Role of Leptin in Inflammation and Vice Versa

 ,
, {kind=link}
{kind=link}
Abstract
:1. Introduction
2. Leptin Signaling in Immune Cells
2.1. Leptin and Innate Immunity
2.1.1. In Monocytes and Macrophages
2.1.2. Polymorphonuclear Cells
2.1.3. Human NK Cells
2.1.4. Other Immune Cells
2.2. Leptin and Adaptive Immunity
3. Leptin as a Mediator of Inflammation
3.1. Leptin Deficiency and Infection Diseases
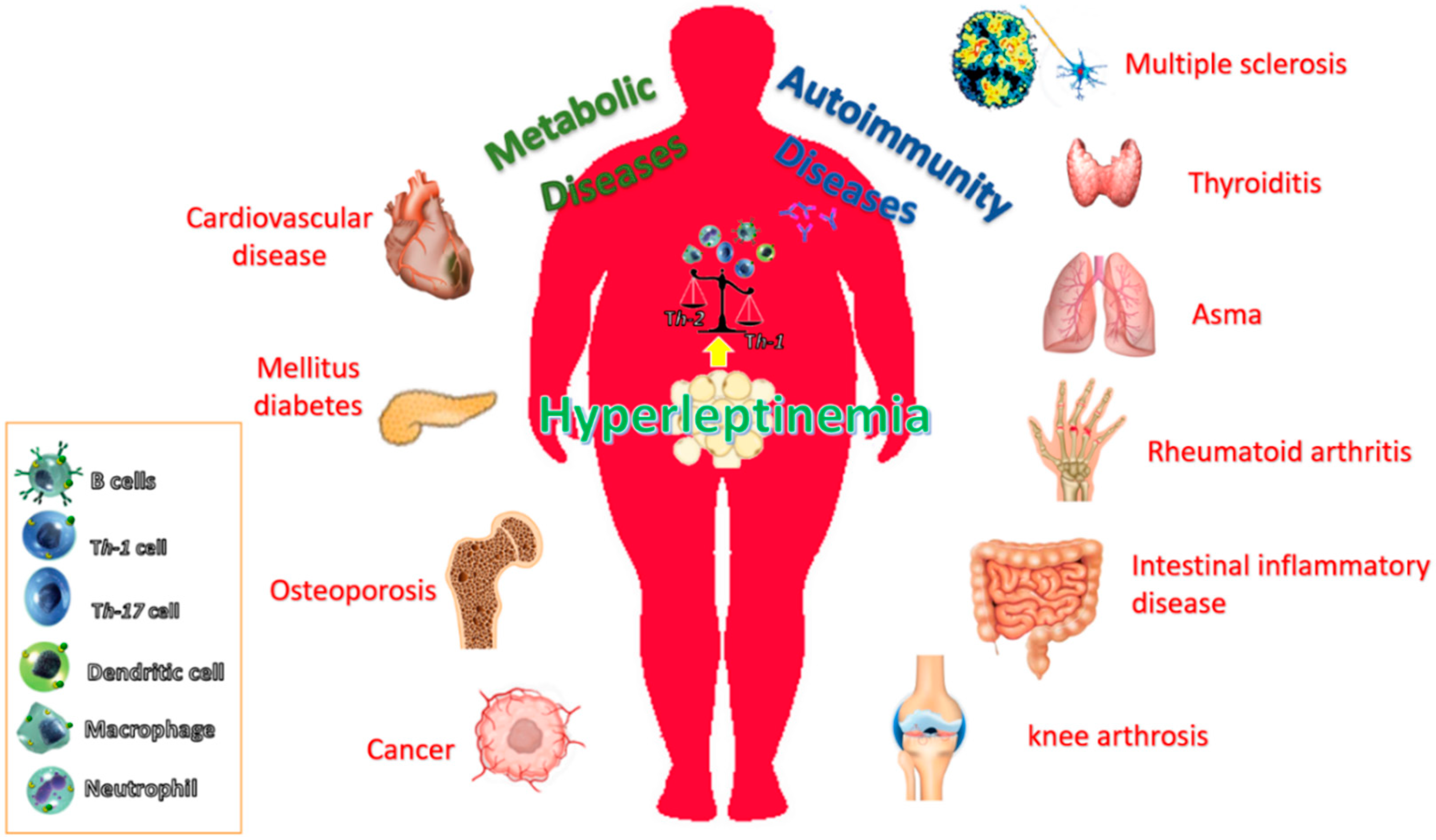
3.2. Leptin as an Inflammatory Mediator in the Obesity-Associated Immuno-Metabolic Disorders: Diabetes, Cardiovascular Disease, Autoinmune Diseases and Cancer
3.2.1. Type 2 Diabetes Mellitus
3.2.2. Cardiovascular Diseases
3.2.3. Autoimmune Diseases
3.2.4. Cancer
3.2.5. Leptin as a Therapeutic Target
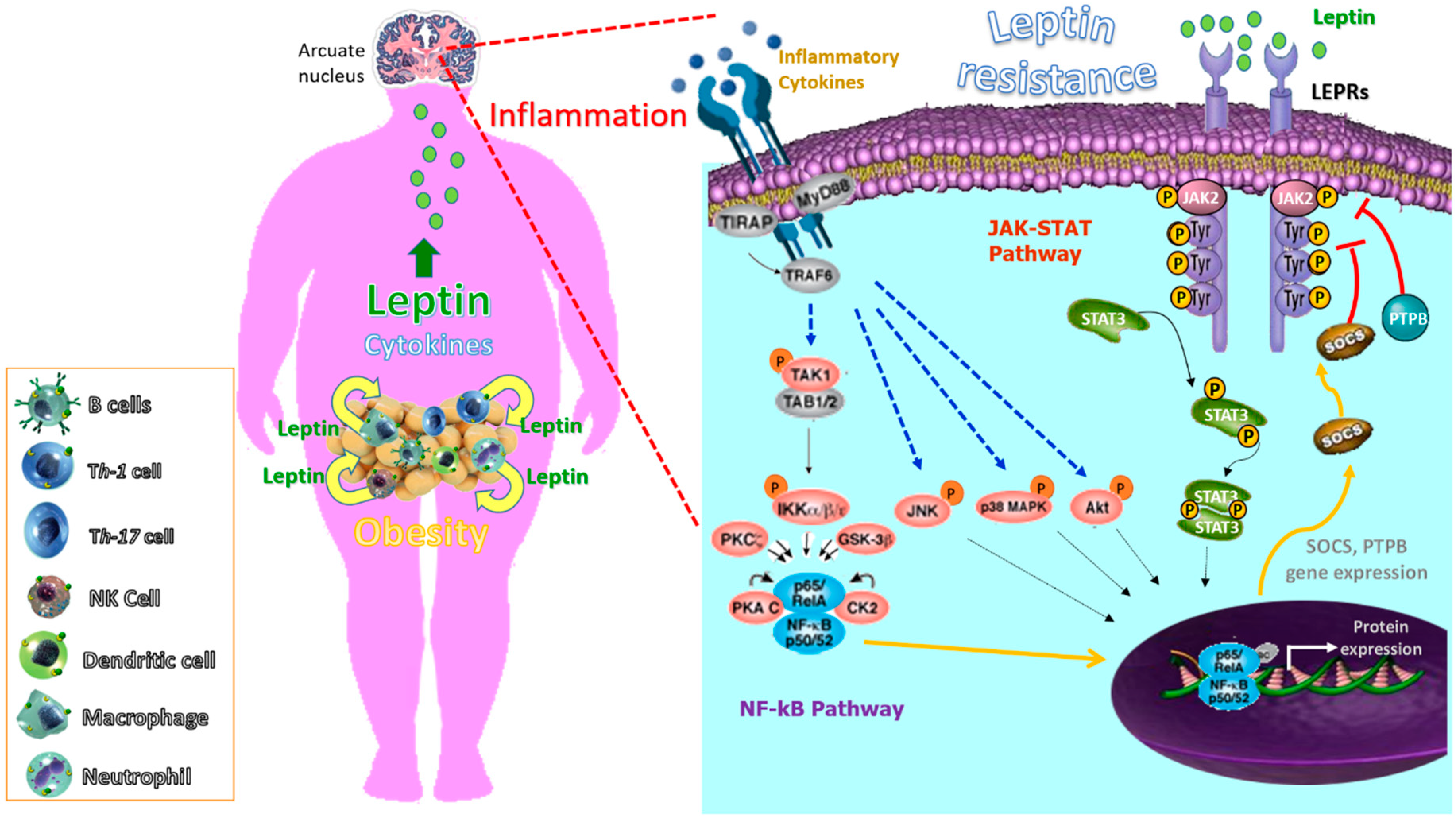
4. Inflammation as a Mediator of Leptin Resistance and Obesity
4.1. Infectious Diseases
4.1.1. Viral Infection
4.1.2. Bacterial Infection
4.2. Microbiota
4.3. The Paradox of Leptin Sensitization by Inflammatory Cytokines
5. Conclusions
Funding
Conflicts of Interest
References
- Gregor, M.F.; Hotamisligil, G.S. Inflammatory mechanisms in obesity. Annu. Rev. Immunol. 2011, 29, 415–445. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Libby, P. Inflammation in atherosclerosis. Nature 2002, 420, 868–874. [Google Scholar] [CrossRef] [PubMed]
- Chronic Inflammatory Systemic Diseases: An Evolutionary Trade-Off Between Acutely Beneficial but Chronically Harmful Programs—PubMed. Available online: https://pubmed.ncbi.nlm.nih.gov/26817483/ (accessed on 8 July 2020).
