Diagnostic Imaging of High-Grade Gliomas. Clin. Nucl. Med. 2018, 43, e419–e421. [Google Scholar] [CrossRef]
- Verma, P.; Malhotra, G.; Goel, A.; Rakshit, S.; Chandak, A.; Chedda, R.; Banerjee, S.; Asopa, R.V. Differential Uptake of 68Ga-PSMA-HBED-CC (PSMA-11) in Low-Grade Versus High-Grade Gliomas in Treatment-Naive Patients. Clin. Nucl. Med. 2019, 44, e318–e322. [Google Scholar] [CrossRef]
- Marafi, F.; Sasikumar, A.; Fathallah, W.; Esmail, A. 18F-PSMA 1007 Brain PET/CT Imaging in Glioma Recurrence. Clin. Nucl. Med. 2020, 45, e61–e62. [Google Scholar] [CrossRef] [Green Version]
- Kunikowska, J.; Kuliński, R.; Muylle, K.; Koziara, H.; Królicki, L. 68Ga-Prostate-Specific Membrane Antigen-11 PET/CT: A New Imaging Option for Recurrent Glioblastoma Multiforme? Clin. Nucl. Med. 2020, 45, 11–18. [Google Scholar] [CrossRef]
- Gupta, M.; Choudhury, P.S.; Premsagar, I.C.; Gairola, M.; Ahlawat, P. Role of 68Ga-Prostate-Specific Membrane Antigen PET/CT in Disease Assessment in Glioblastoma Within 48 Hours of Surgery. Clin. Nucl. Med. 2020, 45, 204–205. [Google Scholar] [CrossRef]
- Van de Wiele, C.; Sathekge, M.; de Spiegeleer, B.; de Jonghe, P.J.; Beels, L.; Maes, A. PSMA-Targeting Positron Emission Agents for Imaging Solid Tumors Other Than Non-Prostate Carcinoma: A Systematic Review. Int. J. Mol. Sci. 2019, 20, 4886. [Google Scholar] [CrossRef] [Green Version]
- Oliveira, D.; Stegmayr, C.; Heinzel, A.; Ermert, J.; Neumaier, B.; Shah, N.J.; Mottaghy, F.M.; Langen, K.-J.; Willuweit, A. High uptake of 68Ga-PSMA and 18F-DCFPyL in the peritumoral area of rat gliomas due to activated astrocytes. EJNMMI Res. 2020, 10, 55. [Google Scholar] [CrossRef]
- de Lucas, A.G.; Schuhmacher, A.J.; Oteo, M.; Romero, E.; Cámara, J.A.; de Martino, A.; Arroyo, A.G.; Morcillo, M.Á.; Squatrito, M.; Martinez-Torrecuadrada, J.L.; et al. Targeting MT1-MMP as an ImmunoPET-Based Strategy for Imaging Gliomas. PLoS ONE 2016, 11, e0158634. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.-J.; Li, H.-S.; Wang, Q.-S.; Wu, H.-B.; Han, Y.-J.; Zhou, W.-L.; Wang, M.; Huang, S. Construction and Evaluation of the Tumor-Targeting, Cell-Penetrating Multifunctional Molecular Probe iCREKA. Contrast Media Mol. Imaging 2018, 2018, 7929617. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, J.; Wang, Y.-L.; Li, X.-B.; Gao, S.-Y.; Liu, S.-Y.; Song, Y.-K.; Wang, J.-Y.; Xiong, Y.; Ma, H.; Jiang, L.; et al. Radiosynthesis and Preliminary Biological Evaluation of 18F-Fluoropropionyl-Chlorotoxin as a Potential PET Tracer for Glioma Imaging. Contrast Media Mol. Imaging 2018, 2018, 8439162. [Google Scholar] [CrossRef] [PubMed]
- Kasten, B.B.; Jiang, K.; Cole, D.; Jani, A.; Udayakumar, N.; Gillespie, G.Y.; Lu, G.; Dai, T.; Rosenthal, E.L.; Markert, J.M.; et al. Targeting MMP-14 for dual PET and fluorescence imaging of glioma in preclinical models. Eur. J. Nucl. Med. Mol. Imaging 2020, 47, 1412–1426. [Google Scholar] [CrossRef] [PubMed]
- Röhrich, M.; Loktev, A.; Wefers, A.K.; Altmann, A.; Paech, D.; Adeberg, S.; Windisch, P.; Hielscher, T.; Flechsig, P.; Floca, R.; et al. IDH-wildtype glioblastomas and grade III/IV IDH-mutant gliomas show elevated tracer uptake in fibroblast activation protein-specific PET/CT. Eur. J. Nucl. Med. Mol. Imaging 2019, 46, 2569–2580. [Google Scholar] [CrossRef]
- Warburg, O. On the Origin of Cancer Cells. Science 1956, 123, 309–314. [Google Scholar] [CrossRef]
- Moreno-Sánchez, R.; Rodríguez-Enríquez, S.; Marín-Hernández, A.; Saavedra, E. Energy metabolism in tumor cells. FEBS J. 2007, 274, 1393–1418. [Google Scholar] [CrossRef]
