T Cell Immunity to Bacterial Pathogens: Mechanisms of Immune Control and Bacterial Evasion


{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. How T Cells Fight Bacteria
2.1. MHC-Restricted αβ T Cells
2.1.1. CD8+ T Cells: The Advent of Tissue Resident Memory T Cell Immunity
2.1.2. HLA-E Restricted CD8+ T Cells: Non-Classical Bacterial Recognition
2.1.3. CD4+ T Cells: Anti-Bacterial Immunity Requires “Help”
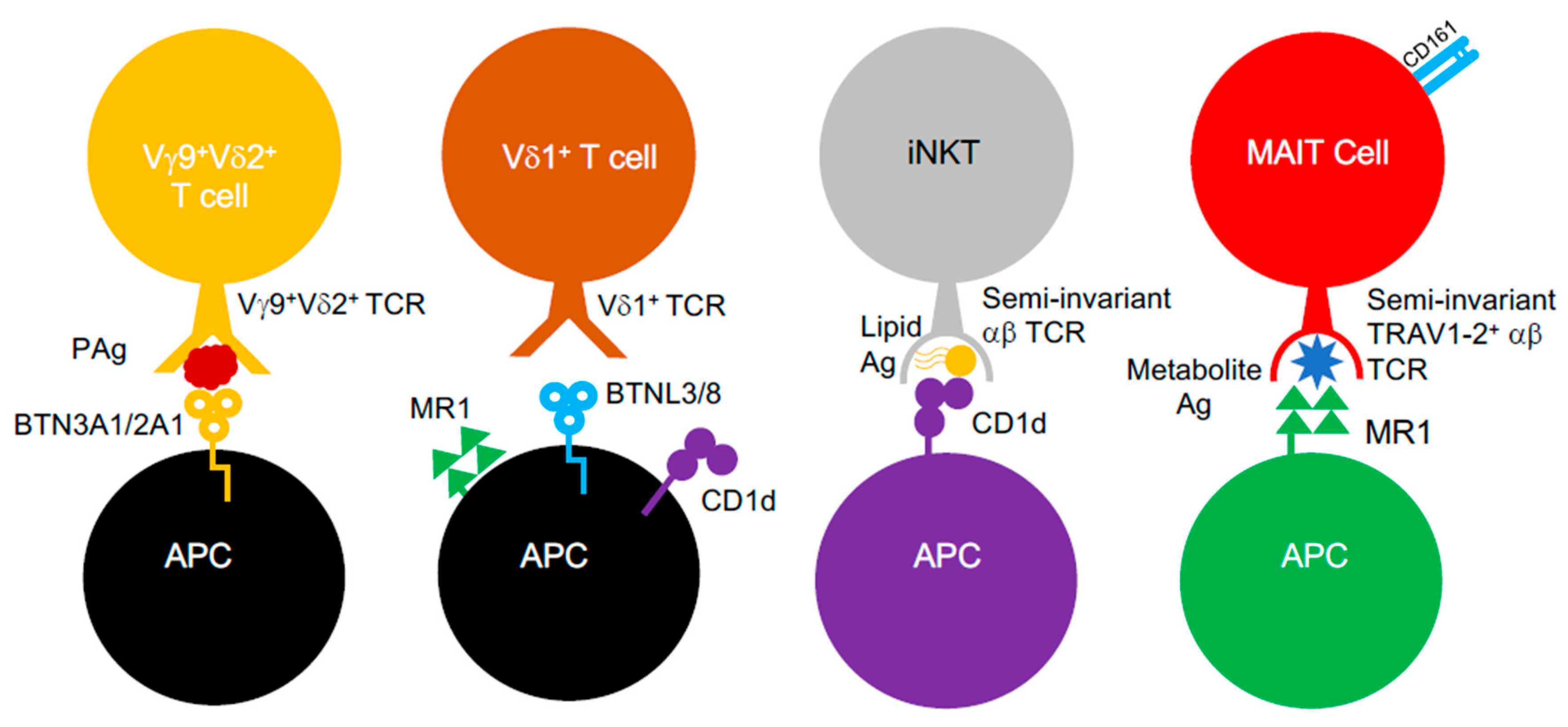
2.2. Unconventional αβ and γδ T Cells
2.2.1. CD1-Restricted T Cells: Lipid-Driven T Cell Immunosurveillance
2.2.2. γδ T Cells: A Story of Anti-Bacterial Immunity Bound by Two Divergent TCR δ-Chains
2.2.3. MAIT Cells: Masters of Riboflavin-Driven T Cell Immunity
3. Bacterial Immune Evasion Strategies
3.1. A Pathogenic Game of ‘Hide and Seek’ and T Cell Evasion by M. tuberculosis
3.2. Type III Secretion Systems and Pore-Forming Toxins: Secretion-Driven Bacterial Immune Defense
3.3. SAgs: Microbial Super-Agents That Manipulate T Cell Immunity
4. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Netea, M.G.; Joosten, L.A.; Latz, E.; Mills, K.H.; Natoli, G.; Stunnenberg, H.G.; O’Neill, L.A.; Xavier, R.J. Trained immunity: A program of innate immune memory in health and disease. Science 2016, 352, aaf1098. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Janssen, E.M.; Lemmens, E.E.; Wolfe, T.; Christen, U.; von Herrath, M.G.; Schoenberger, S.P. CD4+ T cells are required for secondary expansion and memory in CD8+ T lymphocytes. Nature 2003, 421, 852–856. [Google Scholar] [CrossRef] [PubMed]
- Shedlock, D.J.; Shen, H. Requirement for CD4 T cell help in generating functional CD8 T cell memory. Science 2003, 300, 337–339. [Google Scholar] [CrossRef] [PubMed]
- Crotty, S. A brief history of T cell help to B cells. Nat. Rev. Immunol. 2015, 15, 185–189. [Google Scholar] [CrossRef] [PubMed]
- Galperin, M.; Farenc, C.; Mukhopadhyay, M.; Jayasinghe, D.; Decroos, A.; Benati, D.; Tan, L.L.; Ciacchi, L.; Reid, H.H.; Rossjohn, J.; et al. CD4(+) T cell-mediated HLA class II cross-restriction in HIV controllers. Sci. Immunol. 2018, 3, eaat0687. [Google Scholar] [CrossRef] [Green Version]
- Ranasinghe, S.; Lamothe, P.A.; Soghoian, D.Z.; Kazer, S.W.; Cole, M.B.; Shalek, A.K.; Yosef, N.; Jones, R.B.; Donaghey, F.; Nwonu, C.; et al. Antiviral CD8(+) T Cells Restricted by Human Leukocyte Antigen Class II Exist during Natural HIV Infection and Exhibit Clonal Expansion. Immunity 2016, 45, 917–930. [Google Scholar] [CrossRef] [Green Version]
- La Gruta, N.L.; Gras, S.; Daley, S.R.; Thomas, P.G.; Rossjohn, J. Understanding the drivers of MHC restriction of T cell receptors. Nat. Rev. Immunol. 2018, 18, 467–478. [Google Scholar] [CrossRef]
- Arstila, T.P.; Casrouge, A.; Baron, V.; Even, J.; Kanellopoulos, J.; Kourilsky, P. A direct estimate of the human alphabeta T cell receptor diversity. Science 1999, 286, 958–961. [Google Scholar] [CrossRef]
- Rossjohn, J.; Gras, S.; Miles, J.J.; Turner, S.J.; Godfrey, D.I.; McCluskey, J. T cell antigen receptor recognition of antigen-presenting molecules. Annu. Rev. Immunol. 2015, 33, 169–200. [Google Scholar] [CrossRef]
- Jenkins, M.K.; Moon, J.J. The role of naive T cell precursor frequency and recruitment in dictating immune response magnitude. J. Immunol. 2012, 188, 4135–4140. [Google Scholar] [CrossRef] [Green Version]
- Kaech, S.M.; Ahmed, R. Memory CD8+ T cell differentiation: Initial antigen encounter triggers a developmental program in naive cells. Nat. Immunol. 2001, 2, 415–422. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kaech, S.M.; Cui, W. Transcriptional control of effector and memory CD8+ T cell differentiation. Nat. Rev. Immunol. 2012, 12, 749–761. [Google Scholar] [CrossRef] [PubMed]
- Laidlaw, B.J.; Craft, J.E.; Kaech, S.M. The multifaceted role of CD4(+) T cells in CD8(+) T cell memory. Nat. Rev. Immunol. 2016, 16, 102–111. [Google Scholar] [CrossRef] [PubMed]
- Lau, L.L.; Jamieson, B.D.; Somasundaram, T.; Ahmed, R. Cytotoxic T-cell memory without antigen. Nature 1994, 369, 648–652. [Google Scholar] [CrossRef]
- Surh, C.D.; Sprent, J. Homeostasis of naive and memory T cells. Immunity 2008, 29, 848–862. [Google Scholar] [CrossRef] [Green Version]
- Goldrath, A.W.; Sivakumar, P.V.; Glaccum, M.; Kennedy, M.K.; Bevan, M.J.; Benoist, C.; Mathis, D.; Butz, E.A. Cytokine requirements for acute and Basal homeostatic proliferation of naive and memory CD8+ T cells. J. Exp. Med. 2002, 195, 1515–1522. [Google Scholar] [CrossRef]
- Schmidt, C.S.; Mescher, M.F. Peptide antigen priming of naive, but not memory, CD8 T cells requires a third signal that can be provided by IL-12. J. Immunol. 2002, 168, 5521–5529. [Google Scholar] [CrossRef] [Green Version]
- Hosking, M.P.; Flynn, C.T.; Whitton, J.L. Type I IFN Signaling Is Dispensable during Secondary Viral Infection. PLoS. Pathog. 2016, 12, e1005861. [Google Scholar] [CrossRef]
- Bonilla, W.V.; Frohlich, A.; Senn, K.; Kallert, S.; Fernandez, M.; Johnson, S.; Kreutzfeldt, M.; Hegazy, A.N.; Schrick, C.; Fallon, P.G.; et al. The alarmin interleukin-33 drives protective antiviral CD8(+) T cell responses. Science 2012, 335, 984–989. [Google Scholar] [CrossRef]
- McLaren, J.E.; Clement, M.; Marsden, M.; Miners, K.L.; Llewellyn-Lacey, S.; Grant, E.J.; Rubina, A.; Gimeno Brias, S.; Gostick, E.; Stacey, M.A.; et al. IL-33 Augments Virus-Specific Memory T Cell Inflation and Potentiates the Efficacy of an Attenuated Cytomegalovirus-Based Vaccine. J. Immunol. 2019, 202, 943–955. [Google Scholar] [CrossRef] [Green Version]
- Grant, E.J.; Nguyen, A.T.; Lobos, C.A.; Szeto, C.; Chatzileontiadou, D.S.M.; Gras, S. The unconventional role of HLA-E: The road less traveled. Mol. Immunol. 2020, 120, 101–112. [Google Scholar] [CrossRef] [PubMed]
- Beckman, E.M.; Porcelli, S.A.; Morita, C.T.; Behar, S.M.; Furlong, S.T.; Brenner, M.B. Recognition of a lipid antigen by CD1-restricted alpha beta+ T cells. Nature 1994, 372, 691–694. [Google Scholar] [CrossRef]
- Van Rhijn, I.; Godfrey, D.I.; Rossjohn, J.; Moody, D.B. Lipid and small-molecule display by CD1 and MR1. Nat. Rev. Immunol. 2015, 15, 643–654. [Google Scholar] [CrossRef] [PubMed]
- Godfrey, D.I.; Uldrich, A.P.; McCluskey, J.; Rossjohn, J.; Moody, D.B. The burgeoning family of unconventional T cells. Nat. Immunol. 2015, 16, 1114–1123. [Google Scholar] [CrossRef] [PubMed]
- Kjer-Nielsen, L.; Patel, O.; Corbett, A.J.; Le Nours, J.; Meehan, B.; Liu, L.; Bhati, M.; Chen, Z.; Kostenko, L.; Reantragoon, R.; et al. MR1 presents microbial vitamin B metabolites to MAIT cells. Nature 2012, 491, 717–723. [Google Scholar] [CrossRef] [PubMed]
- Willcox, B.E.; Willcox, C.R. gammadelta TCR ligands: The quest to solve a 500-million-year-old mystery. Nat. Immunol. 2019, 20, 121–128. [Google Scholar] [CrossRef]
- Hayday, A.C.; Vantourout, P. The Innate Biologies of Adaptive Antigen Receptors. Annu. Rev. Immunol. 2020, 38, 487–510. [Google Scholar] [CrossRef] [Green Version]
- Rhodes, D.A.; Reith, W.; Trowsdale, J. Regulation of Immunity by Butyrophilins. Annu. Rev. Immunol. 2016, 34, 151–172. [Google Scholar] [CrossRef] [Green Version]
- Le Nours, J.; Gherardin, N.A.; Ramarathinam, S.H.; Awad, W.; Wiede, F.; Gully, B.S.; Khandokar, Y.; Praveena, T.; Wubben, J.M.; Sandow, J.J.; et al. A class of gammadelta T cell receptors recognize the underside of the antigen-presenting molecule MR1. Science 2019, 366, 1522–1527. [Google Scholar] [CrossRef]
- Bai, L.; Picard, D.; Anderson, B.; Chaudhary, V.; Luoma, A.; Jabri, B.; Adams, E.J.; Savage, P.B.; Bendelac, A. The majority of CD1d-sulfatide-specific T cells in human blood use a semiinvariant Vdelta1 TCR. Eur. J. Immunol. 2012, 42, 2505–2510. [Google Scholar] [CrossRef]
- Luoma, A.M.; Castro, C.D.; Mayassi, T.; Bembinster, L.A.; Bai, L.; Picard, D.; Anderson, B.; Scharf, L.; Kung, J.E.; Sibener, L.V.; et al. Crystal structure of Vdelta1 T cell receptor in complex with CD1d-sulfatide shows MHC-like recognition of a self-lipid by human gammadelta T cells. Immunity 2013, 39, 1032–1042. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roy, S.; Ly, D.; Castro, C.D.; Li, N.S.; Hawk, A.J.; Altman, J.D.; Meredith, S.C.; Piccirilli, J.A.; Moody, D.B.; Adams, E.J. Molecular Analysis of Lipid-Reactive Vdelta1 gammadelta T Cells Identified by CD1c Tetramers. J. Immunol. 2016, 196, 1933–1942. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Uldrich, A.P.; Le Nours, J.; Pellicci, D.G.; Gherardin, N.A.; McPherson, K.G.; Lim, R.T.; Patel, O.; Beddoe, T.; Gras, S.; Rossjohn, J.; et al. CD1d-lipid antigen recognition by the gammadelta TCR. Nat. Immunol. 2013, 14, 1137–1145. [Google Scholar] [CrossRef] [PubMed]
- Xu, B.; Pizarro, J.C.; Holmes, M.A.; McBeth, C.; Groh, V.; Spies, T.; Strong, R.K. Crystal structure of a gammadelta T-cell receptor specific for the human MHC class I homolog MICA. Proc. Natl. Acad. Sci. USA 2011, 108, 2414–2419. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Benveniste, P.M.; Roy, S.; Nakatsugawa, M.; Chen, E.L.Y.; Nguyen, L.; Millar, D.G.; Ohashi, P.S.; Hirano, N.; Adams, E.J.; Zuniga-Pflucker, J.C. Generation and molecular recognition of melanoma-associated antigen-specific human gammadelta T cells. Sci. Immunol. 2018, 3. [Google Scholar]
- Liuzzi, A.R.; McLaren, J.E.; Price, D.A.; Eberl, M. Early innate responses to pathogens: Pattern recognition by unconventional human T-cells. Curr. Opin. Immunol. 2015, 36, 31–37. [Google Scholar] [CrossRef] [Green Version]
- Kerksiek, K.M.; Pamer, E.G. T cell responses to bacterial infection. Curr. Opin. Immunol. 1999, 11, 400–405. [Google Scholar] [CrossRef]
