Role of Adipose Tissue-Derived Autotaxin, Lysophosphatidate Signaling, and Inflammation in the Progression and Treatment of Breast Cancer




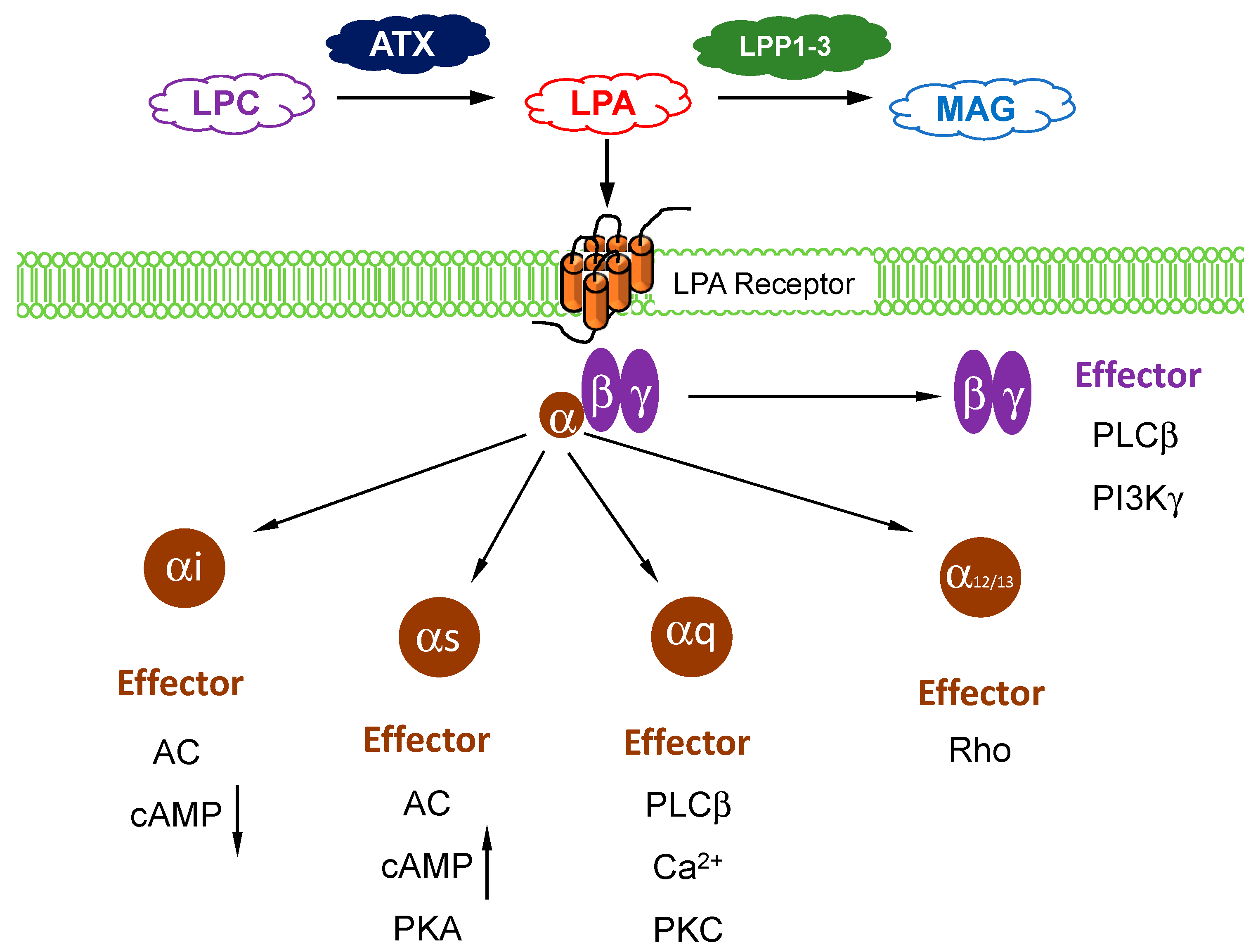{kind=link}
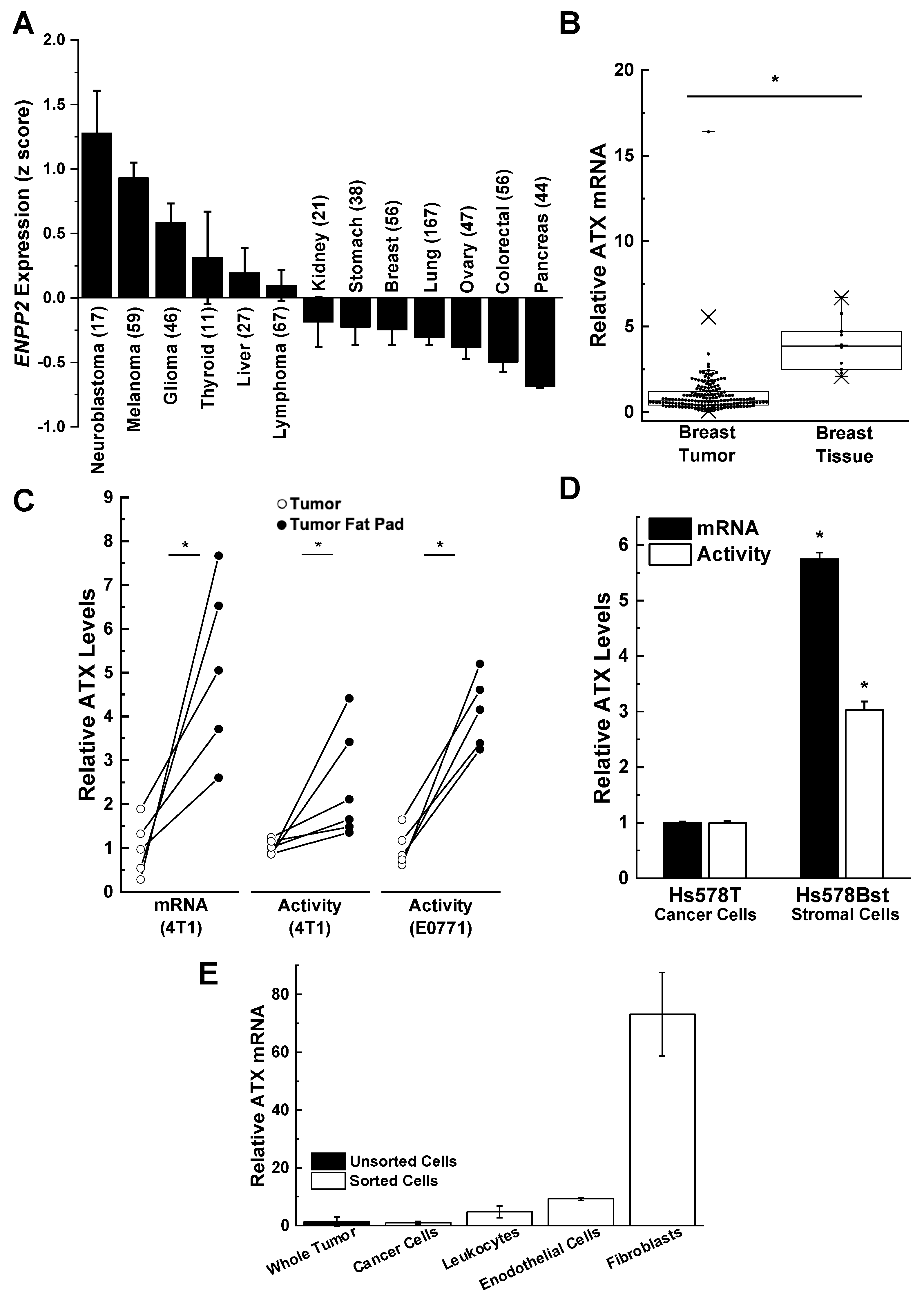{kind=link}
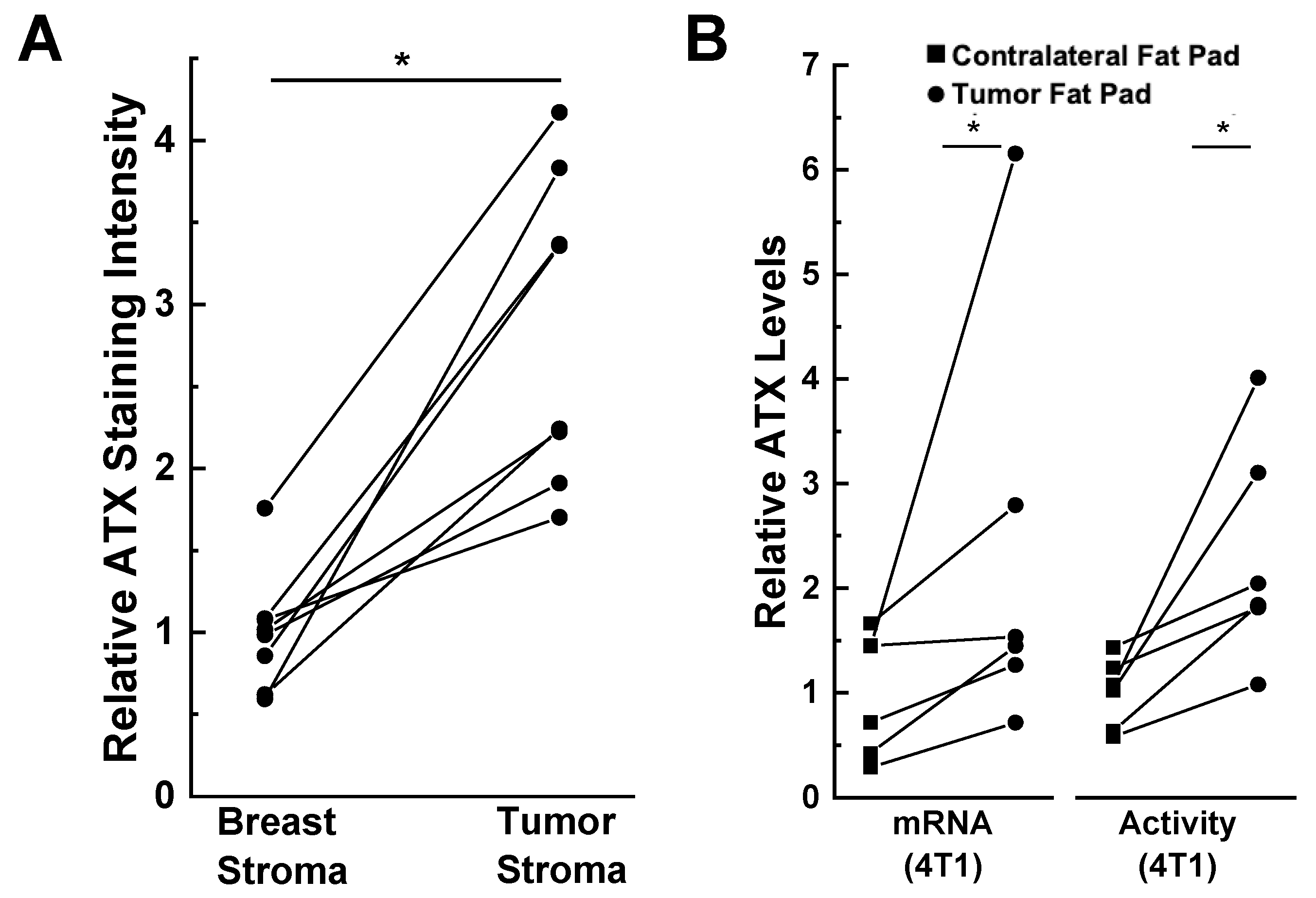{kind=link}
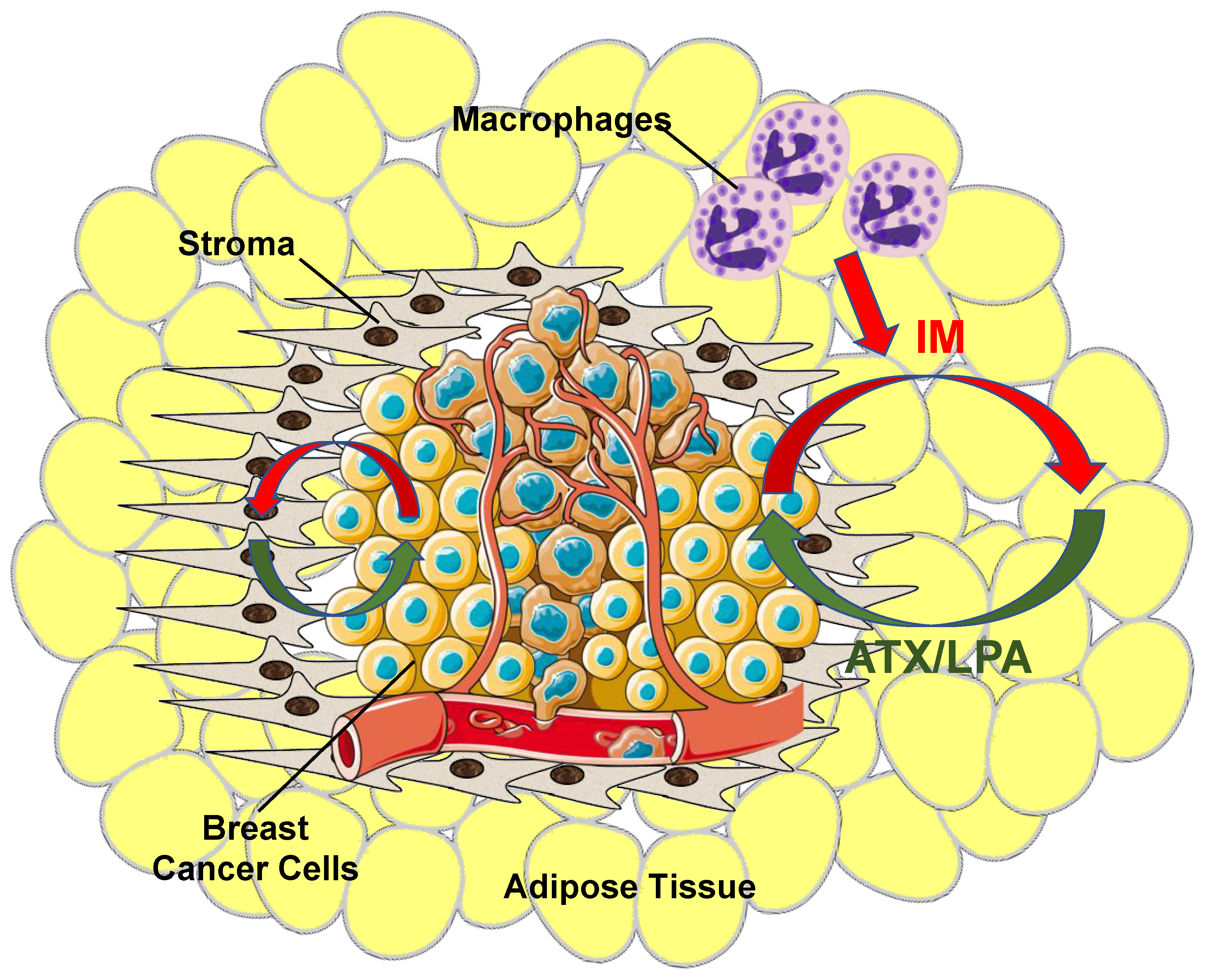{kind=link}
{kind=link}
Abstract
:1. Autotaxin and LPA Metabolism
2. Role of ATX Activity in Wound Healing, Chronic Inflammation, and Cancer
3. Role of Adipose Tissue as a Source of ATX Production in Breast Cancer
4. Influence of Obesity on Breast Cancer
5. Role of Lipid Phosphate Phosphatases in Controlling LPA Signaling and Tumor Progression
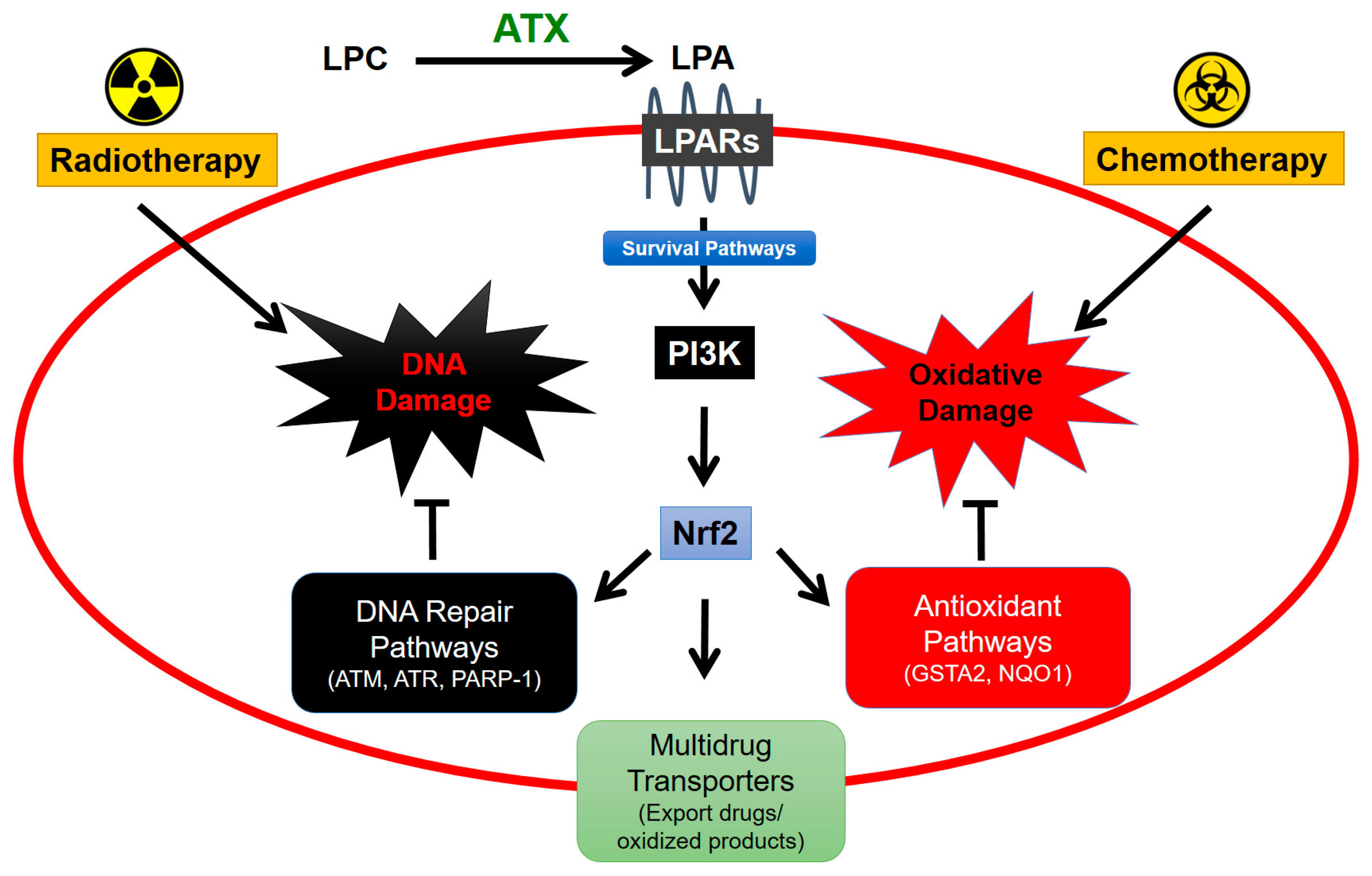
6. Role of Adipose Tissue-Derived ATX in Responses of Breast Tumors to Chemotherapy
7. Role of Adipose Tissue-Derived ATX in Responses to Radiotherapy for Breast Cancer
8. Role of the Anti-Inflammatory Glucocorticoid, Dexamethasone (DEX), in Attenuating ATX and LPA Signaling in Adipose Tissue
9. Effects of DEX on RT-Induced Secretion of ATX and Subsequent LPA Signaling on RT-Induced Fibrosis
10. Future Perspective
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| ATX | autotaxin |
| COX-1,2 | cyclooxygenase-1,2 |
| IL | interleukin |
| IPF | idiopathic pulmonary fibrosis |
| LPA | lysophosphatidate at physiological pH but often referred to as lysophosphatidic acid |
| LPC | lysophosphatidylcholine |
| LPA1–6 | lysophosphatidate 1–6 receptors |
| LPP | lipid phosphate phosphatase |
| NFκB | nuclear factor kappa-light-chain-enhancer of activated B cells |
| Nrf2 | nuclear erythroid 2-like 2 |
| RT | radiotherapy |
| S1P | sphingosine 1-phosphate |
| TNF-α | tumor necrosis factor-α |
| VEGF | vascular endothelial growth factor |
References
- Murata, J.; Lee, H.Y.; Clair, T.; Krutzsch, H.C.; Arestad, A.A.; Sobel, M.E.; Liotta, L.A.; Stracke, M.L. Cdna cloning of the human tumor motility-stimulating protein, autotaxin, reveals a homology with phosphodiesterases. J. Biol. Chem. 1994, 269, 30479–30484. [Google Scholar] [PubMed]
- Stefan, C.; Jansen, S.; Bollen, M. Npp-type ectophosphodiesterases: Unity in diversity. Trends Biochem. Sci. 2005, 30, 542–550. [Google Scholar] [CrossRef] [PubMed]
- Perrakis, A.; Moolenaar, W.H. Autotaxin: Structure-function and signaling. J. Lipid Res. 2014, 55, 1010–1018. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stracke, M.L.; Krutzsch, H.C.; Unsworth, E.J.; Arestad, A.; Cioce, V.; Schiffmann, E.; Liotta, L.A. Identification, purification, and partial sequence analysis of autotaxin, a novel motility-stimulating protein. J. Biol. Chem. 1992, 267, 2524–2529. [Google Scholar] [PubMed]
- Umezu-Goto, M.; Kishi, Y.; Taira, A.; Hama, K.; Dohmae, N.; Takio, K.; Yamori, T.; Mills, G.B.; Inoue, K.; Aoki, J.; et al. Autotaxin has lysophospholipase d activity leading to tumor cell growth and motility by lysophosphatidic acid production. J. Cell Biol. 2002, 158, 227–233. [Google Scholar] [CrossRef]
- Tokumura, A.; Majima, E.; Kariya, Y.; Tominaga, K.; Kogure, K.; Yasuda, K.; Fukuzawa, K. Identification of human plasma lysophospholipase d, a lysophosphatidic acid-producing enzyme, as autotaxin, a multifunctional phosphodiesterase. J. Biol. Chem. 2002, 277, 39436–39442. [Google Scholar] [CrossRef] [Green Version]
- Gaetano, C.G.; Samadi, N.; Tomsig, J.L.; Macdonald, T.L.; Lynch, K.R.; Brindley, D.N. Inhibition of autotaxin production or activity blocks lysophosphatidylcholine-induced migration of human breast cancer and melanoma cells. Mol. Carcinog. 2009, 48, 801–809. [Google Scholar] [CrossRef] [Green Version]
- Benesch, M.G.K.; Ko, Y.M.; McMullen, T.P.W.; Brindley, D.N. Autotaxin in the crosshairs: Taking aim at cancer and other inflammatory conditions. FEBS Lett. 2014, 588, 2712–2727. [Google Scholar] [CrossRef] [Green Version]
- Hemmings, D.G.; Brindley, D.N. Signalling by lysophosphatidate and its health implications. Essays Biochem. 2020, in press. [Google Scholar] [CrossRef]
- Samadi, N.; Bekele, R.; Capatos, D.; Venkatraman, G.; Sariahmetoglu, M.; Brindley, D.N. Regulation of lysophosphatidate signaling by autotaxin and lipid phosphate phosphatases with respect to tumor progression, angiogenesis, metastasis and chemo-resistance. Biochimie 2011, 93, 61–70. [Google Scholar] [CrossRef]
- Brindley, D.N.; Lin, F.T.; Tigyi, G.J. Role of the autotaxin-lysophosphatidate axis in cancer resistance to chemotherapy and radiotherapy. Biochim. Biophys. Acta 2013, 1831, 74–85. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van Meeteren, L.A.; Ruurs, P.; Stortelers, C.; Bouwman, P.; van Rooijen, M.A.; Pradere, J.P.; Pettit, T.R.; Wakelam, M.J.; Saulnier-Blache, J.S.; Mummery, C.L.; et al. Autotaxin, a secreted lysophospholipase d, is essential for blood vessel formation during development. Mol. Cell. Biol. 2006, 26, 5015–5022. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pamuklar, Z.; Federico, L.; Liu, S.; Umezu-Goto, M.; Dong, A.; Panchatcharam, M.; Fulkerson, Z.; Berdyshev, E.; Natarajan, V.; Fang, X.; et al. Autotaxin/lysopholipase d and lysophosphatidic acid regulate murine hemostasis and thrombosis. J. Biol. Chem. 2009, 284, 7385–7394. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Benesch, M.G.K.; Tang, X.; Maeda, T.; Ohhata, A.; Zhao, Y.Y.; Kok, B.P.C.; Dewald, J.; Hitt, M.; Curtis, J.M.; McMullen, T.P.W.; et al. Inhibition of autotaxin delays breast tumor growth and lung metastasis in mice. FASEB J. 2014, 28, 2655–2666. [Google Scholar] [CrossRef] [PubMed]