- Jung, J.; Ahn, B.-C. Current Radiopharmaceuticals for Positron Emission Tomography of Brain Tumors. Brain Tumor Res. Treat. 2018, 6, 47–53. [Google Scholar] [CrossRef]
- Masui, K.; Onizuka, H.; Cavenee, W.K.; Mischel, P.S.; Shibata, N. Metabolic reprogramming in the pathogenesis of glioma: Update. Neuropathology 2019, 39, 3–13. [Google Scholar] [CrossRef] [Green Version]
- Cruzat, V.; Macedo Rogero, M.; Noel Keane, K.; Curi, R.; Newsholme, P. Glutamine: Metabolism and Immune Function, Supplementation and Clinical Translation. Nutrients 2018, 10, 1564. [Google Scholar] [CrossRef] [Green Version]
- Feron, O. Pyruvate into lactate and back: From the Warburg effect to symbiotic energy fuel exchange in cancer cells. Radiother. Oncol. 2009, 92, 329–333. [Google Scholar] [CrossRef]
- Tardito, S.; Oudin, A.; Ahmed, S.U.; Fack, F.; Keunen, O.; Zheng, L.; Miletic, H.; Sakariassen, P.Ø.; Weinstock, A.; Wagner, A.; et al. Glutamine synthetase activity fuels nucleotide biosynthesis and supports growth of glutamine-restricted glioblastoma. Nat. Cell Biol. 2015, 17, 1556–1568. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rosati, A.; Poliani, P.L.; Todeschini, A.; Cominelli, M.; Medicina, D.; Cenzato, M.; Simoncini, E.L.; Magrini, S.M.; Buglione, M.; Grisanti, S.; et al. Glutamine synthetase expression as a valuable marker of epilepsy and longer survival in newly diagnosed glioblastoma multiforme. Neuro-oncology 2013, 15, 618–625. [Google Scholar] [CrossRef] [PubMed]
- Zhou, R.; Pantel, A.R.; Li, S.; Lieberman, B.P.; Ploessl, K.; Choi, H.; Blankemeyer, E.; Lee, H.; Kung, H.F.; Mach, R.H.; et al. [18F](2S,4R)4-Fluoroglutamine PET Detects Glutamine Pool Size Changes in Triple-Negative Breast Cancer in Response to Glutaminase Inhibition. Cancer Res. 2017, 77, 1476–1484. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Price, S.J.; Young, A.M.H.; Scotton, W.J.; Ching, J.; Mohsen, L.A.; Boonzaier, N.R.; Lupson, V.C.; Griffiths, J.R.; McLean, M.A.; Larkin, T.J. Multimodal MRI can identify perfusion and metabolic changes in the invasive margin of glioblastomas. J. Magn. Reson. Imaging 2016, 43, 487–494. [Google Scholar] [CrossRef] [Green Version]
- Nagashima, H.; Sasayama, T.; Tanaka, K.; Kyotani, K.; Sato, N.; Maeyama, M.; Kohta, M.; Sakata, J.; Yamamoto, Y.; Hosoda, K.; et al. Myo-inositol concentration in MR spectroscopy for differentiating high grade glioma from primary central nervous system lymphoma. J. Neurooncol. 2018, 136, 317–326. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Andronesi, O.C.; Arrillaga-Romany, I.C.; Ly, K.I.; Bogner, W.; Ratai, E.M.; Reitz, K.; Iafrate, A.J.; Dietrich, J.; Gerstner, E.R.; Chi, A.S.; et al. Pharmacodynamics of mutant-IDH1 inhibitors in glioma patients probed by in vivo 3D MRS imaging of 2-hydroxyglutarate. Nat. Commun. 2018, 9, 1474. [Google Scholar] [CrossRef]
- Bisdas, S.; Chadzynski, G.L.; Braun, C.; Schittenhelm, J.; Skardelly, M.; Hagberg, G.E.; Ethofer, T.; Pohmann, R.; Shajan, G.; Engelmann, J.; et al. MR spectroscopy for in vivo assessment of the oncometabolite 2-hydroxyglutarate and its effects on cellular metabolism in human brain gliomas at 9.4T. J. Magn. Reson. Imaging 2016, 44, 823–833. [Google Scholar] [CrossRef]
- Heiland, D.H.; Mader, I.; Schlosser, P.; Pfeifer, D.; Carro, M.S.; Lange, T.; Schwarzwald, R.; Vasilikos, I.; Urbach, H.; Weyerbrock, A. Integrative Network-based Analysis of Magnetic Resonance Spectroscopy and Genome Wide Expression in Glioblastoma multiforme. Sci. Rep. 2016, 6, 29052. [Google Scholar] [CrossRef]