- Salgado-Pabon, W.; Celli, S.; Arena, E.T.; Nothelfer, K.; Roux, P.; Sellge, G.; Frigimelica, E.; Bousso, P.; Sansonetti, P.J.; Phalipon, A. Shigella impairs T lymphocyte dynamics in vivo. Proc. Natl. Acad. Sci. USA 2013, 110, 4458–4463. [Google Scholar] [CrossRef] [Green Version]
- Tuffs, S.W.; Haeryfar, S.M.M.; McCormick, J.K. Manipulation of Innate and Adaptive Immunity by Staphylococcal Superantigens. Pathogens 2018, 7, 53. [Google Scholar] [CrossRef] [Green Version]
- Fraunholz, M.; Sinha, B. Intracellular Staphylococcus aureus: Live-in and let die. Front. Cell Infect. Microbiol. 2012, 2, 43. [Google Scholar] [CrossRef] [Green Version]
- Silva, M.T. Classical labeling of bacterial pathogens according to their lifestyle in the host: Inconsistencies and alternatives. Front. Microbiol. 2012, 3, 71. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kaufmann, S.H. Intracellular pathogens: Living in an extreme environment. Immunol. Rev. 2011, 240, 5–10. [Google Scholar] [CrossRef] [PubMed]
- Qiu, Z.; Khairallah, C.; Sheridan, B.S. Listeria Monocytogenes: A Model Pathogen Continues to Refine Our Knowledge of the CD8 T Cell Response. Pathogens 2018, 7, 55. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lin, P.L.; Flynn, J.L. CD8 T cells and Mycobacterium tuberculosis infection. Semin Immunopathol. 2015, 37, 239–249. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, S.J.; Dunmire, S.; McSorley, S.J. MHC class-I-restricted CD8 T cells play a protective role during primary Salmonella infection. Immunol. Lett. 2012, 148, 138–143. [Google Scholar] [CrossRef] [Green Version]
- O’Brien, E.C.; McLoughlin, R.M. Considering the ‘Alternatives’ for Next-Generation Anti-Staphylococcus aureus Vaccine Development. Trends Mol. Med. 2019, 25, 171–184. [Google Scholar] [CrossRef]
- Hess, J.; Ladel, C.; Miko, D.; Kaufmann, S.H. Salmonella typhimurium aroA- infection in gene-targeted immunodeficient mice: Major role of CD4+ TCR-alpha beta cells and IFN-gamma in bacterial clearance independent of intracellular location. J. Immunol. 1996, 156, 3321–3326. [Google Scholar]
- Thakur, A.; Mikkelsen, H.; Jungersen, G. Intracellular Pathogens: Host Immunity and Microbial Persistence Strategies. J. Immunol. Res. 2019, 2019, 1356540. [Google Scholar] [CrossRef]
- Cruz-Adalia, A.; Ramirez-Santiago, G.; Calabia-Linares, C.; Torres-Torresano, M.; Feo, L.; Galan-Diez, M.; Fernandez-Ruiz, E.; Pereiro, E.; Guttmann, P.; Chiappi, M.; et al. T cells kill bacteria captured by transinfection from dendritic cells and confer protection in mice. Cell Host Microbe 2014, 15, 611–622. [Google Scholar] [CrossRef] [Green Version]
- Cruz-Adalia, A.; Ramirez-Santiago, G.; Osuna-Perez, J.; Torres-Torresano, M.; Zorita, V.; Martinez-Riano, A.; Boccasavia, V.; Borroto, A.; Martinez Del Hoyo, G.; Gonzalez-Granado, J.M.; et al. Conventional CD4(+) T cells present bacterial antigens to induce cytotoxic and memory CD8(+) T cell responses. Nat. Commun 2017, 8, 1591. [Google Scholar] [CrossRef]
- Sallusto, F.; Lenig, D.; Forster, R.; Lipp, M.; Lanzavecchia, A. Two subsets of memory T lymphocytes with distinct homing potentials and effector functions. Nature 1999, 401, 708–712. [Google Scholar] [CrossRef] [PubMed]
- Wherry, E.J.; Teichgraber, V.; Becker, T.C.; Masopust, D.; Kaech, S.M.; Antia, R.; von Andrian, U.H.; Ahmed, R. Lineage relationship and protective immunity of memory CD8 T cell subsets. Nat. Immunol. 2003, 4, 225–234. [Google Scholar] [CrossRef] [PubMed]
- Huster, K.M.; Koffler, M.; Stemberger, C.; Schiemann, M.; Wagner, H.; Busch, D.H. Unidirectional development of CD8+ central memory T cells into protective Listeria-specific effector memory T cells. Eur. J. Immunol. 2006, 36, 1453–1464. [Google Scholar] [CrossRef]
- Olson, J.A.; McDonald-Hyman, C.; Jameson, S.C.; Hamilton, S.E. Effector-like CD8(+) T cells in the memory population mediate potent protective immunity. Immunity 2013, 38, 1250–1260. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Masopust, D.; Vezys, V.; Marzo, A.L.; Lefrancois, L. Preferential localization of effector memory cells in nonlymphoid tissue. Science 2001, 291, 2413–2417. [Google Scholar] [CrossRef] [Green Version]
- Gebhardt, T.; Wakim, L.M.; Eidsmo, L.; Reading, P.C.; Heath, W.R.; Carbone, F.R. Memory T cells in nonlymphoid tissue that provide enhanced local immunity during infection with herpes simplex virus. Nat. Immunol. 2009, 10, 524–530. [Google Scholar] [CrossRef]
- Masopust, D.; Choo, D.; Vezys, V.; Wherry, E.J.; Duraiswamy, J.; Akondy, R.; Wang, J.; Casey, K.A.; Barber, D.L.; Kawamura, K.S.; et al. Dynamic T cell migration program provides resident memory within intestinal epithelium. J. Exp. Med. 2010, 207, 553–564. [Google Scholar] [CrossRef] [Green Version]
- Wakim, L.M.; Waithman, J.; van Rooijen, N.; Heath, W.R.; Carbone, F.R. Dendritic cell-induced memory T cell activation in nonlymphoid tissues. Science 2008, 319, 198–202. [Google Scholar] [CrossRef] [Green Version]
- Mueller, S.N.; Mackay, L.K. Tissue-resident memory T cells: Local specialists in immune defence. Nat. Rev. Immunol. 2016, 16, 79–89. [Google Scholar] [CrossRef]
- Ariotti, S.; Hogenbirk, M.A.; Dijkgraaf, F.E.; Visser, L.L.; Hoekstra, M.E.; Song, J.Y.; Jacobs, H.; Haanen, J.B.; Schumacher, T.N. T cell memory. Skin-resident memory CD8(+) T cells trigger a state of tissue-wide pathogen alert. Science 2014, 346, 101–105. [Google Scholar] [CrossRef]
- Mackay, L.K.; Minnich, M.; Kragten, N.A.; Liao, Y.; Nota, B.; Seillet, C.; Zaid, A.; Man, K.; Preston, S.; Freestone, D.; et al. Hobit and Blimp1 instruct a universal transcriptional program of tissue residency in lymphocytes. Science 2016, 352, 459–463. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mackay, L.K.; Rahimpour, A.; Ma, J.Z.; Collins, N.; Stock, A.T.; Hafon, M.L.; Vega-Ramos, J.; Lauzurica, P.; Mueller, S.N.; Stefanovic, T.; et al. The developmental pathway for CD103(+)CD8+ tissue-resident memory T cells of skin. Nat. Immunol. 2013, 14, 1294–1301. [Google Scholar] [CrossRef] [PubMed]
- Schenkel, J.M.; Fraser, K.A.; Beura, L.K.; Pauken, K.E.; Vezys, V.; Masopust, D. T cell memory. Resident memory CD8 T cells trigger protective innate and adaptive immune responses. Science 2014, 346, 98–101. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pan, Y.; Tian, T.; Park, C.O.; Lofftus, S.Y.; Mei, S.; Liu, X.; Luo, C.; O’Malley, J.T.; Gehad, A.; Teague, J.E.; et al. Survival of tissue-resident memory T cells requires exogenous lipid uptake and metabolism. Nature 2017, 543, 252–256. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wakim, L.M.; Woodward-Davis, A.; Liu, R.; Hu, Y.; Villadangos, J.; Smyth, G.; Bevan, M.J. The molecular signature of tissue resident memory CD8 T cells isolated from the brain. J. Immunol. 2012, 189, 3462–3471. [Google Scholar] [CrossRef]
- Mackay, L.K.; Braun, A.; Macleod, B.L.; Collins, N.; Tebartz, C.; Bedoui, S.; Carbone, F.R.; Gebhardt, T. Cutting edge: CD69 interference with sphingosine-1-phosphate receptor function regulates peripheral T cell retention. J. Immunol. 2015, 194, 2059–2063. [Google Scholar] [CrossRef] [Green Version]
- Skon, C.N.; Lee, J.Y.; Anderson, K.G.; Masopust, D.; Hogquist, K.A.; Jameson, S.C. Transcriptional downregulation of S1pr1 is required for the establishment of resident memory CD8+ T cells. Nat. Immunol. 2013, 14, 1285–1293. [Google Scholar] [CrossRef] [Green Version]
- Ma, C.; Mishra, S.; Demel, E.L.; Liu, Y.; Zhang, N. TGF-beta Controls the Formation of Kidney-Resident T Cells via Promoting Effector T Cell Extravasation. J. Immunol. 2017, 198, 749–756. [Google Scholar] [CrossRef] [Green Version]
- Steinert, E.M.; Schenkel, J.M.; Fraser, K.A.; Beura, L.K.; Manlove, L.S.; Igyarto, B.Z.; Southern, P.J.; Masopust, D. Quantifying Memory CD8 T Cells Reveals Regionalization of Immunosurveillance. Cell 2015, 161, 737–749. [Google Scholar] [CrossRef] [Green Version]
- Cheuk, S.; Schlums, H.; Gallais Serezal, I.; Martini, E.; Chiang, S.C.; Marquardt, N.; Gibbs, A.; Detlofsson, E.; Introini, A.; Forkel, M.; et al. CD49a Expression Defines Tissue-Resident CD8(+) T Cells Poised for Cytotoxic Function in Human Skin. Immunity 2017, 46, 287–300. [Google Scholar] [CrossRef]
- Hombrink, P.; Helbig, C.; Backer, R.A.; Piet, B.; Oja, A.E.; Stark, R.; Brasser, G.; Jongejan, A.; Jonkers, R.E.; Nota, B.; et al. Programs for the persistence, vigilance and control of human CD8(+) lung-resident memory T cells. Nat. Immunol. 2016, 17, 1467–1478. [Google Scholar] [CrossRef] [PubMed]
- Kumar, B.V.; Kratchmarov, R.; Miron, M.; Carpenter, D.J.; Senda, T.; Lerner, H.; Friedman, A.; Reiner, S.L.; Farber, D.L. Functional heterogeneity of human tissue-resident memory T cells based on dye efflux capacities. JCI Insight 2018, 3, e123568. [Google Scholar] [CrossRef] [PubMed]
- Pallett, L.J.; Davies, J.; Colbeck, E.J.; Robertson, F.; Hansi, N.; Easom, N.J.W.; Burton, A.R.; Stegmann, K.A.; Schurich, A.; Swadling, L.; et al. IL-2(high) tissue-resident T cells in the human liver: Sentinels for hepatotropic infection. J. Exp. Med. 2017, 214, 1567–1580. [Google Scholar] [CrossRef] [PubMed]
- Milner, J.J.; Toma, C.; Yu, B.; Zhang, K.; Omilusik, K.; Phan, A.T.; Wang, D.; Getzler, A.J.; Nguyen, T.; Crotty, S.; et al. Runx3 programs CD8(+) T cell residency in non-lymphoid tissues and tumours. Nature 2017, 552, 253–257. [Google Scholar] [CrossRef] [PubMed]
- Li, C.; Zhu, B.; Son, Y.M.; Wang, Z.; Jiang, L.; Xiang, M.; Ye, Z.; Beckermann, K.E.; Wu, Y.; Jenkins, J.W.; et al. The Transcription Factor Bhlhe40 Programs Mitochondrial Regulation of Resident CD8(+) T Cell Fitness and Functionality. Immunity 2019, 51, 491–507 e7. [Google Scholar] [CrossRef]