- Albers, H.M.; Dong, A.; van Meeteren, L.A.; Egan, D.A.; Sunkara, M.; van Tilburg, E.W.; Schuurman, K.; van Tellingen, O.; Morris, A.J.; Smyth, S.S.; et al. Boronic acid-based inhibitor of autotaxin reveals rapid turnover of lpa in the circulation. Proc. Natl. Acad. Sci. USA 2010, 107, 7257–7262. [Google Scholar] [CrossRef] [Green Version]
- Koike, S.; Yutoh, Y.; Keino-Masu, K.; Noji, S.; Masu, M.; Ohuchi, H. Autotaxin is required for the cranial neural tube closure and establishment of the midbrain-hindbrain boundary during mouse development. Dev. Dyn. 2011, 240, 413–421. [Google Scholar] [CrossRef]
- Hemrika, W.; Renirie, R.; Dekker, H.L.; Barnett, P.; Wever, R. From phosphatases to vanadium peroxidases: A similar architecture of the active site. Proc. Natl. Acad. Sci. USA 1997, 94, 2145–2149. [Google Scholar] [CrossRef] [Green Version]
- Neuwald, A.F. An unexpected structural relationship between integral membrane phosphatases and soluble haloperoxidases. Protein Sci. 1997, 6, 1764–1767. [Google Scholar] [CrossRef] [Green Version]
- Brindley, D.N.; English, D.; Pilquil, C.; Buri, K.; Ling, Z.C. Lipid phosphate phosphatases regulate signal transduction through glycerolipids and sphingolipids. Biochim. Biophys. Acta 2002, 1582, 33–44. [Google Scholar] [CrossRef]
- Carman, G.M.; Han, G.S. Phosphatidic acid phosphatase, a key enzyme in the regulation of lipid synthesis. J. Biol. Chem. 2009, 284, 2593–2597. [Google Scholar] [CrossRef] [Green Version]
- Tang, X.; Benesch, M.G.; Brindley, D.N. Lipid phosphate phosphatases and their roles in mammalian physiology and pathology. J. Lipid Res. 2015, 56, 2048–2060. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van Meeteren, L.A.; Ruurs, P.; Christodoulou, E.; Goding, J.W.; Takakusa, H.; Kikuchi, K.; Perrakis, A.; Nagano, T.; Moolenaar, W.H. Inhibition of autotaxin by lysophosphatidic acid and sphingosine 1-phosphate. J. Biol. Chem. 2005, 280, 21155–21161. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Benesch, M.G.K.; Zhao, Y.Y.; Curtis, J.M.; McMullen, T.P.; Brindley, D.N. Regulation of autotaxin expression and secretion by lysophosphatidate and sphingosine 1-phosphate. J. Lipid Res. 2015, 56, 1134–1144. [Google Scholar] [CrossRef] [Green Version]
- Jansen, S.; Andries, M.; Vekemans, K.; Vanbilloen, H.; Verbruggen, A.; Bollen, M. Rapid clearance of the circulating metastatic factor autotaxin by the scavenger receptors of liver sinusoidal endothelial cells. Cancer Lett. 2009, 284, 216–221. [Google Scholar] [CrossRef] [PubMed]
- Tang, X.; Zhao, Y.Y.; Dewald, J.; Curtis, J.M.; Brindley, D.N. Tetracyclines increase lipid phosphate phosphatase expression on plasma membranes and turnover of plasma lysophosphatidate. J. Lipid Res. 2016, 57, 597–606. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Salous, A.K.; Panchatcharam, M.; Sunkara, M.; Mueller, P.; Dong, A.; Wang, Y.; Graf, G.A.; Smyth, S.S.; Morris, A.J. Mechanism of rapid elimination of lysophosphatidic acid and related lipids from the circulation of mice. J. Lipid Res. 2013, 54, 2775–2784. [Google Scholar] [CrossRef] [Green Version]
- Tang, X.; McMullen, T.P.W.; Brindley, D.N. Increasing the low lipid phosphate phosphatase 1 activity in breast cancer cells decreases transcription by ap-1 and expressions of matrix metalloproteinases and cyclin d1/d3. Theranostics 2019, 9, 6129–6142. [Google Scholar] [CrossRef]
- Meng, G.; Tang, X.; Yang, Z.; Zhao, Y.; Curtis, J.M.; McMullen, T.P.W.; Brindley, D.N. Dexamethasone decreases the autotaxin-lysophosphatidate-inflammatory axis in adipose tissue: Implications for the metabolic syndrome and breast cancer. FASEB J. 2019, 33, 1899–1910. [Google Scholar] [CrossRef] [Green Version]
- Benesch, M.G.K.; Tang, X.; Brindley, D.N. Autotaxin and breast cancer: Towards overcoming treatment barriers and sequelae. Cancers 2020, 12, 374. [Google Scholar] [CrossRef] [Green Version]
- Ninou, I.; Magkrioti, C.; Aidinis, V. Autotaxin in pathophysiology and pulmonary fibrosis. Front. Med. 2018, 5, 180. [Google Scholar] [CrossRef]
- Van Corven, E.J.; van Rijswijk, A.; Jalink, K.; van der Bend, R.L.; van Blitterswijk, W.J.; Moolenaar, W.H. Mitogenic action of lysophosphatidic acid and phosphatidic acid on fibroblasts. Dependence on acyl-chain length and inhibition by suramin. Biochem. J. 1992, 281 Pt 1, 163–169. [Google Scholar] [CrossRef] [Green Version]
- Chabaud, S.; Marcoux, T.L.; Deschênes-Rompré, M.P.; Rousseau, A.; Morissette, A.; Bouhout, S.; Bernard, G.; Bolduc, S. Lysophosphatidic acid enhances collagen deposition and matrix thickening in engineered tissue. J. Tissue Eng. Regen. Med. 2015, 9, E65–E75. [Google Scholar] [CrossRef] [PubMed]
- Van Corven, E.J.; Groenink, A.; Jalink, K.; Eichholtz, T.; Moolenaar, W.H. Lysophosphatidate-induced cell proliferation: Identification and dissection of signaling pathways mediated by g proteins. Cell 1989, 59, 45–54. [Google Scholar] [CrossRef]
- Panetti, T.S.; Chen, H.; Misenheimer, T.M.; Getzler, S.B.; Mosher, D.F. Endothelial cell mitogenesis induced by lpa: Inhibition by thrombospondin-1 and thrombospondin-2. J. Lab. Clin. Med. 1997, 129, 208–216. [Google Scholar] [CrossRef]
- Knowlden, S.; Georas, S.N. The autotaxin-lpa axis emerges as a novel regulator of lymphocyte homing and inflammation. J. Immunol. 2014, 192, 851–857. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ray, R.; Rai, V. Lysophosphatidic acid converts monocytes into macrophages in both mice and humans. Blood 2017, 129, 1177–1183. [Google Scholar] [CrossRef] [Green Version]