- Hangel, G.; Jain, S.; Springer, E.; Hečková, E.; Strasser, B.; Považan, M.; Gruber, S.; Widhalm, G.; Kiesel, B.; Furtner, J.; et al. High-resolution metabolic mapping of gliomas via patch-based super-resolution magnetic resonance spectroscopic imaging at 7T. Neuroimage 2019, 191, 587–595. [Google Scholar] [CrossRef]
- Kant, S.; Kesarwani, P.; Prabhu, A.; Graham, S.F.; Buelow, K.L.; Nakano, I.; Chinnaiyan, P. Enhanced fatty acid oxidation provides glioblastoma cells metabolic plasticity to accommodate to its dynamic nutrient microenvironment. Cell Death Dis. 2020, 11, 253. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Strickland, M.; Stoll, E.A. Metabolic Reprogramming in Glioma. Front. Cell Dev. Biol. 2017, 5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maher, E.A.; Marin-Valencia, I.; Bachoo, R.M.; Mashimo, T.; Raisanen, J.; Hatanpaa, K.J.; Jindal, A.; Jeffrey, F.M.; Choi, C.; Madden, C.; et al. Metabolism of [U-13C] glucose in human brain tumors in vivo. NMR Biomed. 2012, 25, 1234–1244. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mashimo, T.; Pichumani, K.; Vemireddy, V.; Hatanpaa, K.J.; Singh, D.K.; Sirasanagandla, S.; Nannepaga, S.; Piccirillo, S.G.; Kovacs, Z.; Foong, C.; et al. Acetate Is a Bioenergetic Substrate for Human Glioblastoma and Brain Metastases. Cell 2014, 159, 1603–1614. [Google Scholar] [CrossRef] [Green Version]
- Grube, S.; Göttig, T.; Freitag, D.; Ewald, C.; Kalff, R.; Walter, J. Selection of suitable reference genes for expression analysis in human glioma using RT-qPCR. J. Neurooncol. 2015, 123, 35–42. [Google Scholar] [CrossRef]
- Lin, H.; Patel, S.; Affleck, V.S.; Wilson, I.; Turnbull, D.M.; Joshi, A.R.; Maxwell, R.; Stoll, E.A. Fatty acid oxidation is required for the respiration and proliferation of malignant glioma cells. Neuro-oncology 2017, 19, 43–54. [Google Scholar] [CrossRef] [Green Version]
- Tee, S.-S.; Keshari, K.R. Novel Approaches to Imaging Tumor Metabolism. Cancer J. 2015, 21, 165–173. [Google Scholar] [CrossRef] [Green Version]
- Kesarwani, P.; Prabhu, A.; Kant, S.; Chinnaiyan, P. Metabolic remodeling contributes towards an immune-suppressive phenotype in glioblastoma. Cancer Immunol. Immunother. 2019, 68, 1107–1120. [Google Scholar] [CrossRef]
- Peters, J.C. Tryptophan nutrition and metabolism: An overview. Adv. Exp. Med. Biol. 1991, 294, 345–358. [Google Scholar] [CrossRef]
- Frumento, G.; Rotondo, R.; Tonetti, M.; Damonte, G.; Benatti, U.; Ferrara, G.B. Tryptophan-derived catabolites are responsible for inhibition of T and natural killer cell proliferation induced by indoleamine 2,3-dioxygenase. J. Exp. Med. 2002, 196, 459–468. [Google Scholar] [CrossRef] [Green Version]
- Sordillo, P.P.; Sordillo, L.A.; Helson, L. The Kynurenine Pathway: A Primary Resistance Mechanism in Patients with Glioblastoma. Anticancer Res. 2017, 37, 2159–2171. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ozawa, Y.; Yamamuro, S.; Sano, E.; Tatsuoka, J.; Hanashima, Y.; Yoshimura, S.; Sumi, K.; Hara, H.; Nakayama, T.; Suzuki, Y.; et al. Indoleamine 2,3-dioxygenase 1 is highly expressed in glioma stem cells. Biochem. Biophys. Res. Commun. 2020, 524, 723–729. [Google Scholar] [CrossRef] [PubMed]
- Bosnyák, E.; Kamson, D.O.; Guastella, A.R.; Varadarajan, K.; Robinette, N.L.; Kupsky, W.J.; Muzik, O.; Michelhaugh, S.K.; Mittal, S.; Juhász, C. Molecular imaging correlates of tryptophan metabolism via the kynurenine pathway in human meningiomas. Neuro-oncology 2015, 17, 1284–1292. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Krasikova, R.; Kondrashov, M.; Avagliano, C.; Petukhov, M.; Vazquez-Romero, A.; Revunov, E.; Johnström, P.; Tari, L.; Tóth, M.; Häggkvist, J.; et al. Synthesis and Preclinical Evaluation of 6-[18F] Fluorine-α-methyl-l-tryptophan, a Novel PET Tracer for Measuring Tryptophan Uptake. ACS Chem. Neurosci. 2020, 11, 1756–1761. [Google Scholar] [CrossRef]