- Garn, H.; Bahn, S.; Baune, B.T.; Binder, E.B.; Bisgaard, H.; Chatila, T.A.; Chavakis, T.; Culmsee, C.; Dannlowski, U.; Gay, S.; et al. Current concepts in chronic inflammatory diseases: Interactions between microbes, cellular metabolism, and inflammation. J. Allergy Clin. Immunol. 2016, 138, 47–56. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Y.; Proenca, R.; Maffei, M.; Barone, M.; Leopold, L.; Friedman, J.M. Positional cloning of the mouse obese gene and its human homologue. Nature 1994, 372, 425–432. [Google Scholar] [CrossRef] [PubMed]
- Zavalza-Gómez, A.B.; Anaya-Prado, R.; Rincón-Sánchez, A.R.; Mora-Martínez, J.M. Adipokines and insulin resistance during pregnancy. Diabetes Res. Clin. Pract. 2008, 80, 8–15. [Google Scholar] [CrossRef] [PubMed]
- Maffei, M.; Halaas, J.; Ravussin, E.; Pratley, R.E.; Lee, G.H.; Zhang, Y.; Fei, H.; Kim, S.; Lallone, R.; Ranganathan, S. Leptin levels in human and rodent: Measurement of plasma leptin and ob RNA in obese and weight-reduced subjects. Nat. Med. 1995, 1, 1155–1161. [Google Scholar] [CrossRef] [PubMed]
- Iikuni, N.; Lam, Q.L.K.; Lu, L.; Matarese, G.; La Cava, A. Leptin and Inflammation. Curr. Immunol. Rev. 2008, 4, 70–79. [Google Scholar] [CrossRef]
- Frederich, R.C.; Hamann, A.; Anderson, S.; Löllmann, B.; Lowell, B.B.; Flier, J.S. Leptin levels reflect body lipid content in mice: Evidence for diet-induced resistance to leptin action. Nat. Med. 1995, 1, 1311–1314. [Google Scholar] [CrossRef]
- Maffei, M.; Fei, H.; Lee, G.H.; Dani, C.; Leroy, P.; Zhang, Y.; Proenca, R.; Negrel, R.; Ailhaud, G.; Friedman, J.M. Increased expression in adipocytes of ob RNA in mice with lesions of the hypothalamus and with mutations at the db locus. Proc. Natl. Acad. Sci. USA 1995, 92, 6957–6960. [Google Scholar] [CrossRef] [Green Version]
- Schanton, M.; Maymó, J.L.; Pérez-Pérez, A.; Sánchez-Margalet, V.; Varone, C.L. Involvement of leptin in the molecular physiology of the placenta. Reproduction 2018, 155, R1–R12. [Google Scholar] [CrossRef] [Green Version]
- Lee, G.-H.; Proenca, R.; Montez, J.M.; Carroll, K.M.; Darvishzadeh, J.G.; Lee, J.I.; Friedman, J.M. Abnormal splicing of the leptin receptor in diabetic mice. Nature 1996, 379, 632–635. [Google Scholar] [CrossRef] [PubMed]
- Löllmann, B.; Grüninger, S.; Stricker-Krongrad, A.; Chiesi, M. Detection and quantification of the leptin receptor splice variants Ob-Ra, b, and, e in different mouse tissues. Biochem. Biophys. Res. Commun. 1997, 238, 648–652. [Google Scholar] [CrossRef] [PubMed]
- Tartaglia, L.A.; Dembski, M.; Weng, X.; Deng, N.; Culpepper, J.; Devos, R.; Richards, G.J.; Campfield, L.A.; Clark, F.T.; Deeds, J.; et al. Identification and expression cloning of a leptin receptor, OB-R. Cell 1995, 83, 1263–1271. [Google Scholar] [CrossRef] [Green Version]
- Tartaglia, L.A. The Leptin Receptor. J. Biol. Chem. 1997, 272, 6093–6096. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Myers, M.G. Leptin receptor signaling and the regulation of mammalian physiology. Recent Prog. Horm. Res. 2004, 59, 287–304. [Google Scholar] [CrossRef] [PubMed]
- Bjørbaek, C.; Uotani, S.; da Silva, B.; Flier, J.S. Divergent signaling capacities of the long and short isoforms of the leptin receptor. J. Biol. Chem. 1997, 272, 32686–32695. [Google Scholar] [CrossRef] [Green Version]
- Friedman, J.M.; Halaas, J.L. Leptin and the regulation of body weight in mammals. Nature 1998, 395, 763–770. [Google Scholar] [CrossRef]
- Ducy, P.; Amling, M.; Takeda, S.; Priemel, M.; Schilling, A.F.; Beil, F.T.; Shen, J.; Vinson, C.; Rueger, J.M.; Karsenty, G. Leptin inhibits bone formation through a hypothalamic relay: A central control of bone mass. Cell 2000, 100, 197–207. [Google Scholar] [CrossRef] [Green Version]
- Procaccini, C.; Jirillo, E.; Matarese, G. Leptin as an immunomodulator. Mol. Aspects Med. 2012, 33, 35–45. [Google Scholar] [CrossRef]
- Fernández-Riejos, P.; Najib, S.; Santos-Alvarez, J.; Martín-Romero, C.; Pérez-Pérez, A.; González-Yanes, C.; Sánchez-Margalet, V. Role of Leptin in the Activation of Immune Cells. Mediat. Inflamm. 2010, 2010, 1–8. [Google Scholar] [CrossRef]
- Ahima, R.S.; Osei, S.Y. Leptin signaling. Physiol. Behav. 2004, 81, 223–241. [Google Scholar] [CrossRef] [PubMed]
- Sahu, A. Leptin signaling in the hypothalamus: Emphasis on energy homeostasis and leptin resistance. Front. Neuroendocrinol. 2003, 24, 225–253. [Google Scholar] [CrossRef] [PubMed]
- Sweeney, G. Leptin signalling. Cell. Signal. 2002, 14, 655–663. [Google Scholar] [CrossRef]
- Gualillo, O.; Eiras, S.; White, D.W.; Diéguez, C.; Casanueva, F.F. Leptin promotes the tyrosine phosphorylation of SHC proteins and SHC association with GRB2. Mol. Cell. Endocrinol. 2002, 190, 83–89. [Google Scholar] [CrossRef]
- Santos-Alvarez, J.; Goberna, R.; Sánchez-Margalet, V. Human Leptin Stimulates Proliferation and Activation of Human Circulating Monocytes. Cell. Immunol. 1999, 194, 6–11. [Google Scholar] [CrossRef]
- Najib, S.; Sánchez-Margalet, V. Human leptin promotes survival of human circulating blood monocytes prone to apoptosis by activation of p42/44 MAPK pathway. Cell. Immunol. 2002, 220, 143–149. [Google Scholar] [CrossRef]
- Sanchez-Margalet, V.; Martin-Romero, C. Human Leptin Signaling in Human Peripheral Blood Mononuclear Cells: Activation of the JAK-STAT Pathway. Cell. Immunol. 2001, 211, 30–36. [Google Scholar] [CrossRef]
- Martín-Romero, C.; Santos-Alvarez, J.; Goberna, R.; Sánchez-Margalet, V. Human leptin enhances activation and proliferation of human circulating T lymphocytes. Cell. Immunol. 2000, 199, 15–24. [Google Scholar] [CrossRef]
- Napoleone, E.; DI Santo, A.; Amore, C.; Baccante, G.; di Febbo, C.; Porreca, E.; de Gaetano, G.; Donati, M.B.; Lorenzet, R. Leptin induces tissue factor expression in human peripheral blood mononuclear cells: A possible link between obesity and cardiovascular risk? J. Thromb. Haemost. 2007, 5, 1462–1468. [Google Scholar] [CrossRef]
- Conde, J.; Scotece, M.; Gómez, R.; Gómez-Reino, J.J.; Lago, F.; Gualillo, O. At the crossroad between immunity and metabolism: Focus on leptin. Expert Rev. Clin. Immunol. 2010, 6, 801–808. [Google Scholar] [CrossRef]
- Um, H.D.; Orenstein, J.M.; Wahl, S.M. Fas mediates apoptosis in human monocytes by a reactive oxygen intermediate dependent pathway. J. Immunol. 1996, 156, 3469–3477. [Google Scholar] [PubMed]
- Sánchez-Pozo, C.; Rodriguez-Baño, J.; Domínguez-Castellano, A.; Muniain, M.A.; Goberna, R.; Sánchez-Margalet, V. Leptin stimulates the oxidative burst in control monocytes but attenuates the oxidative burst in monocytes from HIV-infected patients. Clin. Exp. Immunol. 2003, 134, 464–469. [Google Scholar] [CrossRef] [PubMed]
- Zarkesh-Esfahani, H.; Pockley, A.G.; Wu, Z.; Hellewell, P.G.; Weetman, A.P.; Ross, R.J.M. Leptin indirectly activates human neutrophils via induction of TNF-alpha. J. Immunol. 2004, 172, 1809–1814. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bruno, A.; Conus, S.; Schmid, I.; Simon, H.-U. Apoptotic pathways are inhibited by leptin receptor activation in neutrophils. J. Immunol. 2005, 174, 8090–8096. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mancuso, P.; McNish, R.W.; Peters-Golden, M.; Brock, T.G. Evaluation of phagocytosis and arachidonate metabolism by alveolar macrophages and recruited neutrophils from F344xBN rats of different ages. Mech. Ageing Dev. 2001, 122, 1899–1913. [Google Scholar] [CrossRef]
- Matarese, G.; Moschos, S.; Mantzoros, C.S. Leptin in immunology. J. Immunol. 2005, 174, 3137–3142. [Google Scholar] [CrossRef] [Green Version]