- Casey, K.A.; Fraser, K.A.; Schenkel, J.M.; Moran, A.; Abt, M.C.; Beura, L.K.; Lucas, P.J.; Artis, D.; Wherry, E.J.; Hogquist, K.; et al. Antigen-independent differentiation and maintenance of effector-like resident memory T cells in tissues. J. Immunol. 2012, 188, 4866–4875. [Google Scholar] [CrossRef] [Green Version]
- Zhang, N.; Bevan, M.J. Transforming growth factor-beta signaling controls the formation and maintenance of gut-resident memory T cells by regulating migration and retention. Immunity 2013, 39, 687–696. [Google Scholar] [CrossRef] [Green Version]
- Sheridan, B.S.; Pham, Q.M.; Lee, Y.T.; Cauley, L.S.; Puddington, L.; Lefrancois, L. Oral infection drives a distinct population of intestinal resident memory CD8(+) T cells with enhanced protective function. Immunity 2014, 40, 747–757. [Google Scholar] [CrossRef] [Green Version]
- Beura, L.K.; Mitchell, J.S.; Thompson, E.A.; Schenkel, J.M.; Mohammed, J.; Wijeyesinghe, S.; Fonseca, R.; Burbach, B.J.; Hickman, H.D.; Vezys, V.; et al. Intravital mucosal imaging of CD8(+) resident memory T cells shows tissue-autonomous recall responses that amplify secondary memory. Nat. Immunol. 2018, 19, 173–182. [Google Scholar] [CrossRef]
- Park, S.L.; Zaid, A.; Hor, J.L.; Christo, S.N.; Prier, J.E.; Davies, B.; Alexandre, Y.O.; Gregory, J.L.; Russell, T.A.; Gebhardt, T.; et al. Local proliferation maintains a stable pool of tissue-resident memory T cells after antiviral recall responses. Nat. Immunol. 2018, 19, 183–191. [Google Scholar] [CrossRef]
- Perdomo, C.; Zedler, U.; Kuhl, A.A.; Lozza, L.; Saikali, P.; Sander, L.E.; Vogelzang, A.; Kaufmann, S.H.; Kupz, A. Mucosal BCG Vaccination Induces Protective Lung-Resident Memory T Cell Populations against Tuberculosis. mBio 2016, 7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Connor, L.M.; Harvie, M.C.; Rich, F.J.; Quinn, K.M.; Brinkmann, V.; Le Gros, G.; Kirman, J.R. A key role for lung-resident memory lymphocytes in protective immune responses after BCG vaccination. Eur. J. Immunol. 2010, 40, 2482–2492. [Google Scholar] [CrossRef] [PubMed]
- Haddadi, S.; Thanthrige-Don, N.; Afkhami, S.; Khera, A.; Jeyanathan, M.; Xing, Z. Expression and role of VLA-1 in resident memory CD8 T cell responses to respiratory mucosal viral-vectored immunization against tuberculosis. Sci. Rep. 2017, 7, 9525. [Google Scholar] [CrossRef] [PubMed]
- Hansen, S.G.; Zak, D.E.; Xu, G.; Ford, J.C.; Marshall, E.E.; Malouli, D.; Gilbride, R.M.; Hughes, C.M.; Ventura, A.B.; Ainslie, E.; et al. Prevention of tuberculosis in rhesus macaques by a cytomegalovirus-based vaccine. Nat. Med. 2018, 24, 130–143. [Google Scholar] [CrossRef]
- Bergsbaken, T.; Bevan, M.J.; Fink, P.J. Local Inflammatory Cues Regulate Differentiation and Persistence of CD8(+) Tissue-Resident Memory T Cells. Cell Rep. 2017, 19, 114–124. [Google Scholar] [CrossRef]
- Bergsbaken, T.; Bevan, M.J. Proinflammatory microenvironments within the intestine regulate the differentiation of tissue-resident CD8(+) T cells responding to infection. Nat. Immunol. 2015, 16, 406–414. [Google Scholar] [CrossRef]
- Benoun, J.M.; Peres, N.G.; Wang, N.; Pham, O.H.; Rudisill, V.L.; Fogassy, Z.N.; Whitney, P.G.; Fernandez-Ruiz, D.; Gebhardt, T.; Pham, Q.M.; et al. Optimal protection against Salmonella infection requires noncirculating memory. Proc. Natl. Acad. Sci. USA 2018, 115, 10416–10421. [Google Scholar] [CrossRef] [Green Version]
- Lee, S.J.; Benoun, J.; Sheridan, B.S.; Fogassy, Z.; Pham, O.; Pham, Q.M.; Puddington, L.; McSorley, S.J. Dual Immunization with SseB/Flagellin Provides Enhanced Protection against Salmonella Infection Mediated by Circulating Memory Cells. J. Immunol. 2017, 199, 1353–1361. [Google Scholar] [CrossRef] [Green Version]
- Braud, V.M.; Allan, D.S.; O’Callaghan, C.A.; Soderstrom, K.; D’Andrea, A.; Ogg, G.S.; Lazetic, S.; Young, N.T.; Bell, J.I.; Phillips, J.H.; et al. HLA-E binds to natural killer cell receptors CD94/NKG2A, B and C. Nature 1998, 391, 795–799. [Google Scholar] [CrossRef]
- Braud, V.; Jones, E.Y.; McMichael, A. The human major histocompatibility complex class Ib molecule HLA-E binds signal sequence-derived peptides with primary anchor residues at positions 2 and 9. Eur. J. Immunol. 1997, 27, 1164–1169. [Google Scholar] [CrossRef]
- Tomasec, P.; Braud, V.M.; Rickards, C.; Powell, M.B.; McSharry, B.P.; Gadola, S.; Cerundolo, V.; Borysiewicz, L.K.; McMichael, A.J.; Wilkinson, G.W. Surface expression of HLA-E, an inhibitor of natural killer cells, enhanced by human cytomegalovirus gpUL40. Science 2000, 287, 1031. [Google Scholar] [CrossRef] [PubMed]
- Heinzel, A.S.; Grotzke, J.E.; Lines, R.A.; Lewinsohn, D.A.; McNabb, A.L.; Streblow, D.N.; Braud, V.M.; Grieser, H.J.; Belisle, J.T.; Lewinsohn, D.M. HLA-E-dependent presentation of Mtb-derived antigen to human CD8+ T cells. J. Exp. Med. 2002, 196, 1473–1481. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lewinsohn, D.M.; Alderson, M.R.; Briden, A.L.; Riddell, S.R.; Reed, S.G.; Grabstein, K.H. Characterization of human CD8+ T cells reactive with Mycobacterium tuberculosis-infected antigen-presenting cells. J. Exp. Med. 1998, 187, 1633–1640. [Google Scholar] [CrossRef] [PubMed]
- Salerno-Goncalves, R.; Fernandez-Vina, M.; Lewinsohn, D.M.; Sztein, M.B. Identification of a human HLA-E-restricted CD8+ T cell subset in volunteers immunized with Salmonella enterica serovar Typhi strain Ty21a typhoid vaccine. J. Immunol. 2004, 173, 5852–5862. [Google Scholar] [CrossRef] [Green Version]
- Caccamo, N.; Pietra, G.; Sullivan, L.C.; Brooks, A.G.; Prezzemolo, T.; La Manna, M.P.; Di Liberto, D.; Joosten, S.A.; van Meijgaarden, K.E.; Di Carlo, P.; et al. Human CD8 T lymphocytes recognize Mycobacterium tuberculosis antigens presented by HLA-E during active tuberculosis and express type 2 cytokines. Eur. J. Immunol. 2015, 45, 1069–1081. [Google Scholar] [CrossRef] [Green Version]
- Harriff, M.J.; Wolfe, L.M.; Swarbrick, G.; Null, M.; Cansler, M.E.; Canfield, E.T.; Vogt, T.; Toren, K.G.; Li, W.; Jackson, M.; et al. HLA-E Presents Glycopeptides from the Mycobacterium tuberculosis Protein MPT32 to Human CD8(+) T cells. Sci. Rep. 2017, 7, 4622. [Google Scholar] [CrossRef]
- Joosten, S.A.; van Meijgaarden, K.E.; van Weeren, P.C.; Kazi, F.; Geluk, A.; Savage, N.D.; Drijfhout, J.W.; Flower, D.R.; Hanekom, W.A.; Klein, M.R.; et al. Mycobacterium tuberculosis peptides presented by HLA-E molecules are targets for human CD8 T-cells with cytotoxic as well as regulatory activity. PLoS. Pathog. 2010, 6, e1000782. [Google Scholar] [CrossRef] [Green Version]
- Prezzemolo, T.; van Meijgaarden, K.E.; Franken, K.; Caccamo, N.; Dieli, F.; Ottenhoff, T.H.M.; Joosten, S.A. Detailed characterization of human Mycobacterium tuberculosis specific HLA-E restricted CD8(+) T cells. Eur. J. Immunol. 2018, 48, 293–305. [Google Scholar] [CrossRef] [Green Version]
- van Meijgaarden, K.E.; Haks, M.C.; Caccamo, N.; Dieli, F.; Ottenhoff, T.H.; Joosten, S.A. Human CD8+ T-cells recognizing peptides from Mycobacterium tuberculosis (Mtb) presented by HLA-E have an unorthodox Th2-like, multifunctional, Mtb inhibitory phenotype and represent a novel human T-cell subset. PLoS. Pathog. 2015, 11, e1004671. [Google Scholar] [CrossRef]
- Walters, L.C.; Harlos, K.; Brackenridge, S.; Rozbesky, D.; Barrett, J.R.; Jain, V.; Walter, T.S.; O’Callaghan, C.A.; Borrow, P.; Toebes, M.; et al. Pathogen-derived HLA-E bound epitopes reveal broad primary anchor pocket tolerability and conformationally malleable peptide binding. Nat. Commun. 2018, 9, 3137. [Google Scholar] [CrossRef] [Green Version]
- Grotzke, J.E.; Harriff, M.J.; Siler, A.C.; Nolt, D.; Delepine, J.; Lewinsohn, D.A.; Lewinsohn, D.M. The Mycobacterium tuberculosis phagosome is a HLA-I processing competent organelle. PLoS. Pathog. 2009, 5, e1000374. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Harriff, M.J.; Cansler, M.E.; Toren, K.G.; Canfield, E.T.; Kwak, S.; Gold, M.C.; Lewinsohn, D.M. Human lung epithelial cells contain Mycobacterium tuberculosis in a late endosomal vacuole and are efficiently recognized by CD8(+) T cells. PLoS. ONE 2014, 9, e97515. [Google Scholar] [CrossRef] [Green Version]
- Salerno-Goncalves, R.; Wahid, R.; Sztein, M.B. Ex Vivo kinetics of early and long-term multifunctional human leukocyte antigen E-specific CD8+ cells in volunteers immunized with the Ty21a typhoid vaccine. Clin. Vaccine Immunol. 2010, 17, 1305–1314. [Google Scholar] [CrossRef] [Green Version]
- Rudolph, M.E.; McArthur, M.A.; Magder, L.S.; Barnes, R.S.; Chen, W.H.; Sztein, M.B. Age-Associated Heterogeneity of Ty21a-Induced T Cell Responses to HLA-E Restricted Salmonella Typhi Antigen Presentation. Front. Immunol. 2019, 10, 257. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fresnay, S.; McArthur, M.A.; Magder, L.; Darton, T.C.; Jones, C.; Waddington, C.S.; Blohmke, C.J.; Angus, B.; Levine, M.M.; Pollard, A.J.; et al. Salmonella Typhi-specific multifunctional CD8+ T cells play a dominant role in protection from typhoid fever in humans. J. Transl Med. 2016, 14, 62. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martinez-Barricarte, R.; Markle, J.G.; Ma, C.S.; Deenick, E.K.; Ramirez-Alejo, N.; Mele, F.; Latorre, D.; Mahdaviani, S.A.; Aytekin, C.; Mansouri, D.; et al. Human IFN-gamma immunity to mycobacteria is governed by both IL-12 and IL-23. Sci. Immunol. 2018, 3. [Google Scholar]
- Elkins, K.L.; Colombini, S.M.; Meierovics, A.I.; Chu, M.C.; Chou, A.Y.; Cowley, S.C. Survival of secondary lethal systemic Francisella LVS challenge depends largely on interferon gamma. Microbes Infect. 2010, 12, 28–36. [Google Scholar] [CrossRef]