- Lewis, J.S.; Keshari, K.R. Imaging and Metabolism; Springer: Berlin/Heidelberg, Germany, 2017; ISBN 978-3-319-61401-4. [Google Scholar]
- Moreau, A.; Febvey, O.; Mognetti, T.; Frappaz, D.; Kryza, D. Contribution of Different Positron Emission Tomography Tracers in Glioma Management: Focus on Glioblastoma. Front. Oncol. 2019, 9. [Google Scholar] [CrossRef] [Green Version]
- Götz, I.; Grosu, A.L. [18F] FET-PET Imaging for Treatment and Response Monitoring of Radiation Therapy in Malignant Glioma Patients—A Review. Front. Oncol. 2013, 3. [Google Scholar] [CrossRef] [Green Version]
- Nelson, S.J.; Kadambi, A.K.; Park, I.; Li, Y.; Crane, J.; Olson, M.; Molinaro, A.; Roy, R.; Butowski, N.; Cha, S.; et al. Association of early changes in 1H MRSI parameters with survival for patients with newly diagnosed glioblastoma receiving a multimodality treatment regimen. Neuro-oncology 2017, 19, 430–439. [Google Scholar] [CrossRef] [Green Version]
- Lo Dico, A.; Valtorta, S.; Ottobrini, L.; Moresco, R.M. Role of Metformin and AKT Axis Modulation in the Reversion of Hypoxia Induced TMZ-Resistance in Glioma Cells. Front. Oncol. 2019, 9, 463. [Google Scholar] [CrossRef]
- Poon, C.C.; Sarkar, S.; Yong, V.W.; Kelly, J.J.P. Glioblastoma-associated microglia and macrophages: Targets for therapies to improve prognosis. Brain 2017, 140, 1548–1560. [Google Scholar] [CrossRef] [Green Version]
- Roesch, S.; Rapp, C.; Dettling, S.; Herold-Mende, C. When Immune Cells Turn Bad—Tumor-Associated Microglia/Macrophages in Glioma. Int. J. Mol. Sci. 2018, 19, 436. [Google Scholar] [CrossRef] [Green Version]
- Liu, T.; Han, C.; Wang, S.; Fang, P.; Ma, Z.; Xu, L.; Yin, R. Cancer-associated fibroblasts: An emerging target of anti-cancer immunotherapy. J. Hematol. Oncol. 2019, 12, 86. [Google Scholar] [CrossRef]
- Hambardzumyan, D.; Gutmann, D.H.; Kettenmann, H. The role of microglia and macrophages in glioma maintenance and progression. Nat. Neurosci. 2016, 19, 20–27. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Venneti, S.; Lopresti, B.J.; Wiley, C.A. The peripheral benzodiazepine receptor (Translocator protein 18kDa) in microglia: From pathology to imaging. Prog. Neurobiol. 2006, 80, 308–322. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vlodavsky, E.; Soustiel, J.F. Immunohistochemical expression of peripheral benzodiazepine receptors in human astrocytomas and its correlation with grade of malignancy, proliferation, apoptosis and survival. J. Neurooncol. 2007, 81, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Evans, J.C.; Malhotra, M.; Cryan, J.F.; O’Driscoll, C.M. The therapeutic and diagnostic potential of the prostate specific membrane antigen/glutamate carboxypeptidase II (PSMA/GCPII) in cancer and neurological disease. Br. J. Pharmacol. 2016, 173, 3041–3079. [Google Scholar] [CrossRef] [Green Version]
- Pastorino, S.; Riondato, M.; Uccelli, L.; Giovacchini, G.; Giovannini, E.; Duce, V.; Ciarmiello, A. Toward the Discovery and Development of PSMA Targeted Inhibitors for Nuclear Medicine Applications. Curr. Radiopharm. 2020, 13, 63–79. [Google Scholar] [CrossRef]
- Nomura, N.; Pastorino, S.; Jiang, P.; Lambert, G.; Crawford, J.R.; Gymnopoulos, M.; Piccioni, D.; Juarez, T.; Pingle, S.C.; Makale, M.; et al. Prostate specific membrane antigen (PSMA) expression in primary gliomas and breast cancer brain metastases. Cancer Cell Int. 2014, 14, 26. [Google Scholar] [CrossRef] [Green Version]