- Ottonello, L.; Gnerre, P.; Bertolotto, M.; Mancini, M.; Dapino, P.; Russo, R.; Garibotto, G.; Barreca, T.; Dallegri, F. Leptin as a uremic toxin interferes with neutrophil chemotaxis. J. Am. Soc. Nephrol. 2004, 15, 2366–2372. [Google Scholar] [CrossRef] [Green Version]
- CONUS, S.; BRUNO, A.; SIMON, H. Leptin is an eosinophil survival factor. J. Allergy Clin. Immunol. 2005, 116, 1228–1234. [Google Scholar] [CrossRef]
- Park, Y.M.; Bochner, B.S. Eosinophil survival and apoptosis in health and disease. Allergy Asthma Immunol. Res. 2010, 2, 87–101. [Google Scholar] [CrossRef] [Green Version]
- Zhao, Y.; Sun, R.; You, L.; Gao, C.; Tian, Z. Expression of leptin receptors and response to leptin stimulation of human natural killer cell lines. Biochem. Biophys. Res. Commun. 2003, 300, 247–252. [Google Scholar] [CrossRef]
- Loffreda, S.; Yang, S.Q.; Lin, H.Z.; Karp, C.L.; Brengman, M.L.; Wang, D.J.; Klein, A.S.; Bulkley, G.B.; Bao, C.; Noble, P.W.; et al. Leptin regulates proinflammatory immune responses. FASEB J. 1998, 12, 57–65. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Al-Hassi, H.O.; Bernardo, D.; Murugananthan, A.U.; Mann, E.R.; English, N.R.; Jones, A.; Kamm, M.A.; Arebi, N.; Hart, A.L.; Blakemore, A.I.F.; et al. A mechanistic role for leptin in human dendritic cell migration: Differences between ileum and colon in health and Crohn’s disease. Mucosal. Immunol. 2013, 6, 751–761. [Google Scholar] [CrossRef] [Green Version]
- Caldefie-Chezet, F.; Poulin, A.; Tridon, A.; Sion, B.; Vasson, M.P. Leptin: A potential regulator of polymorphonuclear neutrophil bactericidal action? J. Leukoc. Biol. 2001, 69, 414–418. [Google Scholar] [PubMed]
- Mattioli, B.; Straface, E.; Quaranta, M.G.; Giordani, L.; Viora, M. Leptin Promotes Differentiation and Survival of Human Dendritic Cells and Licenses Them for Th1 Priming. J. Immunol. 2005, 174, 6820–6828. [Google Scholar] [CrossRef] [PubMed]
- Mattioli, B.; Straface, E.; Matarrese, P.; Quaranta, M.G.; Giordani, L.; Malorni, W.; Viora, M. Leptin as an immunological adjuvant: Enhanced migratory and CD8+ T cell stimulatory capacity of human dendritic cells exposed to leptin. FASEB J. 2008, 22, 2012–2022. [Google Scholar] [CrossRef] [PubMed]
- Howard, J.K.; Lord, G.M.; Matarese, G.; Vendetti, S.; Ghatei, M.A.; Ritter, M.A.; Lechler, R.I.; Bloom, S.R. Leptin protects mice from starvation-induced lymphoid atrophy and increases thymic cellularity in ob/ob mice. J. Clin. Investig. 1999, 104, 1051–1059. [Google Scholar] [CrossRef] [Green Version]
- Reis, B.S.; Lee, K.; Fanok, M.H.; Mascaraque, C.; Amoury, M.; Cohn, L.B.; Rogoz, A.; Dallner, O.S.; Moraes-Vieira, P.M.; Domingos, A.I.; et al. Leptin receptor signaling in T cells is required for Th17 differentiation. J. Immunol. 2015, 194, 5253–5260. [Google Scholar] [CrossRef] [Green Version]
- Fujita, Y.; Fujii, T.; Mimori, T.; Sato, T.; Nakamura, T.; Iwao, H.; Nakajima, A.; Miki, M.; Sakai, T.; Kawanami, T.; et al. Deficient leptin signaling ameliorates systemic lupus erythematosus lesions in MRL/Mp-Fas lpr mice. J. Immunol. 2014, 192, 979–984. [Google Scholar] [CrossRef] [Green Version]
- Yu, Y.; Liu, Y.; Shi, F.-D.; Zou, H.; Matarese, G.; La Cava, A. Cutting Edge: Leptin-Induced ROR t Expression in CD4+ T Cells Promotes Th17 Responses in Systemic Lupus Erythematosus. J. Immunol. 2013, 190, 3054–3058. [Google Scholar] [CrossRef] [Green Version]
- Procaccini, C.; De Rosa, V.; Galgani, M.; Abanni, L.; Calì, G.; Porcellini, A.; Carbone, F.; Fontana, S.; Horvath, T.L.; La Cava, A.; et al. An oscillatory switch in mTOR kinase activity sets regulatory T cell responsiveness. Immunity 2010, 33, 929–941. [Google Scholar] [CrossRef] [Green Version]
- Kim, S.Y.; Lim, J.H.; Choi, S.W.; Kim, M.; Kim, S.-T.; Kim, M.-S.; Cho, Y.S.; Chun, E.; Lee, K.-Y. Preferential effects of leptin on CD4 T cells in central and peripheral immune system are critically linked to the expression of leptin receptor. Biochem. Biophys. Res. Commun. 2010, 394, 562–568. [Google Scholar] [CrossRef] [PubMed]
- Matarese, G.; Procaccini, C.; De Rosa, V.; Horvath, T.L.; La Cava, A. Regulatory T cells in obesity: The leptin connection. Trends Mol. Med. 2010, 16, 247–256. [Google Scholar] [CrossRef] [PubMed]
- Agrawal, S.; Gollapudi, S.; Su, H.; Gupta, S. Leptin activates human B cells to secrete Tfile:///C:/Users/PC/Desktop/p38 MAPK activation and signaling.pdfNF-??, IL-6, and IL-10 via JAK2/STAT3 and p38MAPK/ERK1/2 signaling pathway. J. Clin. Immunol. 2011, 31, 472–478. [Google Scholar] [CrossRef] [Green Version]
- Claycombe, K.; King, L.E.; Fraker, P.J. A role for leptin in sustaining lymphopoiesis and myelopoiesis. Proc. Natl. Acad. Sci. USA 2008, 105, 2017–2021. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Katona, P.; Katona-Apte, J. The interaction between nutrition and infection. Clin. Infect. Dis. 2008, 46, 1582–1588. [Google Scholar] [CrossRef] [PubMed]
- Woodward, B. Protein, calories, and immune defenses. Nutr. Rev. 1998, 56, S84–S92. [Google Scholar] [CrossRef]
- Schaible, U.E.; Kaufmann, S.H.E. Malnutrition and Infection: Complex Mechanisms and Global Impacts. PLoS Med. 2007, 4, e115. [Google Scholar] [CrossRef] [Green Version]
- Faggioni, R.; Moser, A.; Feingold, K.R.; Grunfeld, C. Reduced Leptin Levels in Starvation Increase Susceptibility to Endotoxic Shock. Am. J. Pathol. 2000, 156, 1781–1787. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.; Olbort, M.; Schwarzer, K.; Nuesslein-Hildesheim, B.; Nicolson, M.; Murphy, E.; Kowalski, T.J.; Schmidt, I.; Leibel, R.L. The Leptin Receptor Mediates Apparent Autocrine Regulation of Leptin Gene Expression. Biochem. Biophys. Res. Commun. 1997, 240, 492–495. [Google Scholar] [CrossRef]
- Ikejima, S.; Sasaki, S.; Sashinami, H.; Mori, F.; Ogawa, Y.; Nakamura, T.; Abe, Y.; Wakabayashi, K.; Suda, T.; Nakane, A. Impairment of host resistance to Listeria monocytogenes infection in liver of db/db and ob/ob mice. Diabetes 2005, 54, 182–189. [Google Scholar] [CrossRef] [Green Version]
- Gainsford, T.; Willson, T.A.; Metcalf, D.; Handman, E.; McFarlane, C.; Ng, A.; Nicola, N.A.; Alexander, W.S.; Hilton, D.J. Leptin can induce proliferation, differentiation, and functional activation of hemopoietic cells. Proc. Natl. Acad. Sci. USA 1996, 93, 14564–14568. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dayakar, A.; Chandrasekaran, S.; Veronica, J.; Maurya, R. Leptin induces the phagocytosis and protective immune response in Leishmania donovani infected THP-1 cell line and human PBMCs. Exp. Parasitol. 2016, 160, 54–59. [Google Scholar] [CrossRef] [PubMed]
- Van Crevel, R.; Ottenhoff, T.H.M.; van der Meer, J.W.M. Innate immunity to Mycobacterium tuberculosis. Clin. Microbiol. Rev. 2002, 15, 294–309. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kaukonen, K.M.; Bailey, M.; Pilcher, D.; Cooper, D.J.; Bellomo, R. Systemic inflammatory response syndrome criteria in defining severe sepsis. N. Engl. J. Med. 2015, 372, 1629–1638. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Takahashi, T.; Imai, K.; Hashizume, K. Generation and characterization of anti-leptin antisera against synthetic peptides and recombinant protein. J. Reprod. Dev. 2004, 50, 717–724. [Google Scholar] [CrossRef] [Green Version]
- Hultgren, O.H.; Stenson, M.; Tarkowski, A. Role of IL-12 in Staphylococcus aureus-triggered arthritis and sepsis. Arthritis Res. 2000, 3, 41. [Google Scholar] [CrossRef] [Green Version]
- Tschöp, J.; Dattilo, J.R.; Prakash, P.S.; Kasten, K.R.; Tschöp, M.H.; Caldwell, C.C. The leptin system: A potential target for sepsis induced immune suppression. Endocr. Metab. Immune Disord. Drug Targets 2010, 10, 336–347. [Google Scholar] [CrossRef]