- Wozniak, T.M.; Saunders, B.M.; Ryan, A.A.; Britton, W.J. Mycobacterium bovis BCG-specific Th17 cells confer partial protection against Mycobacterium tuberculosis infection in the absence of gamma interferon. Infect. Immun 2010, 78, 4187–4194. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Jiang, B.; Guo, Y.; Li, W.; Tian, Y.; Sonnenberg, G.F.; Weiser, J.N.; Ni, X.; Shen, H. Cross-protective mucosal immunity mediated by memory Th17 cells against Streptococcus pneumoniae lung infection. Mucosal Immunol. 2017, 10, 250–259. [Google Scholar] [CrossRef] [Green Version]
- Khader, S.A.; Pearl, J.E.; Sakamoto, K.; Gilmartin, L.; Bell, G.K.; Jelley-Gibbs, D.M.; Ghilardi, N.; deSauvage, F.; Cooper, A.M. IL-23 compensates for the absence of IL-12p70 and is essential for the IL-17 response during tuberculosis but is dispensable for protection and antigen-specific IFN-gamma responses if IL-12p70 is available. J. Immunol. 2005, 175, 788–795. [Google Scholar] [CrossRef] [Green Version]
- Cooper, A.M.; Dalton, D.K.; Stewart, T.A.; Griffin, J.P.; Russell, D.G.; Orme, I.M. Disseminated tuberculosis in interferon gamma gene-disrupted mice. J. Exp. Med. 1993, 178, 2243–2247. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meeks, K.D.; Sieve, A.N.; Kolls, J.K.; Ghilardi, N.; Berg, R.E. IL-23 is required for protection against systemic infection with Listeria monocytogenes. J. Immunol. 2009, 183, 8026–8034. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pepper, M.; Linehan, J.L.; Pagan, A.J.; Zell, T.; Dileepan, T.; Cleary, P.P.; Jenkins, M.K. Different routes of bacterial infection induce long-lived TH1 memory cells and short-lived TH17 cells. Nat. Immunol. 2010, 11, 83–89. [Google Scholar] [CrossRef] [PubMed]
- Ross, P.J.; Sutton, C.E.; Higgins, S.; Allen, A.C.; Walsh, K.; Misiak, A.; Lavelle, E.C.; McLoughlin, R.M.; Mills, K.H. Relative contribution of Th1 and Th17 cells in adaptive immunity to Bordetella pertussis: Towards the rational design of an improved acellular pertussis vaccine. PLoS. Pathog. 2013, 9, e1003264. [Google Scholar] [CrossRef] [Green Version]
- Joshi, A.; Pancari, G.; Cope, L.; Bowman, E.P.; Cua, D.; Proctor, R.A.; McNeely, T. Immunization with Staphylococcus aureus iron regulated surface determinant B (IsdB) confers protection via Th17/IL17 pathway in a murine sepsis model. Hum. Vaccin. Immunother 2012, 8, 336–346. [Google Scholar] [CrossRef] [Green Version]
- Lin, L.; Ibrahim, A.S.; Xu, X.; Farber, J.M.; Avanesian, V.; Baquir, B.; Fu, Y.; French, S.W.; Edwards, J.E., Jr.; Spellberg, B. Th1-Th17 cells mediate protective adaptive immunity against Staphylococcus aureus and Candida albicans infection in mice. PLoS. Pathog. 2009, 5, e1000703. [Google Scholar] [CrossRef]
- Brown, A.F.; Murphy, A.G.; Lalor, S.J.; Leech, J.M.; O’Keeffe, K.M.; Mac Aogain, M.; O’Halloran, D.P.; Lacey, K.A.; Tavakol, M.; Hearnden, C.H.; et al. Memory Th1 Cells Are Protective in Invasive Staphylococcus aureus Infection. PLoS. Pathog. 2015, 11, e1005226. [Google Scholar] [CrossRef]
- Johnston, R.J.; Poholek, A.C.; DiToro, D.; Yusuf, I.; Eto, D.; Barnett, B.; Dent, A.L.; Craft, J.; Crotty, S. Bcl6 and Blimp-1 are reciprocal and antagonistic regulators of T follicular helper cell differentiation. Science 2009, 325, 1006–1010. [Google Scholar] [CrossRef] [Green Version]
- Nurieva, R.I.; Chung, Y.; Martinez, G.J.; Yang, X.O.; Tanaka, S.; Matskevitch, T.D.; Wang, Y.H.; Dong, C. Bcl6 mediates the development of T follicular helper cells. Science 2009, 325, 1001–1005. [Google Scholar] [CrossRef] [Green Version]
- Yu, D.; Rao, S.; Tsai, L.M.; Lee, S.K.; He, Y.; Sutcliffe, E.L.; Srivastava, M.; Linterman, M.; Zheng, L.; Simpson, N.; et al. The transcriptional repressor Bcl-6 directs T follicular helper cell lineage commitment. Immunity 2009, 31, 457–468. [Google Scholar] [CrossRef]
- Vinuesa, C.G.; Linterman, M.A.; Yu, D.; MacLennan, I.C. Follicular Helper T Cells. Annu. Rev. Immunol. 2016, 34, 335–368. [Google Scholar] [CrossRef] [PubMed]
- Breitfeld, D.; Ohl, L.; Kremmer, E.; Ellwart, J.; Sallusto, F.; Lipp, M.; Forster, R. Follicular B helper T cells express CXC chemokine receptor 5, localize to B cell follicles, and support immunoglobulin production. J. Exp. Med. 2000, 192, 1545–1552. [Google Scholar] [CrossRef] [PubMed]
- Ansel, K.M.; Ngo, V.N.; Hyman, P.L.; Luther, S.A.; Forster, R.; Sedgwick, J.D.; Browning, J.L.; Lipp, M.; Cyster, J.G. A chemokine-driven positive feedback loop organizes lymphoid follicles. Nature 2000, 406, 309–314. [Google Scholar] [CrossRef] [PubMed]
- Hatzi, K.; Nance, J.P.; Kroenke, M.A.; Bothwell, M.; Haddad, E.K.; Melnick, A.; Crotty, S. BCL6 orchestrates Tfh cell differentiation via multiple distinct mechanisms. J. Exp. Med. 2015, 212, 539–553. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kusam, S.; Toney, L.M.; Sato, H.; Dent, A.L. Inhibition of Th2 differentiation and GATA-3 expression by BCL-6. J. Immunol. 2003, 170, 2435–2441. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schmitt, N.; Morita, R.; Bourdery, L.; Bentebibel, S.E.; Zurawski, S.M.; Banchereau, J.; Ueno, H. Human dendritic cells induce the differentiation of interleukin-21-producing T follicular helper-like cells through interleukin-12. Immunity 2009, 31, 158–169. [Google Scholar] [CrossRef] [Green Version]
- Batten, M.; Ramamoorthi, N.; Kljavin, N.M.; Ma, C.S.; Cox, J.H.; Dengler, H.S.; Danilenko, D.M.; Caplazi, P.; Wong, M.; Fulcher, D.A.; et al. IL-27 supports germinal center function by enhancing IL-21 production and the function of T follicular helper cells. J. Exp. Med. 2010, 207, 2895–2906. [Google Scholar] [CrossRef]
- Hale, J.S.; Ahmed, R. Memory T follicular helper CD4 T cells. Front. Immunol. 2015, 6, 16. [Google Scholar] [CrossRef] [Green Version]
- Brenna, E.; Davydov, A.N.; Ladell, K.; McLaren, J.E.; Bonaiuti, P.; Metsger, M.; Ramsden, J.D.; Gilbert, S.C.; Lambe, T.; Price, D.A.; et al. CD4(+) T Follicular Helper Cells in Human Tonsils and Blood Are Clonally Convergent but Divergent from Non-Tfh CD4(+) Cells. Cell Rep. 2020, 30, 137–152.e5. [Google Scholar] [CrossRef] [Green Version]
- Bai, X.; Chi, X.; Qiao, Q.; Xie, S.; Wan, S.; Ni, L.; Wang, P.; Jin, W.; Dong, C. T Follicular Helper Cells Regulate Humoral Response for Host Protection against Intestinal Citrobacter rodentium Infection. J. Immunol. 2020, 204, 2754–2761. [Google Scholar] [CrossRef] [Green Version]
- Slight, S.R.; Rangel-Moreno, J.; Gopal, R.; Lin, Y.; Fallert Junecko, B.A.; Mehra, S.; Selman, M.; Becerril-Villanueva, E.; Baquera-Heredia, J.; Pavon, L.; et al. CXCR5(+) T helper cells mediate protective immunity against tuberculosis. J. Clin. Investig. 2013, 123, 712–726. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dan, J.M.; Havenar-Daughton, C.; Kendric, K.; Al-Kolla, R.; Kaushik, K.; Rosales, S.L.; Anderson, E.L.; LaRock, C.N.; Vijayanand, P.; Seumois, G.; et al. Recurrent group A Streptococcus tonsillitis is an immunosusceptibility disease involving antibody deficiency and aberrant TFH cells. Sci. Transl. Med. 2019, 11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mackay, L.K.; Kallies, A. Transcriptional Regulation of Tissue-Resident Lymphocytes. Trends Immunol. 2017, 38, 94–103. [Google Scholar] [CrossRef] [PubMed]
- Kumar, B.V.; Ma, W.; Miron, M.; Granot, T.; Guyer, R.S.; Carpenter, D.J.; Senda, T.; Sun, X.; Ho, S.H.; Lerner, H.; et al. Human Tissue-Resident Memory T Cells Are Defined by Core Transcriptional and Functional Signatures in Lymphoid and Mucosal Sites. Cell Rep. 2017, 20, 2921–2934. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thom, J.T.; Weber, T.C.; Walton, S.M.; Torti, N.; Oxenius, A. The Salivary Gland Acts as a Sink for Tissue-Resident Memory CD8(+) T Cells, Facilitating Protection from Local Cytomegalovirus Infection. Cell Rep. 2015, 13, 1125–1136. [Google Scholar] [CrossRef] [Green Version]
- Collins, N.; Jiang, X.; Zaid, A.; Macleod, B.L.; Li, J.; Park, C.O.; Haque, A.; Bedoui, S.; Heath, W.R.; Mueller, S.N.; et al. Skin CD4(+) memory T cells exhibit combined cluster-mediated retention and equilibration with the circulation. Nat. Commun 2016, 7, 11514. [Google Scholar] [CrossRef]
- Gebhardt, T.; Whitney, P.G.; Zaid, A.; Mackay, L.K.; Brooks, A.G.; Heath, W.R.; Carbone, F.R.; Mueller, S.N. Different patterns of peripheral migration by memory CD4+ and CD8+ T cells. Nature 2011, 477, 216–219. [Google Scholar] [CrossRef]
- Teijaro, J.R.; Turner, D.; Pham, Q.; Wherry, E.J.; Lefrancois, L.; Farber, D.L. Cutting edge: Tissue-retentive lung memory CD4 T cells mediate optimal protection to respiratory virus infection. J. Immunol. 2011, 187, 5510–5514. [Google Scholar] [CrossRef] [Green Version]
- Sakai, S.; Kauffman, K.D.; Schenkel, J.M.; McBerry, C.C.; Mayer-Barber, K.D.; Masopust, D.; Barber, D.L. Cutting edge: Control of Mycobacterium tuberculosis infection by a subset of lung parenchyma-homing CD4 T cells. J. Immunol. 2014, 192, 2965–2969. [Google Scholar] [CrossRef] [Green Version]
- Wilk, M.M.; Misiak, A.; McManus, R.M.; Allen, A.C.; Lynch, M.A.; Mills, K.H.G. Lung CD4 Tissue-Resident Memory T Cells Mediate Adaptive Immunity Induced by Previous Infection of Mice with Bordetella pertussis. J. Immunol. 2017, 199, 233–243. [Google Scholar] [CrossRef] [Green Version]
- Smith, N.M.; Wasserman, G.A.; Coleman, F.T.; Hilliard, K.L.; Yamamoto, K.; Lipsitz, E.; Malley, R.; Dooms, H.; Jones, M.R.; Quinton, L.J.; et al. Regionally compartmentalized resident memory T cells mediate naturally acquired protection against pneumococcal pneumonia. Mucosal Immunol. 2018, 11, 220–235. [Google Scholar] [CrossRef] [PubMed]
- Romagnoli, P.A.; Fu, H.H.; Qiu, Z.; Khairallah, C.; Pham, Q.M.; Puddington, L.; Khanna, K.M.; Lefrancois, L.; Sheridan, B.S. Differentiation of distinct long-lived memory CD4 T cells in intestinal tissues after oral Listeria monocytogenes infection. Mucosal Immunol. 2017, 10, 520–530. [Google Scholar] [CrossRef] [PubMed]