- Sasikumar, A.; Joy, A.; Pillai, M.R.; Bindu, S.; Sudin, S.R. 68Ga-PSMA Uptake in an Incidentally Detected Gastrointestinal Stromal Tumor in a Case of Suspected Carcinoma Prostate. Clin. Nucl. Med. 2017, 42, e447–e448. [Google Scholar] [CrossRef]
- Wang, L.; Yuan, J.; Tu, Y.; Mao, X.; He, S.; Fu, G.; Zong, J.; Zhang, Y. Co-expression of MMP-14 and MMP-19 predicts poor survival in human glioma. Clin. Transl. Oncol. 2013, 15, 139–145. [Google Scholar] [CrossRef]
- Itoh, Y.; Ito, N.; Nagase, H.; Evans, R.D.; Bird, S.A.; Seiki, M. Cell surface collagenolysis requires homodimerization of the membrane-bound collagenase MT1-MMP. Mol. Biol. Cell 2006, 17, 5390–5399. [Google Scholar] [CrossRef] [Green Version]
- Röhrich, M.; Floca, R.; Loi, L.; Adeberg, S.; Windisch, P.; Giesel, F.L.; Kratochwil, C.; Flechsig, P.; Rathke, H.; Lindner, T.; et al. FAP-specific PET signaling shows a moderately positive correlation with relative CBV and no correlation with ADC in 13 IDH wildtype glioblastomas. Eur. J. Radiol. 2020, 127, 109021. [Google Scholar] [CrossRef] [PubMed]
- Chaddad, A.; Kucharczyk, M.J.; Daniel, P.; Sabri, S.; Jean-Claude, B.J.; Niazi, T.; Abdulkarim, B. Radiomics in Glioblastoma: Current Status and Challenges Facing Clinical Implementation. Front. Oncol. 2019, 9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zanfardino, M.; Franzese, M.; Pane, K.; Cavaliere, C.; Monti, S.; Esposito, G.; Salvatore, M.; Aiello, M. Bringing radiomics into a multi-omics framework for a comprehensive genotype–phenotype characterization of oncological diseases. J. Transl. Med. 2019, 17. [Google Scholar] [CrossRef] [PubMed]
- Bell, M.; Turkbey, E.B.; Escorcia, F.E. Radiomics, Radiogenomics, and Next-Generation Molecular Imaging to Augment Diagnosis of Hepatocellular Carcinoma. Cancer J. 2020, 26, 108–115. [Google Scholar] [CrossRef]
- Gallivanone, F.; Cava, C.; Corsi, F.; Bertoli, G.; Castiglioni, I. In Silico Approach for the Definition of radiomiRNomic Signatures for Breast Cancer Differential Diagnosis. Int. J. Mol. Sci. 2019, 20, 5825. [Google Scholar] [CrossRef] [Green Version]
- Prins, R.M.; Soto, H.; Konkankit, V.; Odesa, S.K.; Eskin, A.; Yong, W.H.; Nelson, S.F.; Liau, L.M. Gene Expression Profile Correlates with T-Cell Infiltration and Relative Survival in Glioblastoma Patients Vaccinated with Dendritic Cell Immunotherapy. Clin. Cancer Res. 2011, 17, 1603–1615. [Google Scholar] [CrossRef] [Green Version]
- Klughammer, J.; Kiesel, B.; Roetzer, T.; Fortelny, N.; Nemc, A.; Nenning, K.-H.; Furtner, J.; Sheffield, N.C.; Datlinger, P.; Peter, N.; et al. The DNA methylation landscape of glioblastoma disease progression shows extensive heterogeneity in time and space. Nat. Med. 2018, 24, 1611–1624. [Google Scholar] [CrossRef]
- Elkhaled, A.; Jalbert, L.E.; Phillips, J.J.; Yoshihara, H.A.I.; Parvataneni, R.; Srinivasan, R.; Bourne, G.; Berger, M.S.; Chang, S.M.; Cha, S.; et al. Magnetic resonance of 2-hydroxyglutarate in IDH1-mutated low-grade gliomas. Sci. Transl. Med. 2012, 4, 116ra5. [Google Scholar] [CrossRef] [Green Version]
- Choi, C.; Ganji, S.K.; DeBerardinis, R.J.; Hatanpaa, K.J.; Rakheja, D.; Kovacs, Z.; Yang, X.-L.; Mashimo, T.; Raisanen, J.M.; Marin-Valencia, I.; et al. 2-hydroxyglutarate detection by magnetic resonance spectroscopy in IDH-mutated glioma patients. Nat. Med. 2012, 18, 624–629. [Google Scholar] [CrossRef] [Green Version]
- Kong, D.-S.; Kim, J.; Ryu, G.; You, H.-J.; Sung, J.K.; Han, Y.H.; Shin, H.-M.; Lee, I.-H.; Kim, S.-T.; Park, C.-K.; et al. Quantitative radiomic profiling of glioblastoma represents transcriptomic expression. Oncotarget 2018, 9, 6336–6345. [Google Scholar] [CrossRef] [Green Version]