- Estrada, V.; Serrano-Ríos, M.; Martínez Larrad, M.T.; Villar, N.G.P.; González López, A.; Téllez, M.J.; Fernández, C. Leptin and adipose tissue maldistribution in HIV-infected male patients with predominant fat loss treated with antiretroviral therapy. J. Acquir. Immune Defic. Syndr. 2002, 29, 32–40. [Google Scholar] [CrossRef]
- Kotler, D.P.; Wang, J.; Pierson, R.N. Body composition studies in patients with the acquired immunodeficiency syndrome. Am. J. Clin. Nutr. 1985, 42, 1255–1265. [Google Scholar] [CrossRef] [Green Version]
- Madan, R.; Guo, X.; Naylor, C.; Buonomo, E.L.; Mackay, D.; Noor, Z.; Concannon, P.; Scully, K.W.; Pramoonjago, P.; Kolling, G.L.; et al. Role of leptin-mediated colonic inflammation in defense against Clostridium difficile colitis. Infect. Immun. 2014, 82, 341–349. [Google Scholar] [CrossRef] [Green Version]
- Morgan, T.R.; Ghany, M.G.; Kim, H.-Y.; Snow, K.K.; Shiffman, M.L.; De Santo, J.L.; Lee, W.M.; Di Bisceglie, A.M.; Bonkovsky, H.L.; Dienstag, J.L.; et al. Outcome of sustained virological responders with histologically advanced chronic hepatitis C. Hepatology 2010, 52, 833–844. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Azzoni, L.; Crowther, N.J.; Firnhaber, C.; Foulkes, A.S.; Yin, X.; Glencross, D.; Gross, R.; Kaplan, M.D.; Papasavvas, E.; Schulze, D.; et al. Association between HIV replication and serum leptin levels: An observational study of a cohort of HIV-1-infected South African women. J. Int. AIDS Soc. 2010, 13, 33. [Google Scholar] [CrossRef] [PubMed]
- Pauli, E.-K.; Schmolke, M.; Wolff, T.; Viemann, D.; Roth, J.; Bode, J.G.; Ludwig, S. Influenza A Virus Inhibits Type I IFN Signaling via NF-κB-Dependent Induction of SOCS-3 Expression. PLoS Pathog. 2008, 4, e1000196. [Google Scholar] [CrossRef] [PubMed]
- Akhtar, L.N.; Benveniste, E.N. Viral Exploitation of Host SOCS Protein Functions. J. Virol. 2011, 85, 1912–1921. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Michaud, F.; Coulombe, F.; Gaudreault, E.; Paquet-Bouchard, C.; Rola-Pleszczynski, M.; Gosselin, J. Epstein-Barr Virus Interferes with the Amplification of IFNα Secretion by Activating Suppressor of Cytokine Signaling 3 in Primary Human Monocytes. PLoS ONE 2010, 5, e11908. [Google Scholar] [CrossRef] [Green Version]
- Tian, R.-R.; Guo, H.-X.; Wei, J.-F.; Yang, C.-K.; He, S.-H.; Wang, J.-H. IFN-λ inhibits HIV-1 integration and post-transcriptional events in vitro, but there is only limited in vivo repression of viral production. Antivir. Res. 2012, 95, 57–65. [Google Scholar] [CrossRef]
- Desreumaux, P.; Ernst, O.; Geboes, K.; Gambiez, L.; Berrebi, D.; Müller-Alouf, H.; Hafraoui, S.; Emilie, D.; Ectors, N.; Peuchmaur, M.; et al. Inflammatory alterations in mesenteric adipose tissue in Crohn’s disease. Gastroenterology 1999, 117, 73–81. [Google Scholar] [CrossRef]
- Sánchez-Margalet, V.; Martín-Romero, C.; Santos-Alvarez, J.; Goberna, R.; Najib, S.; Gonzalez-Yanes, C. Role of leptin as an immunomodulator of blood mononuclear cells: Mechanisms of action. Clin. Exp. Immunol. 2003, 133, 11–19. [Google Scholar] [CrossRef]
- Pulido-Mendez, M.; De Sanctis, J.; Rodríguez-Acosta, A. Leptin and leptin receptors during malaria infection in mice. Folia Parasitol. 2002, 49, 249–251. [Google Scholar] [CrossRef] [Green Version]
- Noach, L.A.; Bosma, N.B.; Jansen, J.; Hoek, F.J.; van Deventer, S.J.; Tytgat, G.N. Mucosal tumor necrosis factor-alpha, interleukin-1 beta, and interleukin-8 production in patients with Helicobacter pylori infection. Scand. J. Gastroenterol. 1994, 29, 425–429. [Google Scholar] [CrossRef]
- Sukhotnik, I.; Coran, A.G.; Mogilner, J.G.; Shamian, B.; Karry, R.; Lieber, M.; Shaoul, R. Leptin affects intestinal epithelial cell turnover in correlation with leptin receptor expression along the villus-crypt axis after massive small bowel resection in a rat. Pediatr. Res. 2009, 66, 648–653. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brun, P.; Castagliuolo, I.; Di Leo, V.; Buda, A.; Pinzani, M.; Palù, G.; Martines, D. Increased intestinal permeability in obese mice: New evidence in the pathogenesis of nonalcoholic steatohepatitis. Am. J. Physiol. Gastrointest. Liver Physiol. 2007, 292, G518–G525. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vedantam, G.; Viswanathan, V.K. Leptin signaling protects the gut from Entamoeba histolytica infection. Gut Microbes 2012, 3, 2–3. [Google Scholar] [CrossRef]
- Farooqi, I.S.; Wangensteen, T.; Collins, S.; Kimber, W.; Matarese, G.; Keogh, J.M.; Lank, E.; Bottomley, B.; Lopez-Fernandez, J.; Ferraz-Amaro, I.; et al. Clinical and Molecular Genetic Spectrum of Congenital Deficiency of the Leptin Receptor. N. Engl. J. Med. 2007, 356, 237–247. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Farooqi, I.S.; Matarese, G.; Lord, G.M.; Keogh, J.M.; Lawrence, E.; Agwu, C.; Sanna, V.; Jebb, S.A.; Perna, F.; Fontana, S.; et al. Beneficial effects of leptin on obesity, T cell hyporesponsiveness, and neuroendocrine/metabolic dysfunction of human congenital leptin deficiency. J. Clin. Investig. 2002, 110, 1093–1103. [Google Scholar] [CrossRef]
- White, N.J.; Pukrittayakamee, S.; Hien, T.T.; Faiz, M.A.; Mokuolu, O.A.; Dondorp, A.M. Malaria. Lancet 2014, 383, 723–735. [Google Scholar] [CrossRef]
- Cauchard, S.; Bermúdez-Humarán, L.G.; Blugeon, S.; Laugier, C.; Langella, P.; Cauchard, J. Mucosal co-immunization of mice with recombinant lactococci secreting VapA antigen and leptin elicits a protective immune response against Rhodococcus equi infection. Vaccine 2011, 30, 95–102. [Google Scholar] [CrossRef]
- Jubiz, W.; Draper, R.E.; Gale, J.; Nolan, G. Decreased leukotriene B4 synthesis by polymorphonuclear leukocytes from male patients with diabetes mellitus. Prostaglandins. Leukot. Med. 1984, 14, 305–311. [Google Scholar] [CrossRef]
- Skerrett, S.J.; Henderson, W.R.; Martin, T.R. Alveolar macrophage function in rats with severe protein calorie malnutrition. Arachidonic acid metabolism, cytokine release, and antimicrobial activity. J. Immunol. 1990, 144, 1052–1061. [Google Scholar]
- Cederholm, T.; Lindgren, J.A.; Palmblad, J. Impaired leukotriene C4 generation in granulocytes from protein-energy malnourished chronically ill elderly. J. Intern. Med. 2000, 247, 715–722. [Google Scholar] [CrossRef] [Green Version]
- Coffey, M.J.; Phare, S.M.; Kazanjian, P.H.; Peters-Golden, M. 5-Lipoxygenase metabolism in alveolar macrophages from subjects infected with the human immunodeficiency virus. J. Immunol. 1996, 157, 393–399. [Google Scholar] [PubMed]
- Wellen, K.E.; Hotamisligil, G.S. Inflammation, stress, and diabetes. J. Clin. Investig. 2005, 115, 1111–1119. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Calle, E.E.; Kaaks, R. Overweight, obesity and cancer: Epidemiological evidence and proposed mechanisms. Nat. Rev. Cancer 2004, 4, 579–591. [Google Scholar] [CrossRef] [PubMed]
- Mannino, D.M.; Mott, J.; Ferdinands, J.M.; Camargo, C.A.; Friedman, M.; Greves, H.M.; Redd, S.C. Boys with high body masses have an increased risk of developing asthma: Findings from the National Longitudinal Survey of Youth (NLSY). Int. J. Obes. 2006, 30, 6–13. [Google Scholar] [CrossRef] [Green Version]
- Xu, H.; Barnes, G.T.; Yang, Q.; Tan, G.; Yang, D.; Chou, C.J.; Sole, J.; Nichols, A.; Ross, J.S.; Tartaglia, L.A.; et al. Chronic inflammation in fat plays a crucial role in the development of obesity-related insulin resistance. J. Clin. Investig. 2003, 112, 1821–1830. [Google Scholar] [CrossRef]
- Weisberg, S.P.; McCann, D.; Desai, M.; Rosenbaum, M.; Leibel, R.L.; Ferrante, A.W. Obesity is associated with macrophage accumulation in adipose tissue. J. Clin. Investig. 2003, 112, 1796–1808. [Google Scholar] [CrossRef]