- Mazereeuw-Hautier, J.; Gres, S.; Fanguin, M.; Cariven, C.; Fauvel, J.; Perret, B.; Chap, H.; Salles, J.P.; Saulnier-Blache, J.S. Production of lysophosphatidic acid in blister fluid: Involvement of a lysophospholipase d activity. J. Investig. Dermatol. 2005, 125, 421–427. [Google Scholar] [CrossRef] [Green Version]
- Magkrioti, C.; Galaris, A.; Kanellopoulou, P.; Stylianaki, E.A.; Kaffe, E.; Aidinis, V. Autotaxin and chronic inflammatory diseases. J. Autoimmun. 2019, 104, 102327. [Google Scholar] [CrossRef]
- Dvorak, H.F. Tumors: Wounds that do not heal. Similarities between tumor stroma generation and wound healing. N. Engl. J. Med. 1986, 315, 1650–1659. [Google Scholar]
- Schafer, M.; Werner, S. Cancer as an overhealing wound: An old hypothesis revisited. Nat. Rev. Mol. Cell Biol. 2008, 9, 628–638. [Google Scholar] [CrossRef]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tigyi, G.J.; Yue, J.; Norman, D.D.; Szabo, E.; Balogh, A.; Balazs, L.; Zhao, G.; Lee, S.C. Regulation of tumor cell—Microenvironment interaction by the autotaxin-lysophosphatidic acid receptor axis. Adv. Biol. Regul. 2019, 71, 183–193. [Google Scholar] [CrossRef] [PubMed]
- Colotta, F.; Allavena, P.; Sica, A.; Garlanda, C.; Mantovani, A. Cancer-related inflammation, the seventh hallmark of cancer: Links to genetic instability. Carcinogenesis 2009, 30, 1073–1081. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Benesch, M.G.K.; MacIntyre, I.T.K.; McMullen, T.P.W.; Brindley, D.N. Coming of age for autotaxin and lysophosphatidate signaling: Clinical applications for preventing, detecting and targeting tumor-promoting inflammation. Cancers 2018, 10, 73. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Benesch, M.G.K.; Yang, Z.; Tang, X.; Meng, G.; Brindley, D.N. Lysophosphatidate signaling: The tumor microenvironment’s new nemesis. Trends Cancer 2017, 3, 748–752. [Google Scholar] [CrossRef]
- Lagadari, M.; Truta-Feles, K.; Lehmann, K.; Berod, L.; Ziemer, M.; Idzko, M.; Barz, D.; Kamradt, T.; Maghazachi, A.A.; Norgauer, J. Lysophosphatidic acid inhibits the cytotoxic activity of nk cells: Involvement of gs protein-mediated signaling. Int. Immunol. 2009, 21, 667–677. [Google Scholar] [CrossRef] [Green Version]
- Mathew, D.; Kremer, K.N.; Strauch, P.; Tigyi, G.; Pelanda, R.; Torres, R.M. Lpa5 is an inhibitory receptor that suppresses cd8 t-cell cytotoxic function via disruption of early tcr signaling. Front. Immunol. 2019, 10, 1159. [Google Scholar] [CrossRef] [Green Version]
- Smyth, S.S.; Mueller, P.; Yang, F.; Brandon, J.A.; Morris, A.J. Arguing the case for the autotaxin-lysophosphatidic acid-lipid phosphate phosphatase 3-signaling nexus in the development and complications of atherosclerosis. Arterioscler. Thromb. Vasc. Biol. 2014, 34, 479–486. [Google Scholar] [CrossRef] [Green Version]
- Yang, B.; Zhou, Z.; Li, X.; Niu, J. The effect of lysophosphatidic acid on toll-like receptor 4 expression and the nuclear factor-κb signaling pathway in thp-1 cells. Mol. Cell. Biochem. 2016, 422, 41–49. [Google Scholar] [CrossRef]
- So, J.; Wang, F.Q.; Navari, J.; Schreher, J.; Fishman, D.A. Lpa-induced epithelial ovarian cancer (eoc) in vitro invasion and migration are mediated by vegf receptor-2 (VEGF-R2). Gynecol. Oncol. 2005, 97, 870–878. [Google Scholar] [CrossRef]
- Tang, X.; Benesch, M.G.K.; Brindley, D.N. Role of the autotaxin-lysophosphatidate axis in the development of resistance to cancer therapy. Biochim. Biophys. Acta Mol. Cell Biol. Lipids 2020, 1865, 158716. [Google Scholar] [CrossRef] [PubMed]
- St-Coeur, P.D.; Ferguson, D.; Morin, P., Jr.; Touaibia, M. Pf-8380 and closely related analogs: Synthesis and structure-activity relationship towards autotaxin inhibition and glioma cell viability. Arch. Pharm. 2013, 346, 91–97. [Google Scholar] [CrossRef] [PubMed]
- Euer, N.; Schwirzke, M.; Evtimova, V.; Burtscher, H.; Jarsch, M.; Tarin, D.; Weidle, U.H. Identification of genes associated with metastasis of mammary carcinoma in metastatic versus non-metastatic cell lines. Anticancer Res. 2002, 22, 733–740. [Google Scholar] [PubMed]
- Castellana, B.; Escuin, D.; Peiro, G.; Garcia-Valdecasas, B.; Vazquez, T.; Pons, C.; Perez-Olabarria, M.; Barnadas, A.; Lerma, E. Aspn and gjb2 are implicated in the mechanisms of invasion of ductal breast carcinomas. J. Cancer 2012, 3, 175–183. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vital, A.L.; Tabernero, M.D.; Castrillo, A.; Rebelo, O.; Tao, H.; Gomes, F.; Nieto, A.B.; Resende Oliveira, C.; Lopes, M.C.; Orfao, A. Gene expression profiles of human glioblastomas are associated with both tumor cytogenetics and histopathology. Neuro Oncol. 2010, 12, 991–1003. [Google Scholar] [CrossRef] [PubMed]
- Funes, S.C.; Rios, M.; Escobar-Vera, J.; Kalergis, A.M. Implications of macrophage polarization in autoimmunity. Immunology 2018, 154, 186–195. [Google Scholar] [CrossRef] [Green Version]
- Italiani, P.; Boraschi, D. From monocytes to M1/M2 macrophages: Phenotypical vs. Functional differentiation. Front. Immunol. 2014, 5, 514. [Google Scholar] [CrossRef] [Green Version]
- Ohashi, K.; Parker, J.L.; Ouchi, N.; Higuchi, A.; Vita, J.A.; Gokce, N.; Pedersen, A.A.; Kalthoff, C.; Tullin, S.; Sams, A.; et al. Adiponectin promotes macrophage polarization toward an anti-inflammatory phenotype. J. Biol. Chem. 2010, 285, 6153–6160. [Google Scholar] [CrossRef] [Green Version]
- Reinartz, S.; Lieber, S.; Pesek, J.; Brandt, D.T.; Asafova, A.; Finkernagel, F.; Watzer, B.; Nockher, W.A.; Nist, A.; Stiewe, T.; et al. Cell type-selective pathways and clinical associations of lysophosphatidic acid biosynthesis and signaling in the ovarian cancer microenvironment. Mol. Oncol. 2019, 13, 185–201. [Google Scholar] [CrossRef]