- Verhaak, R.G.W.; Hoadley, K.A.; Purdom, E.; Wang, V.; Qi, Y.; Wilkerson, M.D.; Miller, C.R.; Ding, L.; Golub, T.; Mesirov, J.P.; et al. Integrated Genomic Analysis Identifies Clinically Relevant Subtypes of Glioblastoma Characterized by Abnormalities in PDGFRA, IDH1, EGFR, and NF1. Cancer Cell 2010, 17, 98–110. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Verger, A.; Langen, K.-J. PET Imaging in Glioblastoma: Use in Clinical Practice. In Glioblastoma; De Vleeschouwer, S., Ed.; Codon Publications: Brisbane, Australia, 2017; ISBN 978-0-9944381-2-6. [Google Scholar]
- Kopkova, A.; Sana, J.; Machackova, T.; Vecera, M.; Radova, L.; Trachtova, K.; Vybihal, V.; Smrcka, M.; Kazda, T.; Slaby, O.; et al. Cerebrospinal Fluid MicroRNA Signatures as Diagnostic Biomarkers in Brain Tumors. Cancers 2019, 11, 1546. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ebrahimkhani, S.; Vafaee, F.; Hallal, S.; Wei, H.; Lee, M.Y.T.; Young, P.E.; Satgunaseelan, L.; Beadnall, H.; Barnett, M.H.; Shivalingam, B.; et al. Deep sequencing of circulating exosomal microRNA allows non-invasive glioblastoma diagnosis. NPJ Precis. Oncol. 2018, 2, 1–9. [Google Scholar] [CrossRef] [Green Version]
- Qu, S.; Guan, J.; Liu, Y. Identification of microRNAs as novel biomarkers for glioma detection: A meta-analysis based on 11 articles. J. Neurol. Sci. 2015, 348, 181–187. [Google Scholar] [CrossRef] [PubMed]
- Costa, P.M.; Cardoso, A.L.; Custódia, C.; Cunha, P.; Pereira de Almeida, L.; Pedroso de Lima, M.C. MiRNA-21 silencing mediated by tumor-targeted nanoparticles combined with sunitinib: A new multimodal gene therapy approach for glioblastoma. J. Control. Release 2015, 207, 31–39. [Google Scholar] [CrossRef]
- Shukla, A.; Gupta, P.; Singh, R.; Mishra, D.P. Glycolytic inhibitor 2-Deoxy-d-Glucose activates migration and invasion in glioblastoma cells through modulation of the miR-7-5p/TFF3 signaling pathway. Biochem. Biophys. Res. Commun. 2018, 499, 829–835. [Google Scholar] [CrossRef]
- Siegal, T.; Charbit, H.; Paldor, I.; Zelikovitch, B.; Canello, T.; Benis, A.; Wong, M.L.; Morokoff, A.P.; Kaye, A.H.; Lavon, I. Dynamics of circulating hypoxia-mediated miRNAs and tumor response in patients with high-grade glioma treated with bevacizumab. J. Neurosurg. 2016, 125, 1008–1015. [Google Scholar] [CrossRef] [Green Version]
- Liu, H.; Liu, Z.; Jiang, B.; Huo, L.; Liu, J.; Lu, J. Synthetic miR-145 Mimic Enhances the Cytotoxic Effect of the Antiangiogenic Drug Sunitinib in Glioblastoma. Cell Biochem. Biophys. 2015, 72, 551–557. [Google Scholar] [CrossRef]
- Wei, X.; Schlenkhoff, C.; Schwarz, B.; Essler, M.; Ahmadzadehfar, H. Combination of 177Lu-PSMA-617 and External Radiotherapy for the Treatment of Cerebral Metastases in Patients with Castration-Resistant Metastatic Prostate Cancer. Clin. Nucl. Med. 2017, 42, 704–706. [Google Scholar] [CrossRef]




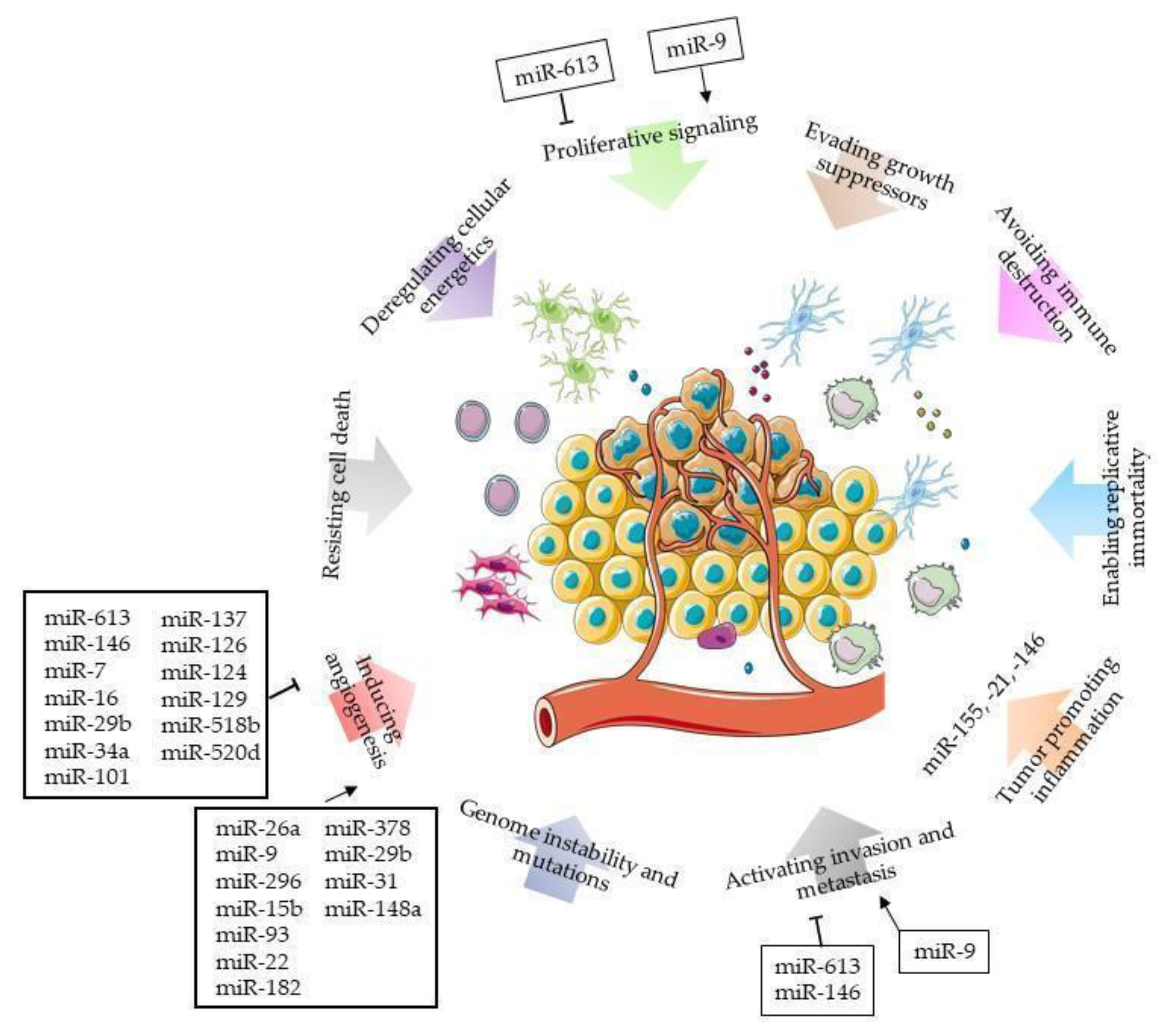{kind=link}
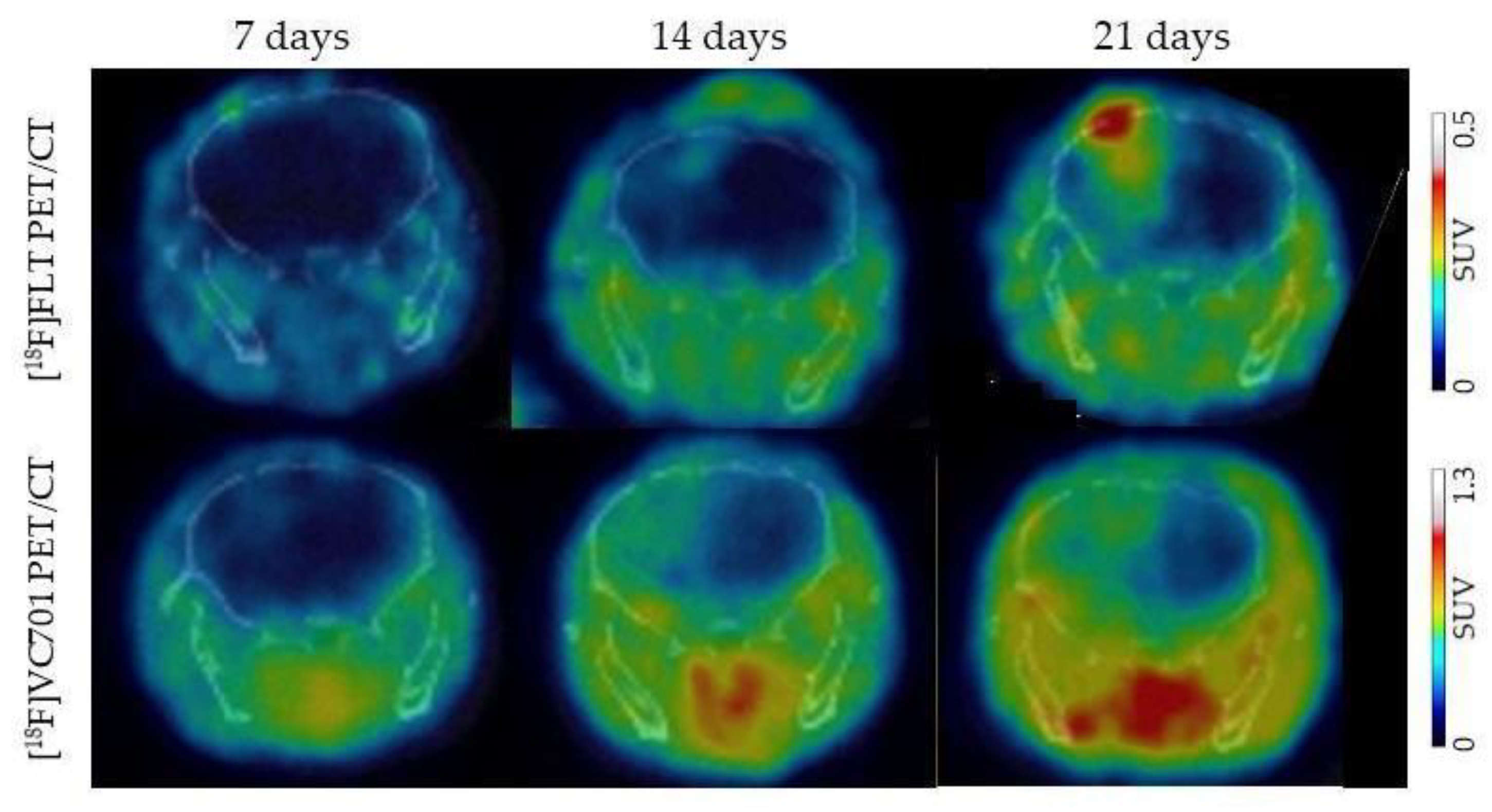{kind=link}
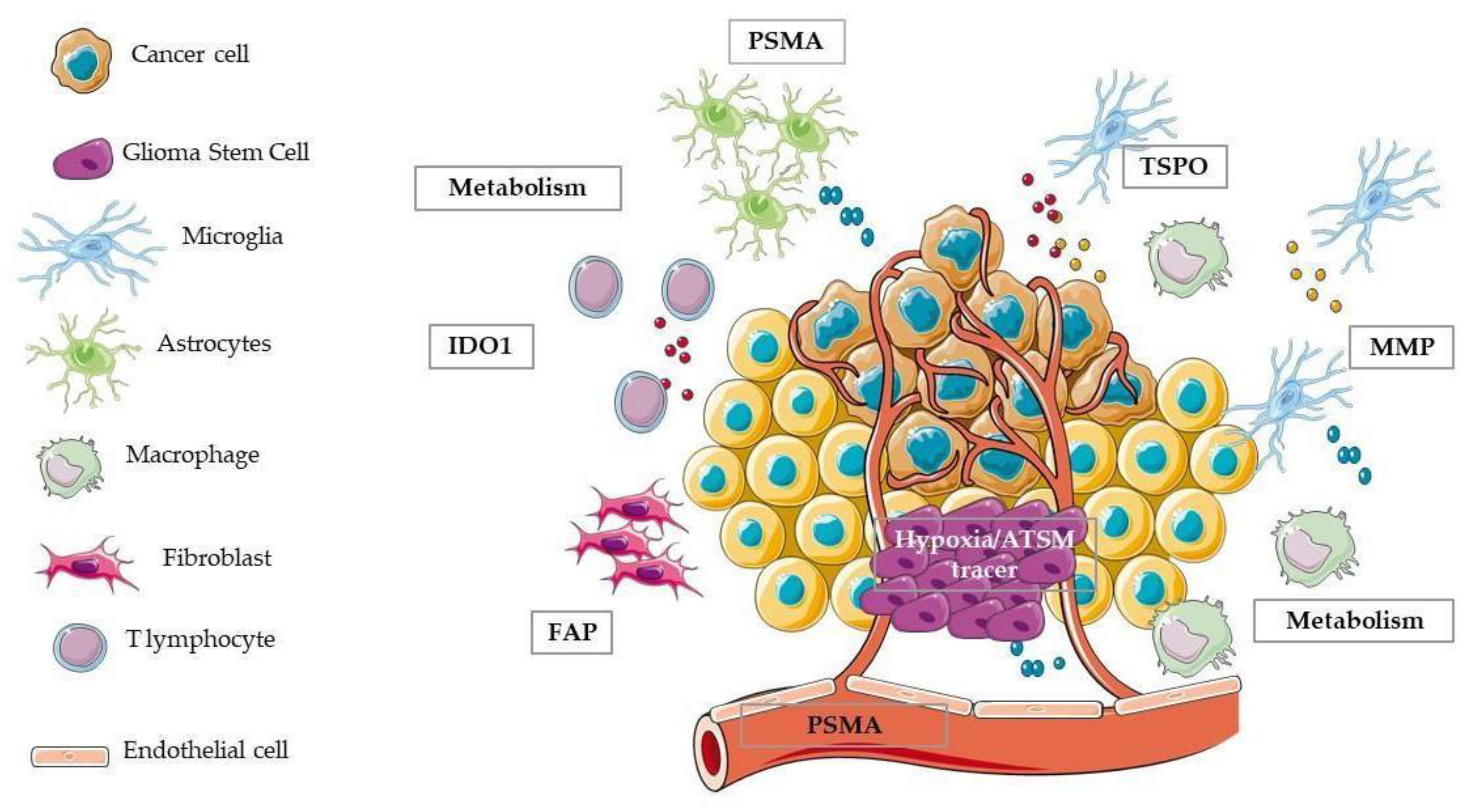{kind=link}
| miRNA | GBM Expression Level | Function | Target | References |
|---|---|---|---|---|
| miR-7 | Down | Inhibits angiogenesis | EGFR, IRS-1, IRS-2, FAK, OGR | [76] |
| miR-296 | Up | Promotes angiogenesis | HGS | [99] |
| miR-15b | Up | Inhibits angiogenesis | NRP-2 | [100,101] |
| miR-93 | Up | Promotes angiogenesis | Integrin B8 | [102] |
| miR-16 | Down | Inhibits angiogenesis | BMI-1 | [103] |
| miR-613 | Down | Inhibits angiogenesis | VEGFA | [84] |