- Sonnenberg, G.E.; Krakower, G.R.; Kissebah, A.H. A Novel Pathway to the Manifestations of Metabolic Syndrome. Obes. Res. 2004, 12, 180–186. [Google Scholar] [CrossRef]
- Mariano, G.; Stilo, R.; Terrazzano, G.; Coccia, E.; Vito, P.; Varricchio, E.; Paolucci, M. Effects of recombinant trout leptin in superoxide production and NF-κB/MAPK phosphorylation in blood leukocytes. Peptides 2013, 48, 59–69. [Google Scholar] [CrossRef]
- Wild, S.; Roglic, G.; Green, A.; Sicree, R.; King, H. Global prevalence of diabetes: Estimates for the year 2000 and projections for 2030. Diabetes Care 2004, 27, 1047–1053. [Google Scholar] [CrossRef] [Green Version]
- DeFronzo, R.A. Pathogenesis of type 2 diabetes mellitus. Med. Clin. N. Am. 2004, 88, 787–835. [Google Scholar] [CrossRef]
- Cefalu, W.T. Animal models of type 2 diabetes: Clinical presentation and pathophysiological relevance to the human condition. ILAR J. 2006, 47, 186–198. [Google Scholar] [CrossRef] [PubMed]
- Srinivasan, K.; Ramarao, P. Animal models in type 2 diabetes research: An overview. Indian J. Med. Res. 2007, 125, 451–472. [Google Scholar]
- Van de Bunt, M.; Gloyn, A.L. From genetic association to molecular mechanism. Curr. Diabetes. Rep. 2010, 10, 452–466. [Google Scholar] [CrossRef] [PubMed]
- Jafar-Mohammadi, B.; McCarthy, M.I. Genetics of type 2 diabetes mellitus and obesity—A review. Ann. Med. 2008, 40, 2–10. [Google Scholar] [CrossRef] [PubMed]
- Poulsen, P.; Kyvik, K.O.; Vaag, A.; Beck-Nielsen, H. Heritability of type II (non-insulin-dependent) diabetes mellitus and abnormal glucose tolerance--a population-based twin study. Diabetologia 1999, 42, 139–145. [Google Scholar] [CrossRef] [PubMed]
- Krebs, D.L.; Hilton, D.J. SOCS Proteins: Negative Regulators of Cytokine Signaling. Stem Cells 2001, 19, 378–387. [Google Scholar] [CrossRef] [PubMed]
- Bjørbæk, C.; Buchholz, R.M.; Davis, S.M.; Bates, S.H.; Pierroz, D.D.; Gu, H.; Neel, B.G.; Myers, M.G.; Flier, J.S. Divergent Roles of SHP-2 in ERK Activation by Leptin Receptors. J. Biol. Chem. 2001, 276, 4747–4755. [Google Scholar] [CrossRef] [Green Version]
- St-Pierre, J.; Tremblay, M.L. Modulation of Leptin Resistance by Protein Tyrosine Phosphatases. Cell Metab. 2012, 15, 292–297. [Google Scholar] [CrossRef] [Green Version]
- Ozcan, L.; Ergin, A.S.; Lu, A.; Chung, J.; Sarkar, S.; Nie, D.; Myers, M.G.; Ozcan, U. Endoplasmic reticulum stress plays a central role in development of leptin resistance. Cell Metab. 2009, 9, 35–51. [Google Scholar] [CrossRef] [Green Version]
- Hosoi, T.; Sasaki, M.; Miyahara, T.; Hashimoto, C.; Matsuo, S.; Yoshii, M.; Ozawa, K. Endoplasmic Reticulum Stress Induces Leptin Resistance. Mol. Pharmacol. 2008, 74, 1610–1619. [Google Scholar] [CrossRef] [Green Version]
- Kawai, T.; Akira, S. The role of pattern-recognition receptors in innate immunity: Update on Toll-like receptors. Nat. Immunol. 2010, 11, 373–384. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.-M.; Chen, L.-L.; Wang, L.; Xu, S.; Wang, X.; Yi, L.-L.; Chen, D.; Wu, Z.-H.; Zhang, J.-Y.; Liao, Y.-F.; et al. Macrophage infiltrates with high levels of Toll-like receptor 4 expression in white adipose tissues of male Chinese. Nutr. Metab. Cardiovasc. Dis. 2009, 19, 736–743. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, M.T.A.; Favelyukis, S.; Nguyen, A.-K.; Reichart, D.; Scott, P.A.; Jenn, A.; Liu-Bryan, R.; Glass, C.K.; Neels, J.G.; Olefsky, J.M. A subpopulation of macrophages infiltrates hypertrophic adipose tissue and is activated by free fatty acids via Toll-like receptors 2 and 4 and JNK-dependent pathways. J. Biol. Chem. 2007, 282, 35279–35292. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liberale, L.; Bonaventura, A.; Vecchiè, A.; Matteo, C.; Dallegri, F.; Montecucco, F.; Carbone, F. The Role of Adipocytokines in Coronary Atherosclerosis. Curr. Atheroscler. Rep. 2017, 19, 10. [Google Scholar] [CrossRef]
- Scheja, L.; Heeren, J. The endocrine function of adipose tissues in health and cardiometabolic disease. Nat. Rev. Endocrinol. 2019, 15, 507–524. [Google Scholar] [CrossRef]
- Dubey, L.; Zeng, H.; Hashmi, S.; Hongjie, W.; Tao, H. Association of plasma leptin levels and complexity of the culprit lesion in patients with unstable angina. Int. J. Cardiol. 2008, 126, 183–189. [Google Scholar] [CrossRef] [PubMed]
- Sweeney, G. Cardiovascular effects of leptin. Nat. Rev. Cardiol. 2010, 7, 22–29. [Google Scholar] [CrossRef]
- Knerr, I.; Wolf, J.; Reinehr, T.; Stachow, R.; Grabert, M.; Schober, E.; Rascher, W.; Holl, R.W. DPV Scientific Initiative of Germany and Austria The ‘accelerator hypothesis’: Relationship between weight, height, body mass index and age at diagnosis in a large cohort of 9,248 German and Austrian children with type 1 diabetes mellitus. Diabetologia 2005, 48, 2501–2504. [Google Scholar] [CrossRef] [Green Version]
- Otero, M.; Lago, R.; Lago, F.; Casanueva, F.F.; Dieguez, C.; Gómez-Reino, J.J.; Gualillo, O. Leptin, from fat to inflammation: Old questions and new insights. FEBS Lett. 2005, 579, 295–301. [Google Scholar] [CrossRef] [Green Version]
- Lee, S.-W.; Park, M.-C.; Park, Y.-B.; Lee, S.-K. Measurement of the serum leptin level could assist disease activity monitoring in rheumatoid arthritis. Rheumatol. Int. 2007, 27, 537–540. [Google Scholar] [CrossRef]
- Targońska-Stepniak, B.; Majdan, M.; Dryglewska, M. Leptin serum levels in rheumatoid arthritis patients: Relation to disease duration and activity. Rheumatol. Int. 2008, 28, 585–591. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Gonzalez, A.; Gonzalez-Lopez, L.; Valera-Gonzalez, I.C.; Cardona-Muñoz, E.G.; Salazar-Paramo, M.; González-Ortiz, M.; Martínez-Abundis, E.; Gamez-Nava, J.I. Serum leptin levels in women with systemic lupus erythematosus. Rheumatol. Int. 2002, 22, 138–141. [Google Scholar] [CrossRef] [PubMed]
- Teichtahl, A.J.; Wluka, A.E.; Proietto, J.; Cicuttini, F.M. Obesity and the female sex, risk factors for knee osteoarthritis that may be attributable to systemic or local leptin biosynthesis and its cellular effects. Med. Hypotheses 2005, 65, 312–315. [Google Scholar] [CrossRef] [PubMed]
- Lourenço, E.V.; Liu, A.; Matarese, G.; La Cava, A. Leptin promotes systemic lupus erythematosus by increasing autoantibody production and inhibiting immune regulation. Proc. Natl. Acad. Sci. USA 2016, 113, 10637–10642. [Google Scholar] [CrossRef] [Green Version]
- Barranco, C. Leptin linked to SLE. Nat. Rev. Rheumatol. 2016, 12, 623. [Google Scholar] [CrossRef]
- Harpsøe, M.C.; Basit, S.; Andersson, M.; Nielsen, N.M.; Frisch, M.; Wohlfahrt, J.; Nohr, E.A.; Linneberg, A.; Jess, T. Body mass index and risk of autoimmune diseases: A study within the Danish National Birth Cohort. Int. J. Epidemiol. 2014, 43, 843–855. [Google Scholar] [CrossRef] [Green Version]
- Hutcheson, J. Adipokines influence the inflammatory balance in autoimmunity. Cytokine 2015, 75, 272–279. [Google Scholar] [CrossRef]
- Evereklioglu, C.; Inalöz, H.S.; Kirtak, N.; Doganay, S.; Bülbül, M.; Ozerol, E.; Er, H.; Ozbek, E. Serum leptin concentration is increased in patients with Behçet’s syndrome and is correlated with disease activity. Br. J. Dermatol. 2002, 147, 331–336. [Google Scholar] [CrossRef]
- Karrasch, T.; Schaeffler, A. Adipokines and the role of visceral adipose tissue in inflammatory bowel disease. Ann. Gastroenterol. 2016, 29, 424–438. [Google Scholar] [CrossRef]
- Marzullo, P.; Minocci, A.; Tagliaferri, M.A.; Guzzaloni, G.; Di Blasio, A.; De Medici, C.; Aimaretti, G.; Liuzzi, A. Investigations of thyroid hormones and antibodies in obesity: Leptin levels are associated with thyroid autoimmunity independent of bioanthropometric, hormonal, and weight-related determinants. J. Clin. Endocrinol. Metab. 2010, 95, 3965–3972. [Google Scholar] [CrossRef] [Green Version]