- Lee, S.C.; Dacheux, M.A.; Norman, D.D.; Balázs, L.; Torres, R.M.; Augelli-Szafran, C.E.; Tigyi, G.J. Regulation of tumor immunity by lysophosphatidic acid. Cancers 2020, 12, 1202. [Google Scholar] [CrossRef]
- Peng, X.H.; Karna, P.; Cao, Z.; Jiang, B.H.; Zhou, M.; Yang, L. Cross-talk between epidermal growth factor receptor and hypoxia-inducible factor-1alpha signal pathways increases resistance to apoptosis by up-regulating survivin gene expression. J. Biol. Chem. 2006, 281, 25903–25914. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brindley, D.N.; Benesch, M.G.K.; Murph, M.M. Autotaxin—An enzymatic augmenter of malignant progression linked to inflammation. In Melanoma—Current Clinical Management and Future Therapeutics; InTech Open: London, UK, 2015; pp. 297–324. [Google Scholar] [CrossRef] [Green Version]
- Cerami, E.; Gao, J.; Dogrusoz, U.; Gross, B.E.; Sumer, S.O.; Aksoy, B.A.; Jacobsen, A.; Byrne, C.J.; Heuer, M.L.; Larsson, E.; et al. The cbio cancer genomics portal: An open platform for exploring multidimensional cancer genomics data. Cancer Discov. 2012, 2, 401–404. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gao, J.; Aksoy, B.A.; Dogrusoz, U.; Dresdner, G.; Gross, B.; Sumer, S.O.; Sun, Y.; Jacobsen, A.; Sinha, R.; Larsson, E.; et al. Integrative analysis of complex cancer genomics and clinical profiles using the cbioportal. Sci. Signal. 2013, 6, pl1. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Samadi, N.; Gaetano, C.; Goping, I.S.; Brindley, D.N. Autotaxin protects mcf-7 breast cancer and mda-mb-435 melanoma cells against taxol-induced apoptosis. Oncogene 2009, 28, 1028–1039. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yung, Y.C.; Stoddard, N.C.; Chun, J. Lpa receptor signaling: Pharmacology, physiology, and pathophysiology. J. Lipid Res. 2014, 55, 1192–1214. [Google Scholar] [CrossRef] [Green Version]
- Benesch, M.G.K.; Tang, X.; Dewald, J.; Dong, W.F.; Mackey, J.R.; Hemmings, D.G.; McMullen, T.P.; Brindley, D.N. Tumor-induced inflammation in mammary adipose tissue stimulates a vicious cycle of autotaxin expression and breast cancer progression. FASEB J. 2015, 29, 3990–4000. [Google Scholar] [CrossRef] [Green Version]
- Leblanc, R.; Sahay, D.; Houssin, A.; Machuca-Gayet, I.; Peyruchaud, O. Autotaxin-beta interaction with the cell surface via syndecan-4 impacts on cancer cell proliferation and metastasis. Oncotarget 2018, 9, 33170–33185. [Google Scholar] [CrossRef]
- Leblanc, R.; Houssin, A.; Peyruchaud, O. Platelets, autotaxin and lysophosphatidic acid signalling: Win-win factors for cancer metastasis. Br. J. Pharmacol. 2018, 175, 3100–3110. [Google Scholar] [CrossRef]
- Fulkerson, Z.; Wu, T.; Sunkara, M.; Kooi, C.V.; Morris, A.J.; Smyth, S.S. Binding of autotaxin to integrins localizes lysophosphatidic acid production to platelets and mammalian cells. J. Biol. Chem. 2011, 286, 34654–34663. [Google Scholar] [CrossRef] [Green Version]
- Hausmann, J.; Kamtekar, S.; Christodoulou, E.; Day, J.E.; Wu, T.; Fulkerson, Z.; Albers, H.M.; van Meeteren, L.A.; Houben, A.J.; van Zeijl, L.; et al. Structural basis of substrate discrimination and integrin binding by autotaxin. Nat. Struct. Mol. Biol. 2011, 18, 198–204. [Google Scholar] [CrossRef] [Green Version]
- Popnikolov, N.K.; Dalwadi, B.H.; Thomas, J.D.; Johannes, G.J.; Imagawa, W.T. Association of autotaxin and lysophosphatidic acid receptor 3 with aggressiveness of human breast carcinoma. Tumour Biol. 2012, 33, 2237–2243. [Google Scholar] [CrossRef] [PubMed]
- Volden, P.A.; Skor, M.N.; Johnson, M.B.; Singh, P.; Patel, F.N.; McClintock, M.K.; Brady, M.J.; Conzen, S.D. Mammary adipose tissue-derived lysophospholipids promote estrogen receptor-negative mammary epithelial cell proliferation. Cancer Prev. Res. 2016, 9, 367–378. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schmid, R.; Wolf, K.; Robering, J.W.; Strauß, S.; Strissel, P.L.; Strick, R.; Rübner, M.; Fasching, P.A.; Horch, R.E.; Kremer, A.E.; et al. Adscs and adipocytes are the main producers in the autotaxin–lysophosphatidic acid axis of breast cancer and healthy mammary tissue in vitro. BMC Cancer 2018, 18, 1273. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fantuzzi, G. Adipose tissue, adipokines, and inflammation. J. Allergy Clin. Immunol. 2005, 115, 911–919, quiz 920. [Google Scholar] [CrossRef] [PubMed]
- Popko, K.; Gorska, E.; Stelmaszczyk-Emmel, A.; Plywaczewski, R.; Stoklosa, A.; Gorecka, D.; Pyrzak, B.; Demkow, U. Proinflammatory cytokines il-6 and tnf-α and the development of inflammation in obese subjects. Eur. J. Med. Res. 2010, 15 (Suppl. 2), 120–122. [Google Scholar] [CrossRef]
- Bastard, J.P.; Jardel, C.; Bruckert, E.; Blondy, P.; Capeau, J.; Laville, M.; Vidal, H.; Hainque, B. Elevated levels of interleukin 6 are reduced in serum and subcutaneous adipose tissue of obese women after weight loss. J. Clin. Endocrinol. Metab. 2000, 85, 3338–3342. [Google Scholar]
- Kanda, H.; Tateya, S.; Tamori, Y.; Kotani, K.; Hiasa, K.; Kitazawa, R.; Kitazawa, S.; Miyachi, H.; Maeda, S.; Egashira, K.; et al. Mcp-1 contributes to macrophage infiltration into adipose tissue, insulin resistance, and hepatic steatosis in obesity. J. Clin. Investig. 2006, 116, 1494–1505. [Google Scholar] [CrossRef]
- Thomas, D.; Apovian, C. Macrophage functions in lean and obese adipose tissue. Metabolism 2017, 72, 120–143. [Google Scholar] [CrossRef]