| miR-26a | Up | Promotes angiogenesis | PTEN and PI3K/Akt | [85] |
| miR-9 | Up | Promotes angiogenesis | COL18A1, THBS2, PTCH1 and PHD3; HIF-1α/VEGF | [86] |
| miR-9-5p, miR-22-3p, miR-182-5p | Up | Promotes angiogenesis | RGS5, SOX7, and ABCB1 | [104] |
| miR-29b, miR-34a, miR-101, and miR-137 | Down | Inhibits angiogenesis | stanniocalcin-1 (STC1) induces eNOS, VEGF, and VEGFR2 | [105] |
| miR-126/126-3p | Down | Inhibits angiogenesis | epidermal growth factor-like protein 7 (EGFL7) | [106] |
| miR-124-3p | Down | Inhibits angiogenesis | NRP-1/GIPC1 pathway | [107] |
| miR-129-5p | Down | Inhibits angiogenesis | WNT5a | [108] |
| miR-378 | Up | Promotes angiogenesis | VEGFR2 | [109] |
| miR-518b | Down | Inhibits angiogenesis | platelet-derived growth factor receptor β (PDGFRB) | [110] |
| miR-29b | Up | Promotes angiogenesis | BCL2L2, which in turn regulates Ang-2 and VEGF | [111] |
| miR-520d-5p | Down | Inhibits angiogenesis | Pituitary Tumour Transforming Gene 1 (PTTG1) | [112] |
| miR-148a and miR-31 | Up | Promotes angiogenesis | factor inhibiting hypoxia-inducible factor 1 (FIH1), and HIF1α and Notch. | [113] |
| Molecule | Cellular/Molecular Target | Clinical Trial in Glioma | References |
|---|---|---|---|
| Bevacizumab * | VEGF | Phase III | [5,155,156,157,158] |
| Lomustine * | Amino acid carbonylation | Phase III | [131,132] |
| Nimotuzumab * | EGFRvIII | Phase II | [135,136] |
| Veliparib/ABT-888 * | PARP | Phase II/III | [137,138,139,140,141,159] |
| Olaparib * | PARP | Phase I | [142,160] |
| DNX-2401/Delta-24-RGD * | Rb pathway-defective glioma cells | Phase I/II | [144,145] |
| Pembrolizumab * | PD-1 | Phase I/II | [146,147] |
| MSC-IFN-β * | MGMT via p53 gene induction | [148] | |
| Momelotinib/MTB/CYT387 * | JAK 1/2 | [149] | |
| Metformin * | AMPK, p53, mTORC1, mtComplex I | Phase I/II | [150,151,152,153,154] |
| Bevacizumab and Lomustine | VEGF and Amino acid carbonylation | Phase III | [130] |
| Lomustine and Regorafenib | Amino acid carbonylation and angiogenic, stromal, and oncogenic receptor tyrosine kinases | Phase II | [133] |
| Nivolumab | PD-1/PD-L1 | Phase II/III | [161,162,163] |
| DS-100b | IDH1 | Phase I | [164,165,166] |
| Bevacizumab and Pembrolizumab | VEGF and PD-1 | Phase II | [167,168] |
| Anti-TIM-3 and Anti-PD-1 | TIM-3 and PD-1 | [169] | |
| Anti-TIGIT and anti-PD-1 | TIGIT and PD-1 | [170] |
| miRNA | Target mRNA or Process | In Chemo-Resistant GBM | References |
|---|---|---|---|
| miR-1238 | CAV1/EGFR pathway | miRNA is UPregulated | [172] |
| miR-151 and miR-151a | XRCC4-mediated DNA repair | miRNA is UPregulated | [173] |
| miR-1587 | NCOR1 in glioma-associated mesenchymal stem cells | miRNA is UPregulated | [82,174] |
| miR-29a | cancer stem cells | miRNA is DOWNregulated | [176] |
| miR-29c | Sp1, MGMT | miRNA is DOWNregulated | [177] |
| miR-423-5p | ING-4, that controls p-AKT and p-ERK1/2; sustain stemness | miRNA is UPregulated | [178] |
| miR-123-3p | SOX2, WNT/beta-Cat pathway | miRNA is DOWNregulated | [180] |
| Let-7i, miR-151-3p, and miR-93 | unknown | miRNAs are DOWNregulated | [179] |
| miR-146b-5p | TRAF6, AKT/NF-κB pathway | miRNA is DOWNregulated | [181,184] |
| miR-223 | PAX, PI3K/Akt pathway | miRNA is UPregulated | [185,186] |
| miR-93; miR-193 | Cyclin D1 and cell cycle progression | Both miRNA are UPregulated | [187] |
| miR-7-5p; miR-186 | YY1, in the RelB pathway | Both miRNA are DOWNregulated | [188,189,190] |
| miR-224-3p | HIF-1α/miR-224-3p/ATG5 pathway | miRNA is UPregulated | [191] |
| miR-519a | STAT3/Bcl-2/Beclin-1 pathway | miRNA is DOWNregulated | [192] |