- Toussirot, E.; Aubin, F.; Dumoulin, G. Relationships between Adipose Tissue and Psoriasis, with or without Arthritis. Front. Immunol. 2014, 5, 368. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tuzun, A.; Uygun, A.; Yesilova, Z.; Ozel, A.M.; Erdil, A.; Yaman, H.; Bagci, S.; Gulsen, M.; Karaeren, N.; Dagalp, K. Leptin levels in the acute stage of ulcerative colitis. J. Gastroenterol. Hepatol. 2004, 19, 429–432. [Google Scholar] [CrossRef] [PubMed]
- Matarese, G.; Di Giacomo, A.; Sanna, V.; Lord, G.M.; Howard, J.K.; Di Tuoro, A.; Bloom, S.R.; Lechler, R.I.; Zappacosta, S.; Fontana, S. Requirement for leptin in the induction and progression of autoimmune encephalomyelitis. J. Immunol. 2001, 166, 5909–5916. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Matarese, G.; Carrieri, P.B.; La Cava, A.; Perna, F.; Sanna, V.; De Rosa, V.; Aufiero, D.; Fontana, S.; Zappacosta, S. Leptin increase in multiple sclerosis associates with reduced number of CD4(+)CD25+ regulatory T cells. Proc. Natl. Acad. Sci. USA 2005, 102, 5150–5155. [Google Scholar] [CrossRef] [Green Version]
- Khandekar, M.J.; Cohen, P.; Spiegelman, B.M. Molecular mechanisms of cancer development in obesity. Nat. Rev. Cancer 2011, 11, 886–895. [Google Scholar] [CrossRef]
- Lin, T.-C.; Huang, K.-W.; Liu, C.-W.; Chang, Y.-C.; Lin, W.-M.; Yang, T.-Y.; Hsiao, M. Leptin signaling axis specifically associates with clinical prognosis and is multifunctional in regulating cancer progression. Oncotarget 2018, 9, 17210–17219. [Google Scholar] [CrossRef] [Green Version]
- Saxena, N.K.; Taliaferro-Smith, L.; Knight, B.B.; Merlin, D.; Anania, F.A.; O’Regan, R.M.; Sharma, D. Bidirectional crosstalk between leptin and insulin-like growth factor-I signaling promotes invasion and migration of breast cancer cells via transactivation of epidermal growth factor receptor. Cancer Res. 2008, 68, 9712–9722. [Google Scholar] [CrossRef] [Green Version]
- Haque, I.; Ghosh, A.; Acup, S.; Banerjee, S.; Dhar, K.; Ray, A.; Sarkar, S.; Kambhampati, S.; Banerjee, S.K. Leptin-induced ER-α-positive breast cancer cell viability and migration is mediated by suppressing CCN5-signaling via activating JAK/AKT/STAT-pathway. BMC Cancer 2018, 18, 99. [Google Scholar] [CrossRef] [Green Version]
- Newman, G.; Gonzalez-Perez, R.R. Leptin–cytokine crosstalk in breast cancer. Mol. Cell. Endocrinol. 2014, 382, 570–582. [Google Scholar] [CrossRef] [Green Version]
- Pérez-Pérez, A.; Sánchez-Jiménez, F.; Vilariño-García, T.; de la Cruz, L.; Virizuela, J.A.; Sánchez-Margalet, V. Sam68 Mediates the Activation of Insulin and Leptin Signalling in Breast Cancer Cells. PLoS ONE 2016, 11, e0158218. [Google Scholar] [CrossRef]
- Kim, H.G.; Jin, S.W.; Kim, Y.A.; Khanal, T.; Lee, G.H.; Kim, S.J.; Rhee, S.D.; Chung, Y.C.; Hwang, Y.J.; Jeong, T.C.; et al. Leptin induces CREB-dependent aromatase activation through COX-2 expression in breast cancer cells. Food Chem. Toxicol. 2017, 106, 232–241. [Google Scholar] [CrossRef] [PubMed]
- Zheng, Q.; Hursting, S.D.; Reizes, O. Leptin regulates cyclin D1 in luminal epithelial cells of mouse MMTV-Wnt-1 mammary tumors. J. Cancer Res. Clin. Oncol. 2012, 138, 1607–1612. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gonzalez-Perez, R.R.; Xu, Y.; Guo, S.; Watters, A.; Zhou, W.; Leibovich, S.J. Leptin upregulates VEGF in breast cancer via canonic and non-canonical signalling pathways and NFκB/HIF-1α activation. Cell. Signal. 2010, 22, 1350–1362. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sánchez-Jiménez, F.; Pérez-Pérez, A.; de la Cruz-Merino, L.; Sánchez-Margalet, V. Obesity and Breast Cancer: Role of Leptin. Front Oncol. 2019, 18, 596. [Google Scholar] [CrossRef]
- Park, J.; Morley, T.S.; Kim, M.; Clegg, D.J.; Scherer, P.E. Obesity and cancer--mechanisms underlying tumour progression and recurrence. Nat. Rev. Endocrinol. 2014, 10, 455–465. [Google Scholar] [CrossRef] [Green Version]
- Kato, S.; Abarzua-Catalan, L.; Trigo, C.; Delpiano, A.; Sanhueza, C.; García, K.; Ibañez, C.; Hormazábal, K.; Diaz, D.; Brañes, J.; et al. Leptin stimulates migration and invasion and maintains cancer stem-like properties in ovarian cancer cells: An explanation for poor outcomes in obese women. Oncotarget 2015, 6, 21100–21119. [Google Scholar] [CrossRef] [Green Version]
- Giordano, C.; Chemi, F.; Panza, S.; Barone, I.; Bonofiglio, D.; Lanzino, M.; Cordella, A.; Campana, A.; Hashim, A.; Rizza, P.; et al. Leptin as a mediator of tumor-stromal interactions promotes breast cancer stem cell activity. Oncotarget 2016, 7, 1262–1275. [Google Scholar] [CrossRef] [Green Version]
- Ghasemi, A.; Saeidi, J.; Azimi-Nejad, M.; Hashemy, S.I. Leptin-induced signaling pathways in cancer cell migration and invasion. Cell. Oncol. 2019. [Google Scholar] [CrossRef]
- Feldman, D.E.; Chen, C.; Punj, V.; Tsukamoto, H.; Machida, K. Pluripotency factor-mediated expression of the leptin receptor (OB-R) links obesity to oncogenesis through tumor-initiating stem cells. Proc. Natl. Acad. Sci. USA 2012, 109, 829–834. [Google Scholar] [CrossRef] [Green Version]
- La Cava, A.; Matarese, G. The weight of leptin in immunity. Nat. Rev. Immunol. 2004, 4, 371–379. [Google Scholar] [CrossRef]
- Li, K.; Wei, L.; Huang, Y.; Wu, Y.; Su, M.; Pang, X.; Wang, N.; Ji, F.; Zhong, C.; Chen, T. Leptin promotes breast cancer cell migration and invasion via IL-18 expression and secretion. Int. J. Oncol. 2016, 48, 2479–2487. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huebner, L.; Engeli, S.; Wrann, C.D.; Goudeva, L.; Laue, T.; Kielstein, H. Human NK Cell Subset Functions Are Differentially Affected by Adipokines. PLoS ONE 2013, 8, e75703. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wrann, C.D.; Laue, T.; Hubner, L.; Kuhlmann, S.; Jacobs, R.; Goudeva, L.; Nave, H. Short-term and long-term leptin exposure differentially affect human natural killer cell immune functions. Am. J. Physiol. Endocrinol. Metab. 2012, 302, E108–E116. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lamas, B.; Goncalves-Mendes, N.; Nachat-Kappes, R.; Rossary, A.; Caldefie-Chezet, F.; Vasson, M.P.; Farges, M.C. Leptin modulates dose-dependently the metabolic and cytolytic activities of NK-92 cells. J. Cell. Physiol. 2013, 228, 1202–1209. [Google Scholar] [CrossRef] [PubMed]
- Cortellini, A.; Bersanelli, M.; Buti, S.; Cannita, K.; Santini, D.; Perrone, F.; Giusti, R.; Tiseo, M.; Michiara, M.; Di Marino, P.; et al. A multicenter study of body mass index in cancer patients treated with anti-PD-1/PD-L1 immune checkpoint inhibitors: When overweight becomes favorable. J. Immunother. Cancer 2019, 7, 57. [Google Scholar] [CrossRef] [PubMed]
- Grivennikov, S.I.; Greten, F.R.; Karin, M. Immunity, inflammation, and cancer. Cell 2010, 140, 883–899. [Google Scholar] [CrossRef] [Green Version]
- Colotta, F.; Allavena, P.; Sica, A.; Garlanda, C.; Mantovani, A. Cancer-related inflammation, the seventh hallmark of cancer: Links to genetic instability. Carcinogenesis 2009, 30, 1073–1081. [Google Scholar] [CrossRef] [Green Version]
- Vesely, M.D.; Kershaw, M.H.; Schreiber, R.D.; Smyth, M.J. Natural Innate and Adaptive Immunity to Cancer. Annu. Rev. Immunol. 2011, 29, 235–271. [Google Scholar] [CrossRef] [Green Version]
- Yoon, Y.S.; Kwon, A.R.; Lee, Y.K.; Oh, S.W. Circulating adipokines and risk of obesity related cancers: A systematic review and meta-analysis. Obes. Res. Clin. Pract. 2019, 13, 329–339. [Google Scholar] [CrossRef]
- Nyasani, E.; Munir, I.; Perez, M.; Payne, K.; Khan, S. Linking obesity-induced leptin-signaling pathways to common endocrine-related cancers in women. Endocrine 2019, 63, 3–17. [Google Scholar] [CrossRef]
