Redox Signaling and Regional Heterogeneity of Endothelial Dysfunction in db/db Mice



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
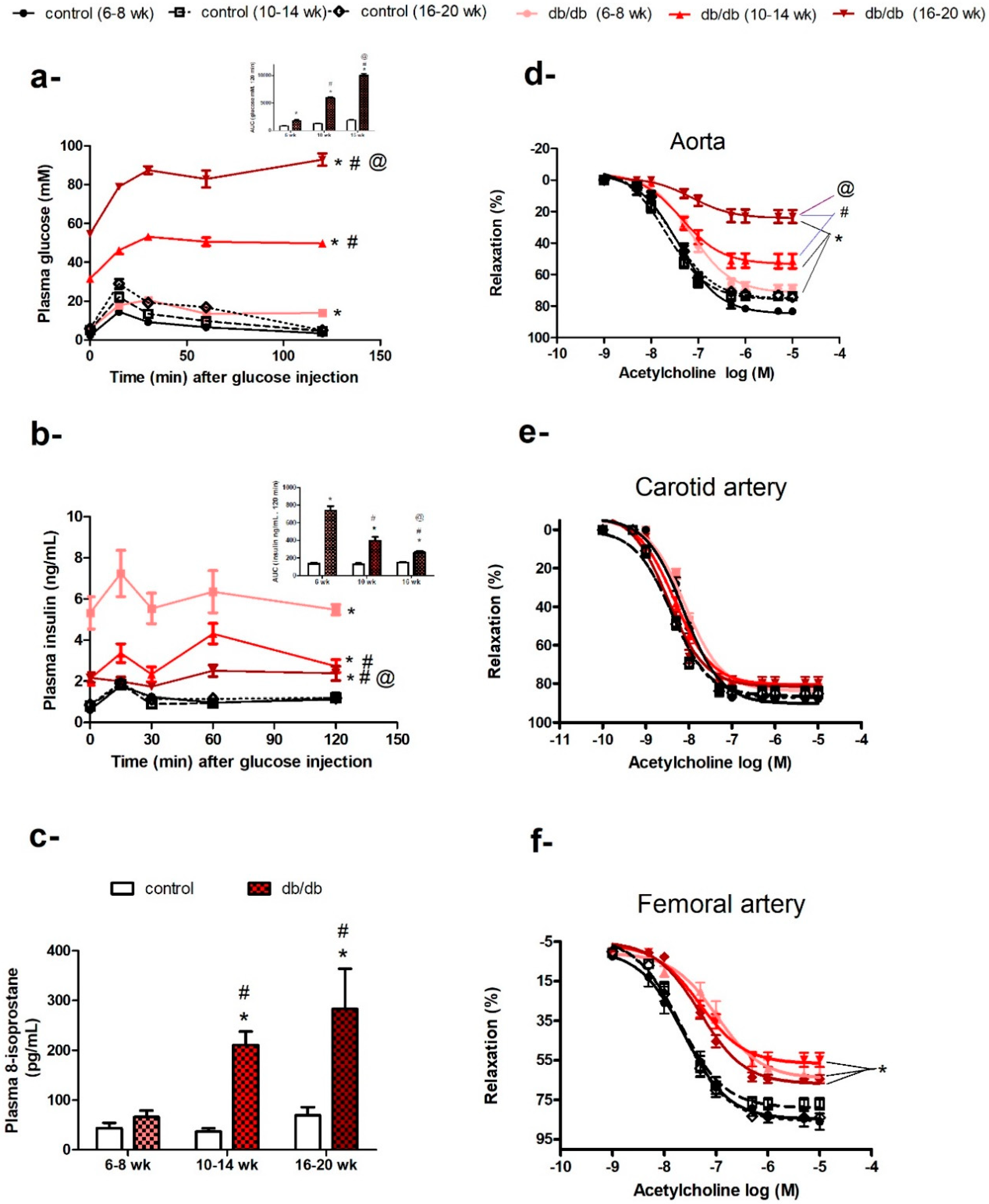
2.1. Age-Related Changes in Body Weight, Glycemic Control, and Systemic Oxidative Stress Biomarkers in db/db Mice