| miR-138 | miR-138/BIM axis and autophagy | miRNA is UPregulated | [193] |
| miR-20a | Under the control of DNMT | miRNA is UPregulated | [194] |
| miR-139 | GFAP in cytoskeleton | miRNA is DOWNregulated | [195,196] |
| miR-221/222 | APE1/miR-221/222 axis regulates SIRT1/MDR1 and PTEN | miRNA is DOWNregulated | [197,198,200,201] |
| miR-191 | Controlled by ER alpha and beta | miRNA is DOWNregulated | [206] |
| miR-101 | GSK3β | miRNA is DOWNregulated | [207] |
| seven miRNAs’ profile: miR-1280, miR-1238, miR-938, and miR-423-5p; let-7i, miR-151-3p, and miR-93 | miRNAs are Upregulated miRNAs are DOWNregulated | [179] | |
| seven miRNAs’ profile: miR-9, -182, and -221; miR-29c, -93, -101, and -130a | miR-101 controls GSK3β | miRNAs are Upregulated; miRNAs are DOWNregulated | [210] |
| Imaging Target | Molecular Target | Radiotracer | References |
|---|---|---|---|
| Glucose metabolism | GLUT-1; HK-1 | [18F]FDG | [217] |
| Glutamine metabolism | ASCT2 | [18F]F-Gln | [218,219,220] |
| Fatty acid synthesis | MCT; ACSS2 | [11C]Acetate | [221,222] |
| Tryptophan pathway/immune tolerance | IDO1 | [11C]AMT; L-1-[18F]FETrp | [212,223,224,225,226,227] |
| Aminoacids | LAT1/2 | [11C]MET; [18F]FET; [18F]FELP; [18F]FIMP; [18F]Fluciclovine | [228,229,230] |
| Cell proliferation | TK1 | [18F]FLT | [231,232,233,234,235,236] |
| Choline metabolism | CK | [11C]Choline | [237,238] |
| Vasculature and hypoxia | Tumor hypoxia | [64Cu]ATSM; nitroimidazole compounds; CAIX agents | [239,240,241,242,243,244] |
| αVβ3 ligand-binding domain | [64Cu]RaftRGD | [242] | |
| Glioma associated macrophages and microglia (GAMMs) | TSPO | [11C](R)PK11195; [18F]DPA-714; [18F]GE-180; [18F]VC701 | [245,246,247,248,249,250,251,252,253] |
| Endothelial cells/astrocytes | PSMA | [68Ga]PSMA; [18F]DCFPyL; [89Zr]Df-IAB2M anti-PSMA minibody | [254,255,256,257,258,259,260,261,262,263,264,265,266] |
| Matrix-metallo proteinases | MT1-MMP (MMP-14); MMP-2; MMP-9 | [89Zr]DFO-LEM2/15; [18F]BR-351; [18F]iCREKA; [18F]-FP-chlorotoxin; [68Ga]/[64Cu]-MMP-14 binding peptide | [246,267,268,269,270] |
| Carcinoma-associated fibroblast | FAP | [117Lu]/[68Ga]FAPI-02/04 | [271] |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Valtorta, S.; Salvatore, D.; Rainone, P.; Belloli, S.; Bertoli, G.; Moresco, R.M. Molecular and Cellular Complexity of Glioma. Focus on Tumour Microenvironment and the Use of Molecular and Imaging Biomarkers to Overcome Treatment Resistance. Int. J. Mol. Sci. 2020, 21, 5631. https://doi.org/10.3390/ijms21165631
Valtorta S, Salvatore D, Rainone P, Belloli S, Bertoli G, Moresco RM. Molecular and Cellular Complexity of Glioma. Focus on Tumour Microenvironment and the Use of Molecular and Imaging Biomarkers to Overcome Treatment Resistance. International Journal of Molecular Sciences. 2020; 21(16):5631. https://doi.org/10.3390/ijms21165631
Chicago/Turabian StyleValtorta, Silvia, Daniela Salvatore, Paolo Rainone, Sara Belloli, Gloria Bertoli, and Rosa Maria Moresco. 2020. "Molecular and Cellular Complexity of Glioma. Focus on Tumour Microenvironment and the Use of Molecular and Imaging Biomarkers to Overcome Treatment Resistance" International Journal of Molecular Sciences 21, no. 16: 5631. https://doi.org/10.3390/ijms21165631
APA StyleValtorta, S., Salvatore, D., Rainone, P., Belloli, S., Bertoli, G., & Moresco, R. M. (2020). Molecular and Cellular Complexity of Glioma. Focus on Tumour Microenvironment and the Use of Molecular and Imaging Biomarkers to Overcome Treatment Resistance. International Journal of Molecular Sciences, 21(16), 5631. https://doi.org/10.3390/ijms21165631