- Pan, H.; Deng, L.-L.; Cui, J.-Q.; Shi, L.; Yang, Y.-C.; Luo, J.-H.; Qin, D.; Wang, L. Association between serum leptin levels and breast cancer risk: An updated systematic review and meta-analysis. Medicine 2018, 97, e11345. [Google Scholar] [CrossRef] [PubMed]
- Andò, S.; Gelsomino, L.; Panza, S.; Giordano, C.; Bonofiglio, D.; Barone, I.; Catalano, S. Obesity, Leptin and Breast Cancer: Epidemiological Evidence and Proposed Mechanisms. Cancers 2019, 11, 62. [Google Scholar] [CrossRef] [Green Version]
- Harbuzariu, A.; Oprea-Ilies, G.M.; Gonzalez-Perez, R.R. The Role of Notch Signaling and Leptin-Notch Crosstalk in Pancreatic Cancer. Medicines 2018, 5, 68. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alshaker, H.; Sacco, K.; Alfraidi, A.; Muhammad, A.; Winkler, M.; Pchejetski, D. Leptin signalling, obesity and prostate cancer: Molecular and clinical perspective on the old dilemma. Oncotarget 2015, 6, 35556–35563. [Google Scholar] [CrossRef] [Green Version]
- Riondino, S.; Roselli, M.; Palmirotta, R.; Della-Morte, D.; Ferroni, P.; Guadagni, F. Obesity and colorectal cancer: Role of adipokines in tumor initiation and progression. World J. Gastroenterol. 2014, 20, 5177–5190. [Google Scholar] [CrossRef]
- He, Y.; Chen, H.; Quon, M.J.; Reitman, M. The mouse obese gene: Genomic organization, promoter activity, and activation by ccaat/enhancer-binding protein α. J. Biol. Chem. 1995, 270, 28887–28891. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Halaas, J.L.; Gajiwala, K.S.; Maffei, M.; Cohen, S.L.; Chait, B.T.; Rabinowitz, D.; Lallone, R.L.; Burley, S.K.; Friedman, J.M. Weight-reducing effects of the plasma protein encoded by the obese gene. Science 1995, 269, 543–546. [Google Scholar] [CrossRef] [PubMed]
- Hamilton, B.S.; Paglia, D.; Kwan, A.Y.M.; Deitel, M. Increased obese mRNA expression in omental fat cells from massively obese humans. Nat. Med. 1995, 1, 953–956. [Google Scholar] [CrossRef]
- Carlsson, B.; Lindell, K.; Gabrielsson, B.; Karlsson, C.; Bjarnason, R.; Westphal, O.; Karlsson, U.; Sjöström, L.; Carlsson, L.M.S. Obese (ob) gene defects are rare in human obesity. Obes. Res. 1997, 5, 30–35. [Google Scholar] [CrossRef]
- Farooqi, I.S.; Jebb, S.A.; Langmack, G.; Lawrence, E.; Cheetham, C.H.; Prentice, A.M.; Hughes, I.A.; McCamish, M.A.; O’Rahilly, S. Effects of recombinant leptin therapy in a child with congenital leptin deficiency. N. Engl. J. Med. 1999, 341, 879–884. [Google Scholar] [CrossRef]
- Strobel, A.; Issad, T.; Camoin, L.; Ozata, M.; Strosberg, A.D. A leptin missense mutation associated with hypogonadism and morbid obesity. Nat. Genet. 1998, 18, 214–215. [Google Scholar] [CrossRef] [PubMed]
- Oral, E.A.; Simha, V.; Ruiz, E.; Andewelt, A.; Premkumar, A.; Snell, P.; Wagner, A.J.; DePaoli, A.M.; Reitman, M.L.; Taylor, S.I.; et al. Leptin-replacement therapy for lipodystrophy. N. Engl. J. Med. 2002, 346, 570–578. [Google Scholar] [CrossRef] [PubMed]
- Meehan, C.A.; Cochran, E.; Kassai, A.; Brown, R.J.; Gorden, P. Metreleptin for injection to treat the complications of leptin deficiency in patients with congenital or acquired generalized lipodystrophy. Expert Rev. Clin. Pharmacol. 2016, 9, 59–68. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chou, S.H.; Chamberland, J.P.; Liu, X.; Matarese, G.; Gao, C.; Stefanakis, R.; Brinkoetter, M.T.; Gong, H.; Arampatzi, K.; Mantzoros, C.S. Leptin is an effective treatment for hypothalamic amenorrhea. Proc. Natl. Acad. Sci. USA 2011, 108, 6585–6590. [Google Scholar] [CrossRef] [Green Version]
- Medzhitov, R. Origin and physiological roles of inflammation. Nature 2008, 454, 428–435. [Google Scholar] [CrossRef]
- Gluckman, P.; Beedle, A.; Buklijas, T.; Low, F.; Hanson, M. Principles of Evolutionary Medicine; Oxford University Press: Oxford, UK, 2016. [Google Scholar]
- Stearns, S.; Koella, J. Evolution in Health and Disease; Oxford University Press: Oxford, UK, 2008. [Google Scholar]
- Fullerton, J.N.; Gilroy, D.W. Resolution of inflammation: A new therapeutic frontier. Nat. Rev. Drug Discov. 2016, 15, 551–567. [Google Scholar] [CrossRef]
- Selye, H. Stress and distress. Compr. Ther. 1975, 1, 9–13. [Google Scholar] [CrossRef]
- Dietrich, M.O.; Horvath, T.L. Hypothalamic control of energy balance: Insights into the role of synaptic plasticity. Trends Neurosci. 2013, 36, 65–73. [Google Scholar] [CrossRef]
- Pan, W.W.; Myers, M.G. Leptin and the maintenance of elevated body weight. Nat. Rev. Neurosci. 2018, 19, 95–105. [Google Scholar] [CrossRef]
- de Git, K.C.G.; Adan, R.A.H. Leptin resistance in diet-induced obesity: The role of hypothalamic inflammation. Obes. Rev. 2015, 16, 207–224. [Google Scholar] [CrossRef]
- Martin, S.S.; Qasim, A.; Reilly, M.P. Leptin Resistance. A Possible Interface of Inflammation and Metabolism in Obesity-Related Cardiovascular Disease. J. Am. Coll. Cardiol. 2008, 52, 1201–1210. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kleinridders, A.; Schenten, D.; Könner, A.C.; Belgardt, B.F.; Mauer, J.; Okamura, T.; Wunderlich, F.T.; Medzhitov, R.; Brüning, J.C. MyD88 Signaling in the CNS Is Required for Development of Fatty Acid-Induced Leptin Resistance and Diet-Induced Obesity. Cell Metab. 2009, 10, 249–259. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mantzoros, C.S. The role of leptin in human obesity and disease: A review of current evidence. Ann. Intern. Med. 1999, 130, 671–680. [Google Scholar] [CrossRef] [PubMed]
- Lord, G. Role of leptin in immunology. In Proceedings of the Nutrition Reviews; International Life Sciences Institute: Washington, DC, USA, 2002; Volume 60. [Google Scholar]
- Hasenkrug, K.J. The Leptin Connection: Regulatory T Cells and Autoimmunity. Immunity 2007, 26, 143–145. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peelman, F.; Iserentant, H.; Eyckerman, S.; Zabeau, L.; Tavernier, J. Leptin, Immune Responses and Autoimmune Disease. Perspectives on the Use of Leptin Antagonists. Curr. Pharm. Des. 2005, 11, 539–548. [Google Scholar] [CrossRef] [PubMed]
- Zlotnikov-Klionsky, Y.; Nathansohn-Levi, B.; Shezen, E.; Rosen, C.; Kagan, S.; Bar-On, L.; Jung, S.; Shifrut, E.; Reich-Zeliger, S.; Friedman, N.; et al. Perforin-Positive Dendritic Cells Exhibit an Immuno-regulatory Role in Metabolic Syndrome and Autoimmunity. Immunity 2015, 43, 776–787. [Google Scholar] [CrossRef] [Green Version]
- Pasarica, M.; Dhurandhar, N.V. Infectobesity: Obesity of Infectious Origin. Adv. Food Nutr. Res. 2007, 52, 61–102. [Google Scholar]
- Tian, Y.; Jennings, J.; Gong, Y.; Sang, Y. Viral infections and interferons in the development of obesity. Biomolecules 2019, 9, 726. [Google Scholar] [CrossRef] [Green Version]
- Voss, J.D.; Dhurandhar, N.V. Viral Infections and Obesity. Curr. Obes. Rep. 2017, 6, 28–37. [Google Scholar] [CrossRef]
- Wu, D.; Sanin, D.E.; Everts, B.; Chen, Q.; Qiu, J.; Buck, M.D.; Patterson, A.; Smith, A.M.; Chang, C.H.; Liu, Z.; et al. Type 1 Interferons Induce Changes in Core Metabolism that Are Critical for Immune Function. Immunity 2016, 44, 1325–1336. [Google Scholar] [CrossRef] [Green Version]
- Iacobellis, G.; Malavazos, A.E.; Ferreira, T. COVID-19 rise in Younger adults with Obesity: Visceral Adiposity can predict the Risk. Obesity 2020, oby.22951. [Google Scholar] [CrossRef] [PubMed]
- Rebello, C.J.; Kirwan, J.P.; Greenway, F.L. Obesity, the most common comorbidity in SARS-CoV-2: Is leptin the link? Int. J. Obes. 2020, 1, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Chaplin, D.D. Overview of the immune response. J. Allergy Clin. Immunol. 2010, 125, S3–S23. [Google Scholar] [CrossRef] [PubMed]