- Bishu, S.; Hou, G.; El Zaatari, M.; Bishu, S.R.; Popke, D.; Zhang, M.; Grasberger, H.; Zou, W.; Stidham, R.W.; Higgins, P.D.R.; et al. Citrobacter rodentium Induces Tissue-Resident Memory CD4(+) T Cells. Infect. Immun. 2019, 87, e00295-19. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Johnson, R.M.; Yu, H.; Strank, N.O.; Karunakaran, K.; Zhu, Y.; Brunham, R.C. B Cell Presentation of Chlamydia Antigen Selects Out Protective CD4gamma13 T Cells: Implications for Genital Tract Tissue-Resident Memory Lymphocyte Clusters. Infect. Immun 2018, 86. [Google Scholar] [CrossRef] [Green Version]
- Schreiner, D.; King, C.G. CD4+ Memory T Cells at Home in the Tissue: Mechanisms for Health and Disease. Front. Immunol. 2018, 9, 2394. [Google Scholar] [CrossRef] [Green Version]
- Park, C.O.; Fu, X.; Jiang, X.; Pan, Y.; Teague, J.E.; Collins, N.; Tian, T.; O’Malley, J.T.; Emerson, R.O.; Kim, J.H.; et al. Staged development of long-lived T-cell receptor alphabeta TH17 resident memory T-cell population to Candida albicans after skin infection. J. Allergy Clin. Immunol. 2018, 142, 647–662. [Google Scholar] [CrossRef] [Green Version]
- Calabi, F.; Jarvis, J.M.; Martin, L.; Milstein, C. Two classes of CD1 genes. Eur. J. Immunol. 1989, 19, 285–292. [Google Scholar] [CrossRef]
- Porcelli, S.; Brenner, M.B.; Greenstein, J.L.; Balk, S.P.; Terhorst, C.; Bleicher, P.A. Recognition of cluster of differentiation 1 antigens by human CD4-CD8-cytolytic T lymphocytes. Nature 1989, 341, 447–450. [Google Scholar] [CrossRef]
- Porcelli, S.; Morita, C.T.; Brenner, M.B. CD1b restricts the response of human CD4-8- T lymphocytes to a microbial antigen. Nature 1992, 360, 593–597. [Google Scholar] [CrossRef]
- Girardi, E.; Maricic, I.; Wang, J.; Mac, T.T.; Iyer, P.; Kumar, V.; Zajonc, D.M. Type II natural killer T cells use features of both innate-like and conventional T cells to recognize sulfatide self antigens. Nat. Immunol. 2012, 13, 851–856. [Google Scholar] [CrossRef] [Green Version]
- Patel, O.; Pellicci, D.G.; Gras, S.; Sandoval-Romero, M.L.; Uldrich, A.P.; Mallevaey, T.; Clarke, A.J.; Le Nours, J.; Theodossis, A.; Cardell, S.L.; et al. Recognition of CD1d-sulfatide mediated by a type II natural killer T cell antigen receptor. Nat. Immunol. 2012, 13, 857–863. [Google Scholar] [CrossRef]
- Pellicci, D.G.; Uldrich, A.P.; Le Nours, J.; Ross, F.; Chabrol, E.; Eckle, S.B.; de Boer, R.; Lim, R.T.; McPherson, K.; Besra, G.; et al. The molecular bases of delta/alphabeta T cell-mediated antigen recognition. J. Exp. Med. 2014, 211, 2599–2615. [Google Scholar] [CrossRef]
- Gras, S.; Van Rhijn, I.; Shahine, A.; Le Nours, J. Molecular recognition of microbial lipid-based antigens by T cells. Cell Mol. Life Sci. 2018, 75, 1623–1639. [Google Scholar] [CrossRef]
- Kawano, T.; Cui, J.; Koezuka, Y.; Toura, I.; Kaneko, Y.; Motoki, K.; Ueno, H.; Nakagawa, R.; Sato, H.; Kondo, E.; et al. CD1d-restricted and TCR-mediated activation of valpha14 NKT cells by glycosylceramides. Science 1997, 278, 1626–1629. [Google Scholar] [CrossRef] [PubMed]
- Fischer, K.; Scotet, E.; Niemeyer, M.; Koebernick, H.; Zerrahn, J.; Maillet, S.; Hurwitz, R.; Kursar, M.; Bonneville, M.; Kaufmann, S.H.; et al. Mycobacterial phosphatidylinositol mannoside is a natural antigen for CD1d-restricted T cells. Proc. Natl. Acad. Sci. USA 2004, 101, 10685–10690. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mattner, J.; Debord, K.L.; Ismail, N.; Goff, R.D.; Cantu, C., 3rd; Zhou, D.; Saint-Mezard, P.; Wang, V.; Gao, Y.; Yin, N.; et al. Exogenous and endogenous glycolipid antigens activate NKT cells during microbial infections. Nature 2005, 434, 525–529. [Google Scholar] [CrossRef] [PubMed]
- Olson, C.M., Jr.; Bates, T.C.; Izadi, H.; Radolf, J.D.; Huber, S.A.; Boyson, J.E.; Anguita, J. Local production of IFN-gamma by invariant NKT cells modulates acute Lyme carditis. J. Immunol. 2009, 182, 3728–3734. [Google Scholar] [CrossRef] [PubMed]
- Kinjo, Y.; Illarionov, P.; Vela, J.L.; Pei, B.; Girardi, E.; Li, X.; Li, Y.; Imamura, M.; Kaneko, Y.; Okawara, A.; et al. Invariant natural killer T cells recognize glycolipids from pathogenic Gram-positive bacteria. Nat. Immunol. 2011, 12, 966–974. [Google Scholar] [CrossRef]
- Tatituri, R.V.; Watts, G.F.; Bhowruth, V.; Barton, N.; Rothchild, A.; Hsu, F.F.; Almeida, C.F.; Cox, L.R.; Eggeling, L.; Cardell, S.; et al. Recognition of microbial and mammalian phospholipid antigens by NKT cells with diverse TCRs. Proc. Natl. Acad. Sci. USA 2013, 110, 1827–1832. [Google Scholar] [CrossRef] [Green Version]
- Kasmar, A.G.; Van Rhijn, I.; Magalhaes, K.G.; Young, D.C.; Cheng, T.Y.; Turner, M.T.; Schiefner, A.; Kalathur, R.C.; Wilson, I.A.; Bhati, M.; et al. Cutting Edge: CD1a tetramers and dextramers identify human lipopeptide-specific T cells ex vivo. J. Immunol. 2013, 191, 4499–4503. [Google Scholar] [CrossRef] [Green Version]
- Kasmar, A.G.; van Rhijn, I.; Cheng, T.Y.; Turner, M.; Seshadri, C.; Schiefner, A.; Kalathur, R.C.; Annand, J.W.; de Jong, A.; Shires, J.; et al. CD1b tetramers bind alphabeta T cell receptors to identify a mycobacterial glycolipid-reactive T cell repertoire in humans. J. Exp. Med. 2011, 208, 1741–1747. [Google Scholar] [CrossRef] [PubMed]
- Ly, D.; Kasmar, A.G.; Cheng, T.Y.; de Jong, A.; Huang, S.; Roy, S.; Bhatt, A.; van Summeren, R.P.; Altman, J.D.; Jacobs, W.R.; et al. CD1c tetramers detect ex vivo T cell responses to processed phosphomycoketide antigens. J. Exp. Med. 2013, 210, 729–741. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van Rhijn, I.; Kasmar, A.; de Jong, A.; Gras, S.; Bhati, M.; Doorenspleet, M.E.; de Vries, N.; Godfrey, D.I.; Altman, J.D.; de Jager, W.; et al. A conserved human T cell population targets mycobacterial antigens presented by CD1b. Nat. Immunol. 2013, 14, 706–713. [Google Scholar] [CrossRef]
- Roy, S.; Ly, D.; Li, N.S.; Altman, J.D.; Piccirilli, J.A.; Moody, D.B.; Adams, E.J. Molecular basis of mycobacterial lipid antigen presentation by CD1c and its recognition by alphabeta T cells. Proc. Natl. Acad. Sci. USA 2014, 111, E4648–E4657. [Google Scholar] [CrossRef] [Green Version]
- Zhao, J.; Siddiqui, S.; Shang, S.; Bian, Y.; Bagchi, S.; He, Y.; Wang, C.R. Mycolic acid-specific T cells protect against Mycobacterium tuberculosis infection in a humanized transgenic mouse model. Elife 2015, 4, e08525. [Google Scholar] [CrossRef]
- Montamat-Sicotte, D.J.; Millington, K.A.; Willcox, C.R.; Hingley-Wilson, S.; Hackforth, S.; Innes, J.; Kon, O.M.; Lammas, D.A.; Minnikin, D.E.; Besra, G.S.; et al. A mycolic acid-specific CD1-restricted T cell population contributes to acute and memory immune responses in human tuberculosis infection. J. Clin. Investig. 2011, 121, 2493–2503. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reinink, P.; Souter, M.N.T.; Cheng, T.Y.; van Gorkom, T.; Lenz, S.; Kubler-Kielb, J.; Strle, K.; Kremer, K.; Thijsen, S.F.T.; Steere, A.C.; et al. CD1b presents self and Borrelia burgdorferi diacylglycerols to human T cells. Eur. J. Immunol. 2019, 49, 737–746. [Google Scholar] [CrossRef] [Green Version]
- Van Rhijn, I.; van Berlo, T.; Hilmenyuk, T.; Cheng, T.Y.; Wolf, B.J.; Tatituri, R.V.; Uldrich, A.P.; Napolitani, G.; Cerundolo, V.; Altman, J.D.; et al. Human autoreactive T cells recognize CD1b and phospholipids. Proc. Natl. Acad. Sci. USA 2016, 113, 380–385. [Google Scholar] [CrossRef] [Green Version]
- Visvabharathy, L.; Genardi, S.; Cao, L.; He, Y.; Alonzo, F., 3rd; Berdyshev, E.; Wang, C.R. Group 1 CD1-restricted T cells contribute to control of systemic Staphylococcus aureus infection. PLoS. Pathog. 2020, 16, e1008443. [Google Scholar] [CrossRef]
- Adams, E.J.; Gu, S.; Luoma, A.M. Human gamma delta T cells: Evolution and ligand recognition. Cell Immunol. 2015, 296, 31–40. [Google Scholar] [CrossRef] [Green Version]
- Khairallah, C.; Chu, T.H.; Sheridan, B.S. Tissue Adaptations of Memory and Tissue-Resident Gamma Delta T Cells. Front. Immunol. 2018, 9, 2636. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; He, Y.; Li, H.; Ai, Q.; Yu, J. The microbiota protects against Pseudomonas aeruginosa pneumonia via gammadelta T cell-neutrophil axis in mice. Microbes Infect. 2020. [Google Scholar] [CrossRef] [PubMed]
- Chennupati, V.; Worbs, T.; Liu, X.; Malinarich, F.H.; Schmitz, S.; Haas, J.D.; Malissen, B.; Forster, R.; Prinz, I. Intra- and intercompartmental movement of gammadelta T cells: Intestinal intraepithelial and peripheral gammadelta T cells represent exclusive nonoverlapping populations with distinct migration characteristics. J. Immunol. 2010, 185, 5160–5168. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Davey, M.S.; Willcox, C.R.; Hunter, S.; Kasatskaya, S.A.; Remmerswaal, E.B.M.; Salim, M.; Mohammed, F.; Bemelman, F.J.; Chudakov, D.M.; Oo, Y.H.; et al. The human Vdelta2(+) T-cell compartment comprises distinct innate-like Vgamma9(+) and adaptive Vgamma9(-) subsets. Nat. Commun. 2018, 9, 1760. [Google Scholar] [CrossRef] [PubMed]
- Karunakaran, M.M.; Gobel, T.W.; Starick, L.; Walter, L.; Herrmann, T. Vgamma9 and Vdelta2 T cell antigen receptor genes and butyrophilin 3 (BTN3) emerged with placental mammals and are concomitantly preserved in selected species like alpaca (Vicugna pacos). Immunogenetics 2014, 66, 243–254. [Google Scholar] [CrossRef] [PubMed]
- Dimova, T.; Brouwer, M.; Gosselin, F.; Tassignon, J.; Leo, O.; Donner, C.; Marchant, A.; Vermijlen, D. Effector Vgamma9Vdelta2 T cells dominate the human fetal gammadelta T-cell repertoire. Proc. Natl. Acad. Sci. USA 2015, 112, E556–E565. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tanaka, Y.; Sano, S.; Nieves, E.; De Libero, G.; Rosa, D.; Modlin, R.L.; Brenner, M.B.; Bloom, B.R.; Morita, C.T. Nonpeptide ligands for human gamma delta T cells. Proc. Natl. Acad. Sci. USA 1994, 91, 8175–8179. [Google Scholar] [CrossRef] [Green Version]