- Sun, H.; Zou, J.; Chen, L.; Zu, X.; Wen, G.; Zhong, J. Triple-negative breast cancer and its association with obesity. Mol. Clin. Oncol. 2017, 7, 935–942. [Google Scholar] [CrossRef]
- Sun, L.; Zhu, Y.; Qian, Q.; Tang, L. Body mass index and prognosis of breast cancer: An analysis by menstruation status when breast cancer diagnosis. Medicine 2018, 97, e11220. [Google Scholar] [CrossRef]
- Blair, C.K.; Wiggins, C.L.; Nibbe, A.M.; Storlie, C.B.; Prossnitz, E.R.; Royce, M.; Lomo, L.C.; Hill, D.A. Obesity and survival among a cohort of breast cancer patients is partially mediated by tumor characteristics. NPJ Breast Cancer 2019, 5, 33. [Google Scholar] [CrossRef]
- Arendt, L.M.; McCready, J.; Keller, P.J.; Baker, D.D.; Naber, S.P.; Seewaldt, V.; Kuperwasser, C. Obesity promotes breast cancer by ccl2-mediated macrophage recruitment and angiogenesis. Cancer Res. 2013, 73, 6080–6093. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cowen, S.; McLaughlin, S.L.; Hobbs, G.; Coad, J.; Martin, K.H.; Olfert, I.M.; Vona-Davis, L. High-fat, high-calorie diet enhances mammary carcinogenesis and local inflammation in mmtv-pymt mouse model of breast cancer. Cancers 2015, 7, 1125–1142. [Google Scholar] [CrossRef] [PubMed]
- Dirat, B.; Bochet, L.; Dabek, M.; Daviaud, D.; Dauvillier, S.; Majed, B.; Wang, Y.Y.; Meulle, A.; Salles, B.; Le Gonidec, S.; et al. Cancer-associated adipocytes exhibit an activated phenotype and contribute to breast cancer invasion. Cancer Res. 2011, 71, 2455–2465. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tahergorabi, Z.; Khazaei, M. The relationship between inflammatory markers, angiogenesis, and obesity. ARYA Atheroscler. 2013, 9, 247–253. [Google Scholar] [PubMed]
- Subbaramaiah, K.; Morris, P.G.; Zhou, X.K.; Morrow, M.; Du, B.; Giri, D.; Kopelovich, L.; Hudis, C.A.; Dannenberg, A.J. Increased levels of cox-2 and prostaglandin e2 contribute to elevated aromatase expression in inflamed breast tissue of obese women. Cancer Discov. 2012, 2, 356–365. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zeyda, M.; Huber, J.; Prager, G.; Stulnig, T.M. Inflammation correlates with markers of t-cell subsets including regulatory t cells in adipose tissue from obese patients. Obesity 2011, 19, 743–748. [Google Scholar] [CrossRef]
- Dusaulcy, R.; Rancoule, C.; Gres, S.; Wanecq, E.; Colom, A.; Guigne, C.; van Meeteren, L.A.; Moolenaar, W.H.; Valet, P.; Saulnier-Blache, J.S. Adipose-specific disruption of autotaxin enhances nutritional fattening and reduces plasma lysophosphatidic acid. J. Lipid Res. 2011, 52, 1247–1255. [Google Scholar] [CrossRef] [Green Version]
- Rancoule, C.; Dusaulcy, R.; Treguer, K.; Gres, S.; Guigne, C.; Quilliot, D.; Valet, P.; Saulnier-Blache, J.S. Depot-specific regulation of autotaxin with obesity in human adipose tissue. J. Physiol. Biochem. 2012, 68, 635–644. [Google Scholar] [CrossRef]
- D’Souza, K.; Paramel, G.V.; Kienesberger, P.C. Lysophosphatidic acid signaling in obesity and insulin resistance. Nutrients 2018, 10, 399. [Google Scholar] [CrossRef] [Green Version]
- Lopez-Jaramillo, P.; Gomez-Arbelaez, D.; Lopez-Lopez, J.; Lopez-Lopez, C.; Martinez-Ortega, J.; Gomez-Rodriguez, A.; Triana-Cubillos, S. The role of leptin/adiponectin ratio in metabolic syndrome and diabetes. Horm. Mol. Biol. Clin. Investig. 2014, 18, 37–45. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sundaram, S.; Johnson, A.R.; Makowski, L. Obesity, metabolism and the microenvironment: Links to cancer. J. Carcinog. 2013, 12, 19. [Google Scholar] [PubMed]
- De Pergola, G.; Silvestris, F. Obesity as a major risk factor for cancer. J. Obes. 2013, 2013, 291546. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, S.; Umezu-Goto, M.; Murph, M.; Lu, Y.; Liu, W.; Zhang, F.; Yu, S.; Stephens, L.C.; Cui, X.; Murrow, G.; et al. Expression of autotaxin and lysophosphatidic acid receptors increases mammary tumorigenesis, invasion, and metastases. Cancer Cell 2009, 15, 539–550. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cha, Y.J.; Koo, J.S. Expression of autotaxin(-)lysophosphatidate signaling-related proteins in breast cancer with adipose stroma. Int. J. Mol. Sci. 2019, 20, 2102. [Google Scholar] [CrossRef] [Green Version]
- Bhattacharjee, A.; Richards, W.G.; Staunton, J.; Li, C.; Monti, S.; Vasa, P.; Ladd, C.; Beheshti, J.; Bueno, R.; Gillette, M.; et al. Classification of human lung carcinomas by mrna expression profiling reveals distinct adenocarcinoma subclasses. Proc. Natl. Acad. Sci. USA 2001, 98, 13790–13795. [Google Scholar] [CrossRef] [Green Version]
- Curtis, C.; Shah, S.P.; Chin, S.F.; Turashvili, G.; Rueda, O.M.; Dunning, M.J.; Speed, D.; Lynch, A.G.; Samarajiwa, S.; Yuan, Y.; et al. The genomic and transcriptomic architecture of 2,000 breast tumours reveals novel subgroups. Nature 2012, 486, 346–352. [Google Scholar] [CrossRef]
- Yoshihara, K.; Tajima, A.; Komata, D.; Yamamoto, T.; Kodama, S.; Fujiwara, H.; Suzuki, M.; Onishi, Y.; Hatae, M.; Sueyoshi, K.; et al. Gene expression profiling of advanced-stage serous ovarian cancers distinguishes novel subclasses and implicates zeb2 in tumor progression and prognosis. Cancer Sci. 2009, 100, 1421–1428. [Google Scholar] [CrossRef]
- Sun, B.; Nishihira, J.; Suzuki, M.; Fukushima, N.; Ishibashi, T.; Kondo, M.; Sato, Y.; Todo, S. Induction of macrophage migration inhibitory factor by lysophosphatidic acid: Relevance to tumor growth and angiogenesis. Int. J. Mol. Med. 2003, 12, 633–641. [Google Scholar] [CrossRef]