- Ren, K.; Dubner, R. Interactions between the immune and nervous systems in pain. Nat. Med. 2010, 16, 1267–1276. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.; Daines, M.O.; Khurana Hershey, G.K. Turning off signal transducer and activator of transcription (STAT): The negative regulation of STAT signaling. J. Allergy Clin. Immunol. 2004, 114, 476–489. [Google Scholar] [CrossRef]
- Duncan, S.A.; Baganizi, D.R.; Sahu, R.; Singh, S.R.; Dennis, V.A. SOCS proteins as regulators of inflammatory responses induced by bacterial infections: A review. Front. Microbiol. 2017, 8, 2431. [Google Scholar] [CrossRef] [Green Version]
- Cani, P.D.; Amar, J.; Iglesias, M.A.; Poggi, M.; Knauf, C.; Bastelica, D.; Neyrinck, A.M.; Fava, F.; Tuohy, K.M.; Chabo, C.; et al. Metabolic endotoxemia initiates obesity and insulin resistance. Diabetes 2007, 56, 1761–1772. [Google Scholar] [CrossRef] [Green Version]
- Hegde, V.; Dhurandhar, N.V. Microbes and obesity-interrelationship between infection, adipose tissue and the immune system. Clin. Microbiol. Infect. 2013, 19, 314–320. [Google Scholar] [CrossRef] [Green Version]
- Cox, L.M.; Blaser, M.J. Pathways in microbe-induced obesity. Cell Metab. 2013, 17, 883–894. [Google Scholar] [CrossRef] [Green Version]
- Dhurandhar, N.V. A framework for identification of infections that contribute to human obesity. Lancet Infect. Dis. 2011, 11, 963–969. [Google Scholar] [CrossRef]
- Jia, W.; Li, H.; Zhao, L.; Nicholson, J.K. Gut microbiota: A potential new territory for drug targeting. Nat. Rev. Drug Discov. 2008, 7, 123–129. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bäckhed, F.; Ding, H.; Wang, T.; Hooper, L.V.; Koh, G.Y.; Nagy, A.; Semenkovich, C.F.; Gordon, J.I. The gut microbiota as an environmental factor that regulates fat storage. Proc. Natl. Acad. Sci. USA 2004, 101, 15718–15723. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Turnbaugh, P.J.; Bäckhed, F.; Fulton, L.; Gordon, J.I. Diet-Induced Obesity Is Linked to Marked but Reversible Alterations in the Mouse Distal Gut Microbiome. Cell Host Microbe 2008, 3, 213–223. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cani, P.D.; Delzenne, N.M. The role of the gut microbiota in energy metabolism and metabolic disease. Curr. Pharm. Des. 2009, 15, 1546–1558. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cani, P.D.; Dewever, C.; Delzenne, N.M. Inulin-type fructans modulate gastrointestinal peptides involved in appetite regulation (glucagon-like peptide-1 and ghrelin) in rats. Br. J. Nutr. 2004, 92, 521–526. [Google Scholar] [CrossRef] [Green Version]
- Li, L.; Messina, J.L. Acute insulin resistance following injury. Trends Endocrinol. Metab. 2009, 20, 429–435. [Google Scholar] [CrossRef] [Green Version]
- Everard, A.; Lazarevic, V.; Derrien, M.; Girard, M.; Muccioli, G.G.; Muccioli, G.M.; Neyrinck, A.M.; Possemiers, S.; Van Holle, A.; François, P.; et al. Responses of gut microbiota and glucose and lipid metabolism to prebiotics in genetic obese and diet-induced leptin-resistant mice. Diabetes 2011, 60, 2775–2786. [Google Scholar] [CrossRef] [Green Version]
- Cani, P.D.; Bibiloni, R.; Knauf, C.; Waget, A.; Neyrinck, A.M.; Delzenne, N.M.; Burcelin, R. Changes in gut microbiota control metabolic endotoxemia-induced inflammation in high-fat diet-induced obesity and diabetes in mice. Diabetes 2008, 57, 1470–1481. [Google Scholar] [CrossRef] [Green Version]
- Cani, P.D.; Neyrinck, A.M.; Fava, F.; Knauf, C.; Burcelin, R.G.; Tuohy, K.M.; Gibson, G.R.; Delzenne, N.M. Selective increases of bifidobacteria in gut microflora improve high-fat-diet-induced diabetes in mice through a mechanism associated with endotoxaemia. Diabetologia 2007, 50, 2374–2383. [Google Scholar] [CrossRef] [Green Version]
- Dewulf, E.M.; Cani, P.D.; Neyrinck, A.M.; Possemiers, S.; Van Holle, A.; Muccioli, G.G.; Deldicque, L.; Bindels, L.B.; Pachikian, B.D.; Sohet, F.M.; et al. Inulin-type fructans with prebiotic properties counteract GPR43 overexpression and PPARγ-related adipogenesis in the white adipose tissue of high-fat diet-fed mice. J. Nutr. Biochem. 2011, 22, 712–722. [Google Scholar] [CrossRef]
- Bäckhed, F.; Normark, S.; Schweda, E.K.H.; Oscarson, S.; Richter-Dahlfors, A. Structural requirements for TLR4-mediated LPS signalling: A biological role for LPS modifications. Microbes Infect. 2003, 5, 1057–1063. [Google Scholar] [CrossRef]
- Vijay-Kumar, M.; Aitken, J.D.; Carvalho, F.A.; Cullender, T.C.; Mwangi, S.; Srinivasan, S.; Sitaraman, S.V.; Knight, R.; Ley, R.E.; Gewirtz, A.T. Metabolic syndrome and altered gut microbiota in mice lacking Toll-like receptor 5. Science 2010, 328, 228–231. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Caricilli, A.M.; Picardi, P.K.; de Abreu, L.L.; Ueno, M.; Prada, P.O.; Ropelle, E.R.; Hirabara, S.M.; Castoldi, Â.; Vieira, P.; Camara, N.O.S.; et al. Gut microbiota is a key modulator of insulin resistance in TLR 2 knockout mice. PLoS Biol. 2011, 9, e1001212. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- García, M.C.; Wernstedt, I.; Berndtsson, A.; Enge, M.; Bell, M.; Hultgren, O.; Horn, M.; Ahrén, B.; Enerback, S.; Ohlsson, C.; et al. Mature-onset obesity in interleukin-1 receptor I knockout mice. Diabetes 2006, 55, 1205–1213. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Glaccum, M.B.; Stocking, K.L.; Charrier, K.; Smith, J.L.; Willis, C.R.; Maliszewski, C.; Livingston, D.J.; Peschon, J.J.; Morrissey, P.J. Phenotypic and functional characterization of mice that lack the type I receptor for IL-1. J. Immunol. 1997, 159, 3364–3371. [Google Scholar]
- O’Neill, L.A.J. The interleukin-1 receptor/Toll-like receptor superfamily: 10 years of progress. Immunol. Rev. 2008, 226, 10–18. [Google Scholar] [CrossRef]
- Feng, X.; Guan, D.; Auen, T.; Choi, J.W.; Salazar Hernández, M.A.; Lee, J.; Chun, H.; Faruk, F.; Kaplun, E.; Herbert, Z.; et al. IL1R1 is required for celastrol’s leptin-sensitization and antiobesity effects. Nat. Med. 2019, 25, 575–582. [Google Scholar] [CrossRef]
- Liu, J.; Lee, J.; Salazar Hernandez, M.A.; Mazitschek, R.; Ozcan, U. Treatment of obesity with celastrol. Cell 2015, 161, 999–1011. [Google Scholar] [CrossRef] [Green Version]
- Saltiel, A.R.; Olefsky, J.M. Inflammatory mechanisms linking obesity and metabolic disease. J. Clin. Investig. 2017, 127, 1–4. [Google Scholar] [CrossRef]
- Lee, J.; Sun, C.; Zhou, Y.; Lee, J.; Gokalp, D.; Herrema, H.; Park, S.W.; Davis, R.J.; Ozcan, U. p38 MAPK-mediated regulation of Xbp1s is crucial for glucose homeostasis. Nat. Med. 2011, 17, 1251–1260. [Google Scholar] [CrossRef] [Green Version]
- Scrivo, R.; Vasile, M.; Bartosiewicz, I.; Valesini, G. Inflammation as “common soil” of the multifactorial diseases. Autoimmun. Rev. 2011, 10, 369–374. [Google Scholar] [CrossRef] [PubMed]


© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pérez-Pérez, A.; Sánchez-Jiménez, F.; Vilariño-García, T.; Sánchez-Margalet, V. Role of Leptin in Inflammation and Vice Versa. Int. J. Mol. Sci. 2020, 21, 5887. https://doi.org/10.3390/ijms21165887
Pérez-Pérez A, Sánchez-Jiménez F, Vilariño-García T, Sánchez-Margalet V. Role of Leptin in Inflammation and Vice Versa. International Journal of Molecular Sciences. 2020; 21(16):5887. https://doi.org/10.3390/ijms21165887
Chicago/Turabian StylePérez-Pérez, Antonio, Flora Sánchez-Jiménez, Teresa Vilariño-García, and Víctor Sánchez-Margalet. 2020. "Role of Leptin in Inflammation and Vice Versa" International Journal of Molecular Sciences 21, no. 16: 5887. https://doi.org/10.3390/ijms21165887
APA StylePérez-Pérez, A., Sánchez-Jiménez, F., Vilariño-García, T., & Sánchez-Margalet, V. (2020). Role of Leptin in Inflammation and Vice Versa. International Journal of Molecular Sciences, 21(16), 5887. https://doi.org/10.3390/ijms21165887