- Morita, C.T.; Jin, C.; Sarikonda, G.; Wang, H. Nonpeptide antigens, presentation mechanisms, and immunological memory of human Vgamma2Vdelta2 T cells: Discriminating friend from foe through the recognition of prenyl pyrophosphate antigens. Immunol. Rev. 2007, 215, 59–76. [Google Scholar] [CrossRef]
- Hintz, M.; Reichenberg, A.; Altincicek, B.; Bahr, U.; Gschwind, R.M.; Kollas, A.K.; Beck, E.; Wiesner, J.; Eberl, M.; Jomaa, H. Identification of (E)-4-hydroxy-3-methyl-but-2-enyl pyrophosphate as a major activator for human gammadelta T cells in Escherichia coli. Febs. Lett. 2001, 509, 317–322. [Google Scholar] [CrossRef] [Green Version]
- Rhodes, D.A.; Chen, H.C.; Price, A.J.; Keeble, A.H.; Davey, M.S.; James, L.C.; Eberl, M.; Trowsdale, J. Activation of human gammadelta T cells by cytosolic interactions of BTN3A1 with soluble phosphoantigens and the cytoskeletal adaptor periplakin. J. Immunol. 2015, 194, 2390–2398. [Google Scholar] [CrossRef] [Green Version]
- Sandstrom, A.; Peigne, C.M.; Leger, A.; Crooks, J.E.; Konczak, F.; Gesnel, M.C.; Breathnach, R.; Bonneville, M.; Scotet, E.; Adams, E.J. The intracellular B30.2 domain of butyrophilin 3A1 binds phosphoantigens to mediate activation of human Vgamma9Vdelta2 T cells. Immunity 2014, 40, 490–500. [Google Scholar] [CrossRef] [Green Version]
- Vavassori, S.; Kumar, A.; Wan, G.S.; Ramanjaneyulu, G.S.; Cavallari, M.; El Daker, S.; Beddoe, T.; Theodossis, A.; Williams, N.K.; Gostick, E.; et al. Butyrophilin 3A1 binds phosphorylated antigens and stimulates human gammadelta T cells. Nat. Immunol. 2013, 14, 908–916. [Google Scholar] [CrossRef] [PubMed]
- Vantourout, P.; Laing, A.; Woodward, M.J.; Zlatareva, I.; Apolonia, L.; Jones, A.W.; Snijders, A.P.; Malim, M.H.; Hayday, A.C. Heteromeric interactions regulate butyrophilin (BTN) and BTN-like molecules governing gammadelta T cell biology. Proc. Natl. Acad. Sci. USA 2018, 115, 1039–1044. [Google Scholar] [CrossRef] [Green Version]
- Karunakaran, M.M.; Willcox, C.R.; Salim, M.; Paletta, D.; Fichtner, A.S.; Noll, A.; Starick, L.; Nohren, A.; Begley, C.R.; Berwick, K.A.; et al. Butyrophilin-2A1 Directly Binds Germline-Encoded Regions of the Vgamma9Vdelta2 TCR and Is Essential for Phosphoantigen Sensing. Immunity 2020, 52, 487–498.e6. [Google Scholar] [CrossRef] [PubMed]
- Rigau, M.; Ostrouska, S.; Fulford, T.S.; Johnson, D.N.; Woods, K.; Ruan, Z.; McWilliam, H.E.G.; Hudson, C.; Tutuka, C.; Wheatley, A.K.; et al. Butyrophilin 2A1 is essential for phosphoantigen reactivity by gammadelta T cells. Science 2020, 367, eaay5516. [Google Scholar] [CrossRef] [PubMed]
- Davey, M.S.; Willcox, C.R.; Joyce, S.P.; Ladell, K.; Kasatskaya, S.A.; McLaren, J.E.; Hunter, S.; Salim, M.; Mohammed, F.; Price, D.A.; et al. Clonal selection in the human Vdelta1 T cell repertoire indicates gammadelta TCR-dependent adaptive immune surveillance. Nat. Commun. 2017, 8, 14760. [Google Scholar] [CrossRef]
- Deusch, K.; Luling, F.; Reich, K.; Classen, M.; Wagner, H.; Pfeffer, K. A major fraction of human intraepithelial lymphocytes simultaneously expresses the gamma/delta T cell receptor, the CD8 accessory molecule and preferentially uses the V delta 1 gene segment. Eur. J. Immunol. 1991, 21, 1053–1059. [Google Scholar] [CrossRef]
- Di Marco Barros, R.; Roberts, N.A.; Dart, R.J.; Vantourout, P.; Jandke, A.; Nussbaumer, O.; Deban, L.; Cipolat, S.; Hart, R.; Iannitto, M.L.; et al. Epithelia Use Butyrophilin-like Molecules to Shape Organ-Specific gammadelta T Cell Compartments. Cell 2016, 167, 203–218.e17. [Google Scholar] [CrossRef] [Green Version]
- Willcox, C.R.; Vantourout, P.; Salim, M.; Zlatareva, I.; Melandri, D.; Zanardo, L.; George, R.; Kjaer, S.; Jeeves, M.; Mohammed, F.; et al. Butyrophilin-like 3 Directly Binds a Human Vgamma4(+) T Cell Receptor Using a Modality Distinct from Clonally-Restricted Antigen. Immunity 2019, 51, 813–825.e4. [Google Scholar] [CrossRef] [Green Version]
- Godfrey, D.I.; Koay, H.F.; McCluskey, J.; Gherardin, N.A. The biology and functional importance of MAIT cells. Nat. Immunol. 2019, 20, 1110–1128. [Google Scholar] [CrossRef]
- Porcelli, S.; Yockey, C.E.; Brenner, M.B.; Balk, S.P. Analysis of T cell antigen receptor (TCR) expression by human peripheral blood CD4-8- alpha/beta T cells demonstrates preferential use of several V beta genes and an invariant TCR alpha chain. J. Exp. Med. 1993, 178, 1–16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tilloy, F.; Treiner, E.; Park, S.H.; Garcia, C.; Lemonnier, F.; de la Salle, H.; Bendelac, A.; Bonneville, M.; Lantz, O. An invariant T cell receptor alpha chain defines a novel TAP-independent major histocompatibility complex class Ib-restricted alpha/beta T cell subpopulation in mammals. J. Exp. Med. 1999, 189, 1907–1921. [Google Scholar] [CrossRef] [PubMed]
- Treiner, E.; Duban, L.; Bahram, S.; Radosavljevic, M.; Wanner, V.; Tilloy, F.; Affaticati, P.; Gilfillan, S.; Lantz, O. Selection of evolutionarily conserved mucosal-associated invariant T cells by MR1. Nature 2003, 422, 164–169. [Google Scholar] [CrossRef] [PubMed]
- Martin, E.; Treiner, E.; Duban, L.; Guerri, L.; Laude, H.; Toly, C.; Premel, V.; Devys, A.; Moura, I.C.; Tilloy, F.; et al. Stepwise development of MAIT cells in mouse and human. PLoS. Biol. 2009, 7, e54. [Google Scholar] [CrossRef] [PubMed]
- Gold, M.C.; Cerri, S.; Smyk-Pearson, S.; Cansler, M.E.; Vogt, T.M.; Delepine, J.; Winata, E.; Swarbrick, G.M.; Chua, W.J.; Yu, Y.Y.; et al. Human mucosal associated invariant T cells detect bacterially infected cells. PLoS. Biol. 2010, 8, e1000407. [Google Scholar] [CrossRef] [Green Version]
- Le Bourhis, L.; Martin, E.; Peguillet, I.; Guihot, A.; Froux, N.; Core, M.; Levy, E.; Dusseaux, M.; Meyssonnier, V.; Premel, V.; et al. Antimicrobial activity of mucosal-associated invariant T cells. Nat. Immunol. 2010, 11, 701–708. [Google Scholar] [CrossRef] [Green Version]
- Corbett, A.J.; Eckle, S.B.; Birkinshaw, R.W.; Liu, L.; Patel, O.; Mahony, J.; Chen, Z.; Reantragoon, R.; Meehan, B.; Cao, H.; et al. T-cell activation by transitory neo-antigens derived from distinct microbial pathways. Nature 2014, 509, 361–365. [Google Scholar] [CrossRef]
- Harriff, M.J.; McMurtrey, C.; Froyd, C.A.; Jin, H.; Cansler, M.; Null, M.; Worley, A.; Meermeier, E.W.; Swarbrick, G.; Nilsen, A.; et al. MR1 displays the microbial metabolome driving selective MR1-restricted T cell receptor usage. Sci. Immunol. 2018, 3, eaao2556. [Google Scholar] [CrossRef]
- Meermeier, E.W.; Laugel, B.F.; Sewell, A.K.; Corbett, A.J.; Rossjohn, J.; McCluskey, J.; Harriff, M.J.; Franks, T.; Gold, M.C.; Lewinsohn, D.M. Human TRAV1-2-negative MR1-restricted T cells detect S. pyogenes and alternatives to MAIT riboflavin-based antigens. Nat. Commun. 2016, 7, 12506. [Google Scholar] [CrossRef]
- Keller, A.N.; Eckle, S.B.; Xu, W.; Liu, L.; Hughes, V.A.; Mak, J.Y.; Meehan, B.S.; Pediongco, T.; Birkinshaw, R.W.; Chen, Z.; et al. Drugs and drug-like molecules can modulate the function of mucosal-associated invariant T cells. Nat. Immunol. 2017, 18, 402–411. [Google Scholar] [CrossRef]
- Rahimpour, A.; Koay, H.F.; Enders, A.; Clanchy, R.; Eckle, S.B.; Meehan, B.; Chen, Z.; Whittle, B.; Liu, L.; Fairlie, D.P.; et al. Identification of phenotypically and functionally heterogeneous mouse mucosal-associated invariant T cells using MR1 tetramers. J. Exp. Med. 2015, 212, 1095–1108. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reantragoon, R.; Corbett, A.J.; Sakala, I.G.; Gherardin, N.A.; Furness, J.B.; Chen, Z.; Eckle, S.B.; Uldrich, A.P.; Birkinshaw, R.W.; Patel, O.; et al. Antigen-loaded MR1 tetramers define T cell receptor heterogeneity in mucosal-associated invariant T cells. J. Exp. Med. 2013, 210, 2305–2320. [Google Scholar] [CrossRef] [PubMed]
- Koay, H.F.; Gherardin, N.A.; Xu, C.; Seneviratna, R.; Zhao, Z.; Chen, Z.; Fairlie, D.P.; McCluskey, J.; Pellicci, D.G.; Uldrich, A.P.; et al. Diverse MR1-restricted T cells in mice and humans. Nat. Commun. 2019, 10, 2243. [Google Scholar] [CrossRef] [Green Version]
- Dusseaux, M.; Martin, E.; Serriari, N.; Peguillet, I.; Premel, V.; Louis, D.; Milder, M.; Le Bourhis, L.; Soudais, C.; Treiner, E.; et al. Human MAIT cells are xenobiotic-resistant, tissue-targeted, CD161hi IL-17-secreting T cells. Blood 2011, 117, 1250–1259. [Google Scholar] [CrossRef]
- Ussher, J.E.; Bilton, M.; Attwod, E.; Shadwell, J.; Richardson, R.; de Lara, C.; Mettke, E.; Kurioka, A.; Hansen, T.H.; Klenerman, P.; et al. CD161++ CD8+ T cells, including the MAIT cell subset, are specifically activated by IL-12+IL-18 in a TCR-independent manner. Eur. J. Immunol. 2014, 44, 195–203. [Google Scholar] [CrossRef]
- van Wilgenburg, B.; Scherwitzl, I.; Hutchinson, E.C.; Leng, T.; Kurioka, A.; Kulicke, C.; de Lara, C.; Cole, S.; Vasanawathana, S.; Limpitikul, W.; et al. MAIT cells are activated during human viral infections. Nat. Commun. 2016, 7, 11653. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, H.; Kjer-Nielsen, L.; Shi, M.; D’Souza, C.; Pediongco, T.J.; Cao, H.; Kostenko, L.; Lim, X.Y.; Eckle, S.B.G.; Meehan, B.S.; et al. IL-23 costimulates antigen-specific MAIT cell activation and enables vaccination against bacterial infection. Sci. Immunol. 2019, 4, eaaw0402. [Google Scholar] [CrossRef]
- Koay, H.F.; Gherardin, N.A.; Enders, A.; Loh, L.; Mackay, L.K.; Almeida, C.F.; Russ, B.E.; Nold-Petry, C.A.; Nold, M.F.; Bedoui, S.; et al. A three-stage intrathymic development pathway for the mucosal-associated invariant T cell lineage. Nat. Immunol. 2016, 17, 1300–1311. [Google Scholar] [CrossRef]