- Fang, X.; Schummer, M.; Mao, M.; Yu, S.; Tabassam, F.H.; Swaby, R.; Hasegawa, Y.; Tanyi, J.L.; LaPushin, R.; Eder, A.; et al. Lysophosphatidic acid is a bioactive mediator in ovarian cancer. Biochim. Biophys. Acta 2002, 1582, 257–264. [Google Scholar] [CrossRef]
- Baker, D.L.; Morrison, P.; Miller, B.; Riely, C.A.; Tolley, B.; Westermann, A.M.; Bonfrer, J.M.; Bais, E.; Moolenaar, W.H.; Tigyi, G. Plasma lysophosphatidic acid concentration and ovarian cancer. JAMA 2002, 287, 3081–3082. [Google Scholar] [CrossRef] [PubMed]
- Mao, X.Y.; Lee, M.J.; Zhu, J.; Zhu, C.; Law, S.M.; Snijders, A.M. Genome-wide screen identifies a novel prognostic signature for breast cancer survival. Oncotarget 2017, 8, 14003–14016. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kok, B.P.; Venkatraman, G.; Capatos, D.; Brindley, D.N. Unlike two peas in a pod: Lipid phosphate phosphatases and phosphatidate phosphatases. Chem. Rev. 2012, 112, 5121–5146. [Google Scholar] [CrossRef] [PubMed]
- Morris, A.J.; Smyth, S.S. Lipid phosphate phosphatases: More than one way to put the brakes on lpa signaling? J. Lipid Res. 2014, 55, 2195–2197. [Google Scholar] [CrossRef] [Green Version]
- Tang, X.; Benesch, M.G.; Dewald, J.; Zhao, Y.Y.; Patwardhan, N.; Santos, W.L.; Curtis, J.M.; McMullen, T.P.; Brindley, D.N. Lipid phosphate phosphatase-1 expression in cancer cells attenuates tumor growth and metastasis in mice. J. Lipid Res. 2014, 55, 2389–2400. [Google Scholar] [CrossRef] [Green Version]
- Pilquil, C.; Dewald, J.; Cherney, A.; Gorshkova, I.; Tigyi, G.; English, D.; Natarajan, V.; Brindley, D.N. Lipid phosphate phosphatase-1 regulates lysophosphatidate-induced fibroblast migration by controlling phospholipase d2-dependent phosphatidate generation. J. Biol. Chem. 2006, 281, 38418–38429. [Google Scholar] [CrossRef] [Green Version]
- Alderton, F.; Darroch, P.; Sambi, B.; McKie, A.; Ahmed, I.S.; Pyne, N.; Pyne, S. G-protein-coupled receptor stimulation of the p42/p44 mitogen-activated protein kinase pathway is attenuated by lipid phosphate phosphatases 1, 1a, and 2 in human embryonic kidney 293 cells. J. Biol. Chem. 2001, 276, 13452–13460. [Google Scholar] [CrossRef] [Green Version]
- Nakayama, J.; Raines, T.A.; Lynch, K.R.; Slack-Davis, J.K. Decreased peritoneal ovarian cancer growth in mice lacking expression of lipid phosphate phosphohydrolase 1. PLoS ONE 2015, 10, e0120071. [Google Scholar] [CrossRef] [Green Version]
- Flanagan, J.M.; Funes, J.M.; Henderson, S.; Wild, L.; Carey, N.; Boshoff, C. Genomics screen in transformed stem cells reveals RNASEH2A, PPAP2C, and ADARB1 as putative anticancer drug targets. Mol. Cancer Ther. 2009, 8, 249–260. [Google Scholar] [CrossRef] [Green Version]
- Morris, K.E.; Schang, L.M.; Brindley, D.N. Lipid phosphate phosphatase-2 activity regulates s-phase entry of the cell cycle in rat2 fibroblasts. J. Biol. Chem. 2006, 281, 9297–9306. [Google Scholar] [CrossRef] [Green Version]
- Clarke, N.; Arenzana, N.; Hai, T.; Minden, A.; Prywes, R. Epidermal growth factor induction of the c-jun promoter by a rac pathway. Mol. Cell. Biol. 1998, 18, 1065–1073. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Crowe, D.L.; Tsang, K.J.; Shemirani, B. Jun n-terminal kinase 1 mediates transcriptional induction of matrix metalloproteinase 9 expression. Neoplasia 2001, 3, 27–32. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Balogh, A.; Shimizu, Y.; Lee, S.C.; Norman, D.D.; Gangwar, R.; Bavaria, M.; Moon, C.; Shukla, P.; Rao, R.; Ray, R.; et al. The autotaxin-LPA2 GPCR axis is modulated by gamma-irradiation and facilitates DNA damage repair. Cell Signal. 2015, 27, 1751–1762. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Venkatraman, G.; Benesch, M.G.; Tang, X.; Dewald, J.; McMullen, T.P.; Brindley, D.N. Lysophosphatidate signaling stabilizes Nrf2 and increases the expression of genes involved in drug resistance and oxidative stress responses: Implications for cancer treatment. FASEB J. 2015, 29, 772–785. [Google Scholar] [CrossRef] [Green Version]
- Smith, B.D.; Bellon, J.R.; Blitzblau, R.; Freedman, G.; Haffty, B.; Hahn, C.; Halberg, F.; Hoffman, K.; Horst, K.; Moran, J.; et al. Radiation therapy for the whole breast: Executive summary of an american society for radiation oncology (astro) evidence-based guideline. Pract. Radiat. Oncol. 2018, 8, 145–152. [Google Scholar] [CrossRef] [Green Version]
- Meng, G.; Tang, X.; Yang, Z.; Benesch, M.G.K.; Marshall, A.; Murray, D.; Hemmings, D.G.; Wuest, F.; McMullen, T.P.W.; Brindley, D.N. Implications for breast cancer treatment from increased autotaxin production in adipose tissue after radiotherapy. FASEB J. 2017, 31, 4064–4077. [Google Scholar] [CrossRef] [Green Version]
- Meng, G.; Wuest, M.; Tang, X.; Dufour, J.; Zhao, Y.; Curtis, J.M.; McMullen, T.P.W.; Murray, D.; Wuest, F.; Brindley, D.N. Repeated fractions of x-radiation to the breast fat pads of mice augment activation of the autotaxin-lysophosphatidate-inflammatory cycle. Cancers 2019, 11, 1816. [Google Scholar] [CrossRef] [Green Version]
- Jayakumar, S.; Pal, D.; Sandur, S.K. Nrf2 facilitates repair of radiation induced DNA damage through homologous recombination repair pathway in a ros independent manner in cancer cells. Mutat. Res. 2015, 779, 33–45. [Google Scholar] [CrossRef]
- Sekhar, K.R.; Freeman, M.L. Nrf2 promotes survival following exposure to ionizing radiation. Free Radic. Biol. Med. 2015, 88, 268–274. [Google Scholar] [CrossRef] [Green Version]
- Zhou, S.; Ye, W.; Shao, Q.; Zhang, M.; Liang, J. Nrf2 is a potential therapeutic target in radioresistance in human cancer. Crit. Rev. Oncol. Hematol. 2013, 88, 706–715. [Google Scholar] [CrossRef]
- Dalamaga, M.; Diakopoulos, K.N.; Mantzoros, C.S. The role of adiponectin in cancer: A review of current evidence. Endocr. Rev. 2012, 33, 547–594. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Deng, W.; Balazs, L.; Wang, D.A.; Van Middlesworth, L.; Tigyi, G.; Johnson, L.R. Lysophosphatidic acid protects and rescues intestinal epithelial cells from radiation- and chemotherapy-induced apoptosis. Gastroenterology 2002, 123, 206–216. [Google Scholar] [CrossRef] [PubMed]
- Deng, W.; Shuyu, E.; Tsukahara, R.; Valentine, W.J.; Durgam, G.; Gududuru, V.; Balazs, L.; Manickam, V.; Arsura, M.; VanMiddlesworth, L.; et al. The lysophosphatidic acid type 2 receptor is required for protection against radiation-induced intestinal injury. Gastroenterology 2007, 132, 1834–1851. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brown, J.M.; Attardi, L.D. The role of apoptosis in cancer development and treatment response. Nat. Rev. Cancer 2005, 5, 231–237. [Google Scholar] [CrossRef]
- Tang, X.; Wuest, M.; Benesch, M.G.K.; Dufour, J.; Zhao, Y.; Curtis, J.M.; Monjardet, A.; Heckmann, B.; Murray, D.; Wuest, F.; et al. Inhibition of autotaxin with GLPG1690 increases the efficacy of radiotherapy and chemotherapy in a mouse model of breast cancer. Mol. Cancer Ther. 2020, 19, 63–74. [Google Scholar] [CrossRef] [Green Version]
- Brindley, D.N.; Rolland, Y. Possible connections between stress, diabetes, obesity, hypertension and altered lipoprotein metabolism that may result in atherosclerosis. Clin. Sci. 1989, 77, 453–461. [Google Scholar] [CrossRef]
- Brindley, D.N.; Wang, C.-N.; O’Brien, L.; Mei, J. Insulin resistance: The role of glucocorticoids, fatty acids and tumor necrosis factor-a. Can. J. Diabetes Care 1998, 22, S57–S61. [Google Scholar]
- Cao, P.; Aoki, Y.; Badri, L.; Walker, N.M.; Manning, C.M.; Lagstein, A.; Fearon, E.R.; Lama, V.N. Autocrine lysophosphatidic acid signaling activates beta-catenin and promotes lung allograft fibrosis. J. Clin. Investig. 2017, 127, 1517–1530. [Google Scholar] [CrossRef]
- Erstad, D.J.; Tager, A.M.; Hoshida, Y.; Fuchs, B.C. The autotaxin-lysophosphatidic acid pathway emerges as a therapeutic target to prevent liver cancer. Mol. Cell Oncol. 2017, 4, e1311827. [Google Scholar] [CrossRef]
- Farquhar, M.J.; Humphreys, I.S.; Rudge, S.A.; Wilson, G.K.; Bhattacharya, B.; Ciaccia, M.; Hu, K.; Zhang, Q.; Mailly, L.; Reynolds, G.M.; et al. Autotaxin-lysophosphatidic acid receptor signalling regulates hepatitis c virus replication. J. Hepatol. 2017, 66, 919–929. [Google Scholar] [CrossRef] [Green Version]
- Gan, L.; Xue, J.X.; Li, X.; Liu, D.S.; Ge, Y.; Ni, P.Y.; Deng, L.; Lu, Y.; Jiang, W. Blockade of lysophosphatidic acid receptors LPAR1/3 ameliorates lung fibrosis induced by irradiation. Biochem. Biophys. Res. Commun. 2011, 409, 7–13. [Google Scholar] [CrossRef] [PubMed]
- Kaffe, E.; Katsifa, A.; Xylourgidis, N.; Ninou, I.; Zannikou, M.; Harokopos, V.; Foka, P.; Dimitriadis, A.; Evangelou, K.; Moulas, A.N.; et al. Hepatocyte autotaxin expression promotes liver fibrosis and cancer. Hepatology 2017, 65, 1369–1383. [Google Scholar] [CrossRef] [PubMed]
- Oikonomou, N.; Mouratis, M.A.; Tzouvelekis, A.; Kaffe, E.; Valavanis, C.; Vilaras, G.; Karameris, A.; Prestwich, G.D.; Bouros, D.; Aidinis, V. Pulmonary autotaxin expression contributes to the pathogenesis of pulmonary fibrosis. Am. J. Respir. Cell Mol. Biol. 2012, 47, 566–574. [Google Scholar] [CrossRef] [PubMed]
- Okudaira, S.; Yukiura, H.; Aoki, J. Biological roles of lysophosphatidic acid signaling through its production by autotaxin. Biochimie 2010, 92, 698–706. [Google Scholar] [CrossRef]
- Pradere, J.P.; Klein, J.; Gres, S.; Guigne, C.; Neau, E.; Valet, P.; Calise, D.; Chun, J.; Bascands, J.L.; Saulnier-Blache, J.S.; et al. LPA1 receptor activation promotes renal interstitial fibrosis. J. Am. Soc. Nephrol. 2007, 18, 3110–3118. [Google Scholar] [CrossRef] [Green Version]
- Rancoule, C.; Pradere, J.P.; Gonzalez, J.; Klein, J.; Valet, P.; Bascands, J.L.; Schanstra, J.P.; Saulnier-Blache, J.S. Lysophosphatidic acid-1-receptor targeting agents for fibrosis. Expert Opin. Investig. Drugs 2011, 20, 657–667. [Google Scholar] [CrossRef]
- Sevastou, I.; Kaffe, E.; Mouratis, M.A.; Aidinis, V. Lysoglycerophospholipids in chronic inflammatory disorders: The PLA(2)/LPC and ATX/LPA axes. Biochim. Biophys. Acta 2013, 1831, 42–60. [Google Scholar] [CrossRef]
- Swaney, J.S.; Chapman, C.; Correa, L.D.; Stebbins, K.J.; Bundey, R.A.; Prodanovich, P.C.; Fagan, P.; Baccei, C.S.; Santini, A.M.; Hutchinson, J.H.; et al. A novel, orally active lpa(1) receptor antagonist inhibits lung fibrosis in the mouse bleomycin model. Br. J. Pharmacol. 2010, 160, 1699–1713. [Google Scholar] [CrossRef] [Green Version]
- Zhao, Y.; Natarajan, V. Lysophosphatidic acid (lpa) and its receptors: Role in airway inflammation and remodeling. Biochim. Biophys. Acta 2013, 1831, 86–92. [Google Scholar] [CrossRef] [Green Version]
- Tager, A.M.; LaCamera, P.; Shea, B.S.; Campanella, G.S.; Selman, M.; Zhao, Z.; Polosukhin, V.; Wain, J.; Karimi-Shah, B.A.; Kim, N.D.; et al. The lysophosphatidic acid receptor lpa1 links pulmonary fibrosis to lung injury by mediating fibroblast recruitment and vascular leak. Nat. Med. 2008, 14, 45–54. [Google Scholar] [CrossRef]
- Nikolaou, A.; Ninou, I.; Kokotou, M.G.; Kaffe, E.; Afantitis, A.; Aidinis, V.; Kokotos, G. Hydroxamic acids constitute a novel class of autotaxin inhibitors that exhibit in vivo efficacy in a pulmonary fibrosis model. J. Med. Chem. 2018, 61, 3697–3711. [Google Scholar] [PubMed]