- Legoux, F.; Bellet, D.; Daviaud, C.; El Morr, Y.; Darbois, A.; Niort, K.; Procopio, E.; Salou, M.; Gilet, J.; Ryffel, B.; et al. Microbial metabolites control the thymic development of mucosal-associated invariant T cells. Science 2019, 366, 494–499. [Google Scholar] [CrossRef]
- Constantinides, M.G.; Link, V.M.; Tamoutounour, S.; Wong, A.C.; Perez-Chaparro, P.J.; Han, S.J.; Chen, Y.E.; Li, K.; Farhat, S.; Weckel, A.; et al. MAIT cells are imprinted by the microbiota in early life and promote tissue repair. Science 2019, 366, eaax6624. [Google Scholar] [CrossRef]
- D’Souza, C.; Pediongco, T.; Wang, H.; Scheerlinck, J.Y.; Kostenko, L.; Esterbauer, R.; Stent, A.W.; Eckle, S.B.G.; Meehan, B.S.; Strugnell, R.A.; et al. Mucosal-Associated Invariant T Cells Augment Immunopathology and Gastritis in Chronic Helicobacter pylori Infection. J. Immunol. 2018, 200, 1901–1916. [Google Scholar] [PubMed] [Green Version]
- Ankley, L.; Thomas, S.; Olive, A.J. Fighting persistence; how chronic infections with Mycobacterium tuberculosis evade T cell-mediated clearance and new strategies to defeat them. Infect. Immun. 2020, 88. [Google Scholar] [CrossRef] [PubMed]
- Hingley-Wilson, S.M.; Sambandamurthy, V.K.; Jacobs, W.R., Jr. Survival perspectives from the world’s most successful pathogen, Mycobacterium tuberculosis. Nat. Immunol. 2003, 4, 949–955. [Google Scholar] [CrossRef] [PubMed]
- Miller, J.D.; van der Most, R.G.; Akondy, R.S.; Glidewell, J.T.; Albott, S.; Masopust, D.; Murali-Krishna, K.; Mahar, P.L.; Edupuganti, S.; Lalor, S.; et al. Human effector and memory CD8+ T cell responses to smallpox and yellow fever vaccines. Immunity 2008, 28, 710–722. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lawrence, C.W.; Braciale, T.J. Activation, differentiation, and migration of naive virus-specific CD8+ T cells during pulmonary influenza virus infection. J. Immunol. 2004, 173, 1209–1218. [Google Scholar] [CrossRef] [Green Version]
- Zhai, W.; Wu, F.; Zhang, Y.; Fu, Y.; Liu, Z. The Immune Escape Mechanisms of Mycobacterium Tuberculosis. Int. J. Mol. Sci. 2019, 20, 340. [Google Scholar] [CrossRef] [Green Version]
- Chen, Z.; Wang, T.; Liu, Z.; Zhang, G.; Wang, J.; Feng, S.; Liang, J. Inhibition of Autophagy by MiR-30A Induced by Mycobacteria tuberculosis as a Possible Mechanism of Immune Escape in Human Macrophages. Jpn. J. Infect. Dis. 2015, 68, 420–424. [Google Scholar] [CrossRef] [Green Version]
- Briken, V.; Porcelli, S.A.; Besra, G.S.; Kremer, L. Mycobacterial lipoarabinomannan and related lipoglycans: From biogenesis to modulation of the immune response. Mol. Microbiol. 2004, 53, 391–403. [Google Scholar] [CrossRef]
- Chen, M.; Divangahi, M.; Gan, H.; Shin, D.S.; Hong, S.; Lee, D.M.; Serhan, C.N.; Behar, S.M.; Remold, H.G. Lipid mediators in innate immunity against tuberculosis: Opposing roles of PGE2 and LXA4 in the induction of macrophage death. J. Exp. Med. 2008, 205, 2791–2801. [Google Scholar] [CrossRef] [Green Version]
- Urdahl, K.B.; Shafiani, S.; Ernst, J.D. Initiation and regulation of T-cell responses in tuberculosis. Mucosal Immunol. 2011, 4, 288–293. [Google Scholar] [CrossRef]
- Moguche, A.O.; Musvosvi, M.; Penn-Nicholson, A.; Plumlee, C.R.; Mearns, H.; Geldenhuys, H.; Smit, E.; Abrahams, D.; Rozot, V.; Dintwe, O.; et al. Antigen Availability Shapes T Cell Differentiation and Function during Tuberculosis. Cell Host Microbe 2017, 21, 695–706.e5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bold, T.D.; Banaei, N.; Wolf, A.J.; Ernst, J.D. Suboptimal activation of antigen-specific CD4+ effector cells enables persistence of M. tuberculosis in vivo. PLoS. Pathog. 2011, 7, e1002063. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Egen, J.G.; Rothfuchs, A.G.; Feng, C.G.; Horwitz, M.A.; Sher, A.; Germain, R.N. Intravital imaging reveals limited antigen presentation and T cell effector function in mycobacterial granulomas. Immunity 2011, 34, 807–819. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Raghuvanshi, S.; Sharma, P.; Singh, S.; Van Kaer, L.; Das, G. Mycobacterium tuberculosis evades host immunity by recruiting mesenchymal stem cells. Proc. Natl. Acad. Sci. USA 2010, 107, 21653–21658. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, X.; Zhou, B.; Li, M.; Deng, Q.; Wu, X.; Le, X.; Wu, C.; Larmonier, N.; Zhang, W.; Zhang, H.; et al. CD4(+)CD25(+)FoxP3(+) regulatory T cells suppress Mycobacterium tuberculosis immunity in patients with active disease. Clin. Immunol. 2007, 123, 50–59. [Google Scholar] [CrossRef]
- Shafiani, S.; Tucker-Heard, G.; Kariyone, A.; Takatsu, K.; Urdahl, K.B. Pathogen-specific regulatory T cells delay the arrival of effector T cells in the lung during early tuberculosis. J. Exp. Med. 2010, 207, 1409–1420. [Google Scholar] [CrossRef] [Green Version]
- Comas, I.; Chakravartti, J.; Small, P.M.; Galagan, J.; Niemann, S.; Kremer, K.; Ernst, J.D.; Gagneux, S. Human T cell epitopes of Mycobacterium tuberculosis are evolutionarily hyperconserved. Nat. Genet. 2010, 42, 498–503. [Google Scholar] [CrossRef] [Green Version]
- Ernst, J.D. Mechanisms of M. tuberculosis Immune Evasion as Challenges to TB Vaccine Design. Cell Host Microbe 2018, 24, 34–42. [Google Scholar] [CrossRef] [Green Version]
- Pickar-Oliver, A.; Gersbach, C.A. The next generation of CRISPR-Cas technologies and applications. Nat. Rev. Mol. Cell Biol. 2019, 20, 490–507. [Google Scholar] [CrossRef]
- Coburn, B.; Sekirov, I.; Finlay, B.B. Type III secretion systems and disease. Clin. Microbiol. Rev. 2007, 20, 535–549. [Google Scholar] [CrossRef] [Green Version]
- Deng, W.; Marshall, N.C.; Rowland, J.L.; McCoy, J.M.; Worrall, L.J.; Santos, A.S.; Strynadka, N.C.J.; Finlay, B.B. Assembly, structure, function and regulation of type III secretion systems. Nat. Rev. Microbiol. 2017, 15, 323–337. [Google Scholar] [CrossRef] [PubMed]
- Sansonetti, P.J.; Di Santo, J.P. Debugging how bacteria manipulate the immune response. Immunity 2007, 26, 149–161. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cornelis, G.R. The type III secretion injectisome. Nat. Rev. Microbiol. 2006, 4, 811–825. [Google Scholar] [CrossRef]
- Ashida, H.; Ogawa, M.; Mimuro, H.; Sasakawa, C. Shigella Infection of Intestinal Epithelium and Circumvention of the Host Innate Defense System. In Molecular Mechanisms of Bacterial Infection via the Gut; Sasakawa, C., Ed.; Springer: Berlin/Heidelberg, Germany, 2009; pp. 231–255. [Google Scholar]
- Hilbi, H.; Moss, J.E.; Hersh, D.; Chen, Y.; Arondel, J.; Banerjee, S.; Flavell, R.A.; Yuan, J.; Sansonetti, P.J.; Zychlinsky, A. Shigella-induced apoptosis is dependent on caspase-1 which binds to IpaB. J. Biol. Chem. 1998, 273, 32895–32900. [Google Scholar] [CrossRef] [Green Version]
- Senerovic, L.; Tsunoda, S.P.; Goosmann, C.; Brinkmann, V.; Zychlinsky, A.; Meissner, F.; Kolbe, M. Spontaneous formation of IpaB ion channels in host cell membranes reveals how Shigella induces pyroptosis in macrophages. Cell Death Dis. 2012, 3, e384. [Google Scholar] [CrossRef] [PubMed]
- Ashida, H.; Kim, M.; Sasakawa, C. Manipulation of the host cell death pathway by Shigella. Cell Microbiol. 2014, 16, 1757–1766. [Google Scholar] [CrossRef]
- Phalipon, A.; Sansonetti, P.J. Shigella’s ways of manipulating the host intestinal innate and adaptive immune system: A tool box for survival? Immunol. Cell Biol. 2007, 85, 119–129. [Google Scholar] [CrossRef]
- Islam, D.; Bandholtz, L.; Nilsson, J.; Wigzell, H.; Christensson, B.; Agerberth, B.; Gudmundsson, G. Downregulation of bactericidal peptides in enteric infections: A novel immune escape mechanism with bacterial DNA as a potential regulator. Nat. Med. 2001, 7, 180–185. [Google Scholar] [CrossRef]
- Cerny, O.; Holden, D.W. Salmonella SPI-2 type III secretion system-dependent inhibition of antigen presentation and T cell function. Immunol. Lett. 2019, 215, 35–39. [Google Scholar] [CrossRef]
- Carden, S.E.; Walker, G.T.; Honeycutt, J.; Lugo, K.; Pham, T.; Jacobson, A.; Bouley, D.; Idoyaga, J.; Tsolis, R.M.; Monack, D. Pseudogenization of the Secreted Effector Gene sseI Confers Rapid Systemic Dissemination of S. Typhimurium ST313 within Migratory Dendritic Cells. Cell Host Microbe 2017, 21, 182–194. [Google Scholar] [CrossRef] [Green Version]
- van der Velden, A.W.; Copass, M.K.; Starnbach, M.N. Salmonella inhibit T cell proliferation by a direct, contact-dependent immunosuppressive effect. Proc. Natl. Acad. Sci. USA 2005, 102, 17769–17774. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Patel, R.; Kim, K.; Shutinoski, B.; Wachholz, K.; Krishnan, L.; Sad, S. Culling of APCs by inflammatory cell death pathways restricts TIM3 and PD-1 expression and promotes the survival of primed CD8 T cells. Cell Death Differ. 2017, 24, 1900–1911. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Broker, B.M.; Mrochen, D.; Peton, V. The T Cell Response to Staphylococcus aureus. Pathogens 2016, 5, 31. [Google Scholar] [CrossRef] [PubMed]
- Alonzo, F., 3rd; Kozhaya, L.; Rawlings, S.A.; Reyes-Robles, T.; DuMont, A.L.; Myszka, D.G.; Landau, N.R.; Unutmaz, D.; Torres, V.J. CCR5 is a receptor for Staphylococcus aureus leukotoxin ED. Nature 2013, 493, 51–55. [Google Scholar] [CrossRef] [Green Version]
- Ziegler, C.; Goldmann, O.; Hobeika, E.; Geffers, R.; Peters, G.; Medina, E. The dynamics of T cells during persistent Staphylococcus aureus infection: From antigen-reactivity to in vivo anergy. EMBO Mol. Med. 2011, 3, 652–666. [Google Scholar] [CrossRef]
- White, J.; Herman, A.; Pullen, A.M.; Kubo, R.; Kappler, J.W.; Marrack, P. The V-Beta-Specific Superantigen Staphylococcal Enterotoxin-B-Stimulation of Mature T-Cells and Clonal Deletion in Neonatal Mice. Cell 1989, 56, 27–35. [Google Scholar] [CrossRef]
- McCormick, J.K.; Yarwood, J.M.; Schlievert, P.M. Toxic shock syndrome and bacterial superantigens: An update. Annu. Rev. Microbiol. 2001, 55, 77–104. [Google Scholar] [CrossRef]
- Zeppa, J.J.; Kasper, K.J.; Mohorovic, I.; Mazzuca, D.M.; Haeryfar, S.M.M.; McCormick, J.K. Nasopharyngeal infection by Streptococcus pyogenes requires superantigen-responsive Vbeta-specific T cells. Proc. Natl. Acad. Sci. USA 2017, 114, 10226–10231. [Google Scholar] [CrossRef] [Green Version]
- Dellabona, P.; Peccoud, J.; Kappler, J.; Marrack, P.; Benoist, C.; Mathis, D. Superantigens interact with MHC class II molecules outside of the antigen groove. Cell 1990, 62, 1115–1121. [Google Scholar] [CrossRef]
- Gunther, S.; Varma, A.K.; Moza, B.; Kasper, K.J.; Wyatt, A.W.; Zhu, P.; Rahman, A.K.M.N.U.; Li, Y.L.; Mariuzza, R.A.; McCormick, J.K.; et al. A novel loop domain in superantigens extends their T cell receptor recognition site. J. Mol. Biol. 2007, 371, 210–221. [Google Scholar] [CrossRef] [Green Version]
- Moza, B.; Varma, A.K.; Buonpane, R.A.; Zhu, P.; Herfst, C.A.; Nicholson, M.J.; Wilbuer, A.K.; Seth, N.P.; Wucherpfennig, K.W.; McCormick, J.K.; et al. Structural basis of T-cell specificity and activation by the bacterial superantigen TSST-1. Embo J. 2007, 26, 1187–1197. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pumphrey, N.; Vuidepot, A.; Jakobsen, B.; Forsberg, G.; Walse, B.; Lindkvist-Petersson, K. Cutting edge: Evidence of direct TCR alpha-chain interaction with superantigen. J. Immunol. 2007, 179, 2700–2704. [Google Scholar] [CrossRef] [PubMed]
- Saline, M.; Rodstrom, K.E.; Fischer, G.; Orekhov, V.Y.; Karlsson, B.G.; Lindkvist-Petersson, K. The structure of superantigen complexed with TCR and MHC reveals novel insights into superantigenic T cell activation. Nat. Commun 2010, 1, 119. [Google Scholar] [CrossRef]
- Thomas, D.; Dauwalder, O.; Brun, V.; Badiou, C.; Ferry, T.; Etienne, J.; Vandenesch, F.; Lina, G. Staphylococcus aureus superantigens elicit redundant and extensive human Vbeta patterns. Infect. Immun 2009, 77, 2043–2050. [Google Scholar] [CrossRef] [Green Version]
- Abdurrahman, G.; Schmiedeke, F.; Bachert, C.; Broker, B.M.; Holtfreter, S. Allergy-A New Role for T Cell Superantigens of Staphylococcus aureus? Toxins 2020, 12, 176. [Google Scholar] [CrossRef] [Green Version]
- Seo, K.S.; Park, J.Y.; Terman, D.S.; Bohach, G.A. A quantitative real time PCR method to analyze T cell receptor Vbeta subgroup expansion by staphylococcal superantigens. J. Transl Med. 2010, 8, 2. [Google Scholar] [CrossRef] [Green Version]
- Commons, R.J.; Smeesters, P.R.; Proft, T.; Fraser, J.D.; Robins-Browne, R.; Curtis, N. Streptococcal superantigens: Categorization and clinical associations. Trends Mol. Med. 2014, 20, 48–62. [Google Scholar] [CrossRef]
- Petersson, K.; Pettersson, H.; Skartved, N.J.; Walse, B.; Forsberg, G. Staphylococcal enterotoxin H induces V alpha-specific expansion of T cells. J. Immunol. 2003, 170, 4148–4154. [Google Scholar] [CrossRef]
- Llewelyn, M.; Sriskandan, S.; Peakman, M.; Ambrozak, D.R.; Douek, D.C.; Kwok, W.W.; Cohen, J.; Altmann, D.M. HLA class II polymorphisms determine responses to bacterial superantigens. J. Immunol. 2004, 172, 1719–1726. [Google Scholar] [CrossRef] [Green Version]
- Kotb, M.; Norrby-Teglund, A.; McGeer, A.; El-Sherbini, H.; Dorak, M.T.; Khurshid, A.; Green, K.; Peeples, J.; Wade, J.; Thomson, G.; et al. An immunogenetic and molecular basis for differences in outcomes of invasive group A streptococcal infections. Nat. Med. 2002, 8, 1398–1404. [Google Scholar] [CrossRef]
- Arad, G.; Levy, R.; Nasie, I.; Hillman, D.; Rotfogel, Z.; Barash, U.; Supper, E.; Shpilka, T.; Minis, A.; Kaempfer, R. Binding of superantigen toxins into the CD28 homodimer interface is essential for induction of cytokine genes that mediate lethal shock. PLoS. Biol. 2011, 9, e1001149. [Google Scholar] [CrossRef] [PubMed]
- Levy, R.; Rotfogel, Z.; Hillman, D.; Popugailo, A.; Arad, G.; Supper, E.; Osman, F.; Kaempfer, R. Superantigens hyperinduce inflammatory cytokines by enhancing the B7-2/CD28 costimulatory receptor interaction. Proc. Natl. Acad. Sci. USA 2016, 113, E6437–E6446. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kaempfer, R. Bacterial Superantigen Toxins, CD28, and Drug Development. Toxins 2018, 10, 459. [Google Scholar] [CrossRef] [Green Version]
- Kawabe, Y.; Ochi, A. Selective anergy of V beta 8+,CD4+ T cells in Staphylococcus enterotoxin B-primed mice. J. Exp. Med. 1990, 172, 1065–1070. [Google Scholar] [CrossRef]
- Janik, D.K.; Lee, W.T. Staphylococcal Enterotoxin B (SEB) Induces Memory CD4 T Cell Anergy in vivo and Impairs Recall Immunity to Unrelated Antigens. J. Clin. Cell Immunol. 2015, 6, 1–8. [Google Scholar]
- Watson, A.R.; Janik, D.K.; Lee, W.T. Superantigen-induced CD4 memory T cell anergy. I. Staphylococcal enterotoxin B induces Fyn-mediated negative signaling. Cell Immunol. 2012, 276, 16–25. [Google Scholar] [CrossRef] [Green Version]
- Spaulding, A.R.; Salgado-Pabon, W.; Kohler, P.L.; Horswill, A.R.; Leung, D.Y.; Schlievert, P.M. Staphylococcal and streptococcal superantigen exotoxins. Clin. Microbiol. Rev. 2013, 26, 422–447. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Radcliff, F.J.; Clow, F.; Mahadevan, M.; Johnston, J.; Proft, T.; Douglas, R.G.; Fraser, J.D. A potential role for staphylococcal and streptococcal superantigens in driving skewing of TCR Vbeta subsets in tonsillar hyperplasia. Med. Microbiol. Immunol. 2017, 206, 337–346. [Google Scholar] [CrossRef]
- Huang, C.C.; Shah, S.; Nguyen, P.; Altman, J.D.; Blackman, M.A. Bacterial superantigen exposure after resolution of influenza virus infection perturbs the virus-specific memory CD8(+)-T-cell repertoire. J. Virol. 2002, 76, 6852–6856. [Google Scholar] [CrossRef] [Green Version]
- Zhang, W.J.; Sarawar, S.; Nguyen, P.; Daly, K.; Rehg, J.E.; Doherty, P.C.; Woodland, D.L.; Blackman, M.A. Lethal synergism between influenza infection and staphylococcal enterotoxin B in mice. J. Immunol. 1996, 157, 5049–5060. [Google Scholar]
- Meilleur, C.E.; Memarnejadian, A.; Shivji, A.N.; Benoit, J.M.; Tuffs, S.W.; Mele, T.S.; Singh, B.; Dikeakos, J.D.; Topham, D.J.; Mu, H.H.; et al. Discordant rearrangement of primary and anamnestic CD8+ T cell responses to influenza A viral epitopes upon exposure to bacterial superantigens: Implications for prophylactic vaccination, heterosubtypic immunity and superinfections. PLoS. Pathog. 2020, 16, e1008393. [Google Scholar] [CrossRef] [PubMed]
- Shaler, C.R.; Choi, J.; Rudak, P.T.; Memarnejadian, A.; Szabo, P.A.; Tun-Abraham, M.E.; Rossjohn, J.; Corbett, A.J.; McCluskey, J.; McCormick, J.K.; et al. MAIT cells launch a rapid, robust and distinct hyperinflammatory response to bacterial superantigens and quickly acquire an anergic phenotype that impedes their cognate antimicrobial function: Defining a novel mechanism of superantigen-induced immunopathology and immunosuppression. PLoS. Biol. 2017, 15, e2001930. [Google Scholar]
- Emgard, J.; Bergsten, H.; McCormick, J.K.; Barrantes, I.; Skrede, S.; Sandberg, J.K.; Norrby-Teglund, A. MAIT Cells Are Major Contributors to the Cytokine Response in Group A Streptococcal Toxic Shock Syndrome. Proc. Natl. Acad. Sci. USA 2019, 116, 25923–25931. [Google Scholar] [CrossRef] [Green Version]
- Szabo, P.A.; Rudak, P.T.; Choi, J.; Xu, S.X.; Schaub, R.; Singh, B.; McCormick, J.K.; Haeryfar, S.M.M. Invariant Natural Killer T Cells Are Pathogenic in the HLA-DR4-Transgenic Humanized Mouse Model of Toxic Shock Syndrome and Can Be Targeted to Reduce Morbidity. J. Infect. Dis. 2017, 215, 824–829. [Google Scholar]
- Hayworth, J.L.; Mazzuca, D.M.; Maleki Vareki, S.; Welch, I.; McCormick, J.K.; Haeryfar, S.M. CD1d-independent activation of mouse and human iNKT cells by bacterial superantigens. Immunol. Cell Biol. 2012, 90, 699–709. [Google Scholar] [CrossRef]
- Kalyan, S.; Chow, A.W. Human peripheral gamma delta T cells potentiate the early proinflammatory cytokine response to staphylococcal toxic shock syndrome toxin-1. J. Infect. Dis. 2004, 189, 1892–1896. [Google Scholar] [CrossRef] [Green Version]
- Morita, C.T.; Li, H.M.; Lamphear, J.G.; Rich, R.R.; Fraser, J.D.; Mariuzza, R.A.; Lee, H.K. Superantigen recognition by gamma delta T cells: SEA recognition site for human V gamma 2 T cell receptors. Immunity 2001, 14, 331–344. [Google Scholar] [CrossRef] [Green Version]
- Rust, C.J.J.; Verreck, F.; Vietor, H.; Koning, F. Specific Recognition of Staphylococcal Enterotoxin-a by Human T-Cells Bearing Receptors with the V-Gamma-9 Region. Nature 1990, 346, 572–574. [Google Scholar] [CrossRef]
- Stinissen, P.; Vandevyver, C.; Raus, J.; Zhang, J.W. Superantigen Reactivity of Gamma-Delta T-Cell Clones Isolated from Patients with Multiple-Sclerosis and Controls. Cell Immunol. 1995, 166, 227–235. [Google Scholar] [CrossRef]



© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Shepherd, F.R.; McLaren, J.E. T Cell Immunity to Bacterial Pathogens: Mechanisms of Immune Control and Bacterial Evasion. Int. J. Mol. Sci. 2020, 21, 6144. https://doi.org/10.3390/ijms21176144
Shepherd FR, McLaren JE. T Cell Immunity to Bacterial Pathogens: Mechanisms of Immune Control and Bacterial Evasion. International Journal of Molecular Sciences. 2020; 21(17):6144. https://doi.org/10.3390/ijms21176144
Chicago/Turabian StyleShepherd, Freya R., and James E. McLaren. 2020. "T Cell Immunity to Bacterial Pathogens: Mechanisms of Immune Control and Bacterial Evasion" International Journal of Molecular Sciences 21, no. 17: 6144. https://doi.org/10.3390/ijms21176144
APA StyleShepherd, F. R., & McLaren, J. E. (2020). T Cell Immunity to Bacterial Pathogens: Mechanisms of Immune Control and Bacterial Evasion. International Journal of Molecular Sciences, 21(17), 6144. https://doi.org/10.3390/ijms21176144