- Povirk, L.F. DNA damage and mutagenesis by radiomimetic DNA-cleaving agents: Bleomycin, neocarzinostatin and other enediynes. Mutat. Res. 1996, 355, 71–89. [Google Scholar] [CrossRef]
- Maher, T.M.; Kreuter, M.; Lederer, D.J.; Brown, K.K.; Wuyts, W.; Verbruggen, N.; Stutvoet, S.; Fieuw, A.; Ford, P.; Abi-Saab, W.; et al. Rationale, design and objectives of two phase iii, randomised, placebo-controlled studies of GLPG1690, a novel autotaxin inhibitor, in idiopathic pulmonary fibrosis (ISABELA 1 and 2). BMJ Open Respir. Res. 2019, 6, e000422. [Google Scholar] [CrossRef] [PubMed]
- Palmer, S.M.; Snyder, L.; Todd, J.L.; Soule, B.; Christian, R.; Anstrom, K.; Luo, Y.; Gagnon, R.; Rosen, G. Randomized, double-blind, placebo-controlled, phase 2 trial of bms-986020, a lysophosphatidic acid receptor antagonist for the treatment of idiopathic pulmonary fibrosis. Chest 2018, 154, 1061–1069. [Google Scholar] [CrossRef] [Green Version]
- Meng, G.; Wuest, M.; Tang, X.; Dufour, J.; McMullen, T.P.W.; Wuest, F.; Murray, D.; Brindley, D.N. Dexamethasone attenuates x-ray-induced activation of the autotaxin-lysophosphatidate-inflammatory cycle in breast tissue and subsequent breast fibrosis. Cancers 2020, 12, 999. [Google Scholar] [CrossRef] [Green Version]
- Van Meeteren, L.A.; Moolenaar, W.H. Regulation and biological activities of the autotaxin-lpa axis. Prog. Lipid Res. 2007, 46, 145–160. [Google Scholar] [CrossRef]
- Shea, B.S.; Tager, A.M. Role of the lysophospholipid mediators lysophosphatidic acid and sphingosine 1-phosphate in lung fibrosis. Proc. Am. Thorac. Soc. 2012, 9, 102–110. [Google Scholar] [CrossRef] [Green Version]
- Miyabe, C.; Miyabe, Y.; Nagai, J.; Miura, N.N.; Ohno, N.; Chun, J.; Tsuboi, R.; Ueda, H.; Miyasaka, M.; Miyasaka, N.; et al. Abrogation of lysophosphatidic acid receptor 1 ameliorates murine vasculitis. Arthritis Res. Ther. 2019, 21, 191. [Google Scholar] [CrossRef] [Green Version]
- Funke, M.; Zhao, Z.; Xu, Y.; Chun, J.; Tager, A.M. The lysophosphatidic acid receptor lpa1 promotes epithelial cell apoptosis after lung injury. Am. J. Respir. Cell Mol. Biol. 2012, 46, 355–364. [Google Scholar] [CrossRef] [Green Version]
- Nincheri, P.; Bernacchioni, C.; Cencetti, F.; Donati, C.; Bruni, P. Sphingosine kinase-1/S1P1 signalling axis negatively regulates mitogenic response elicited by PDGF in mouse myoblasts. Cell Signal. 2010, 22, 1688–1699. [Google Scholar] [CrossRef]
- Tager, A.M. Autotaxin emerges as a therapeutic target for idiopathic pulmonary fibrosis: Limiting fibrosis by limiting lysophosphatidic acid synthesis. Am. J. Respir. Cell Mol. Biol. 2012, 47, 563–565. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hong, J.H.; Chiang, C.S.; Tsao, C.Y.; Lin, P.Y.; McBride, W.H.; Wu, C.J. Rapid induction of cytokine gene expression in the lung after single and fractionated doses of radiation. Int. J. Radiat. Biol. 1999, 75, 1421–1427. [Google Scholar] [PubMed]
- Hong, J.H.; Chiang, C.S.; Tsao, C.Y.; Lin, P.Y.; Wu, C.J.; McBride, W.H. Can short-term administration of dexamethasone abrogate radiation-induced acute cytokine gene response in lung and modify subsequent molecular responses? Int. J. Radiat. Oncol. Biol. Phys. 2001, 51, 296–303. [Google Scholar] [CrossRef]
- Wang, L.P.; Wang, Y.W.; Wang, B.Z.; Sun, G.M.; Wang, X.Y.; Xu, J.L. Expression of interleukin-17a in lung tissues of irradiated mice and the influence of dexamethasone. Sci. World J. 2014, 2014, 251067. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.H.; Fan, W.; Chen, R.C. 3,4-dihydroxyphenylethanol suppresses irradiation-induced pulmonary fibrosis in adult rats. Int. J. Clin. Exp. Pathol. 2015, 8, 3441–3450. [Google Scholar]
- Ward, H.E.; Kemsley, L.; Davies, L.; Holecek, M.; Berend, N. The effect of steroids on radiation-induced lung disease in the rat. Radiat. Res. 1993, 136, 22–28. [Google Scholar] [CrossRef]
- Allanore, Y.; Distler, O.; Jagerschmidt, A.; Illiano, S.; Ledein, L.; Boitier, E.; Agueusop, I.; Denton, C.P.; Khanna, D. Lysophosphatidic acid receptor 1 antagonist sar100842 for patients with diffuse cutaneous systemic sclerosis: A double-blind, randomized, eight-week placebo-controlled study followed by a sixteen-week open-label extension study. Arthritis Rheumatol. 2018, 70, 1634–1643. [Google Scholar] [CrossRef] [Green Version]





© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Brindley, D.N.; Tang, X.; Meng, G.; Benesch, M.G.K. Role of Adipose Tissue-Derived Autotaxin, Lysophosphatidate Signaling, and Inflammation in the Progression and Treatment of Breast Cancer. Int. J. Mol. Sci. 2020, 21, 5938. https://doi.org/10.3390/ijms21165938
Brindley DN, Tang X, Meng G, Benesch MGK. Role of Adipose Tissue-Derived Autotaxin, Lysophosphatidate Signaling, and Inflammation in the Progression and Treatment of Breast Cancer. International Journal of Molecular Sciences. 2020; 21(16):5938. https://doi.org/10.3390/ijms21165938
Chicago/Turabian StyleBrindley, David N., Xiaoyun Tang, Guanmin Meng, and Matthew G. K. Benesch. 2020. "Role of Adipose Tissue-Derived Autotaxin, Lysophosphatidate Signaling, and Inflammation in the Progression and Treatment of Breast Cancer" International Journal of Molecular Sciences 21, no. 16: 5938. https://doi.org/10.3390/ijms21165938
APA StyleBrindley, D. N., Tang, X., Meng, G., & Benesch, M. G. K. (2020). Role of Adipose Tissue-Derived Autotaxin, Lysophosphatidate Signaling, and Inflammation in the Progression and Treatment of Breast Cancer. International Journal of Molecular Sciences, 21(16), 5938. https://doi.org/10.3390/ijms21165938