2.2. Age-Related Changes in Endothelium-Dependent Vasodilatation in Aortae but not in Carotid or Femoral Arteries of db/db Mice
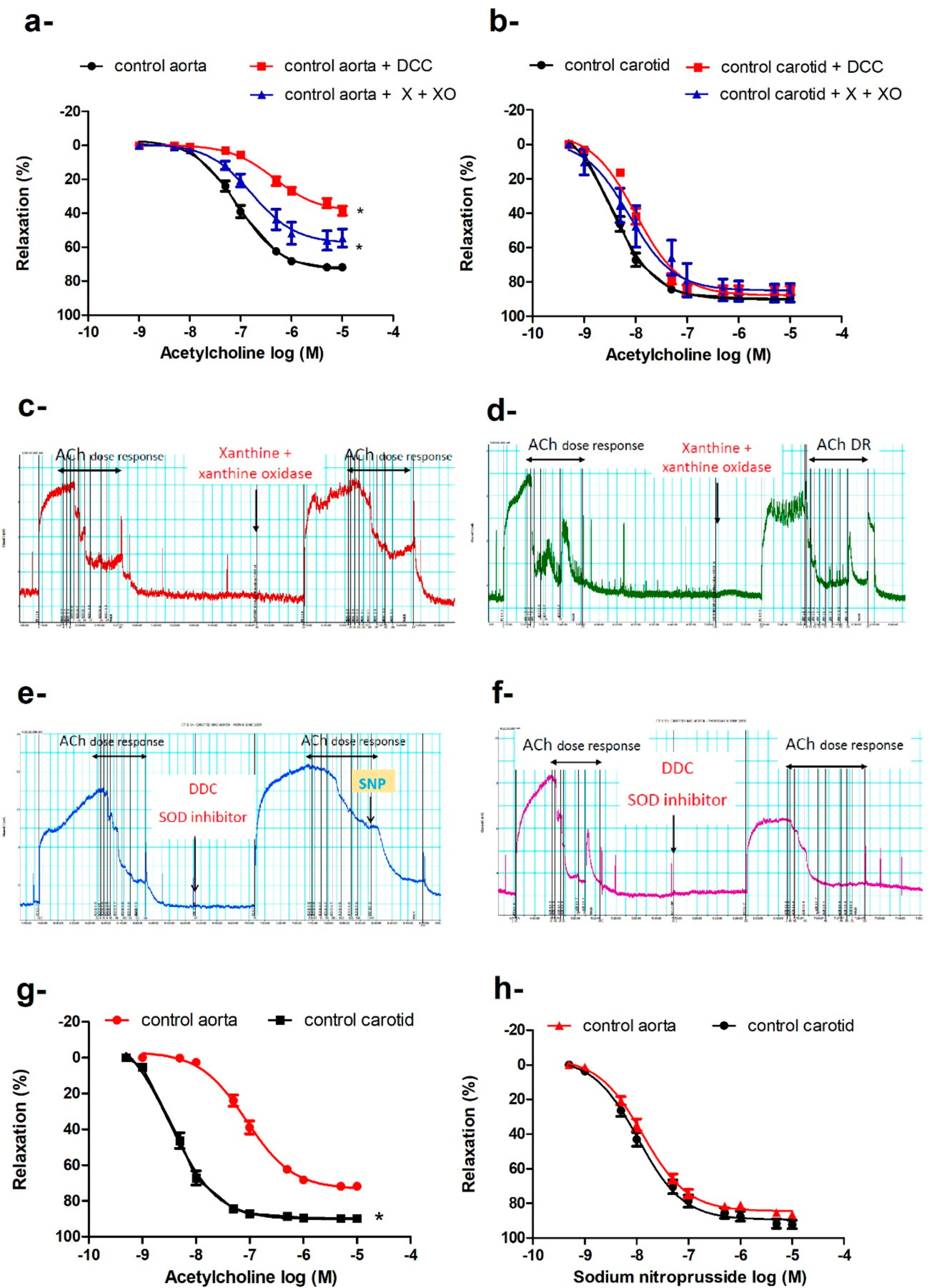
2.3. Preserved Endothelium-Dependent Vasodilatation in Carotid Arteries of db/db Mice in the Presence of Increased Oxidative Burden
2.4. Signaling Pathway of Endothelium-Dependent Vasodilatation in the Carotid Artery
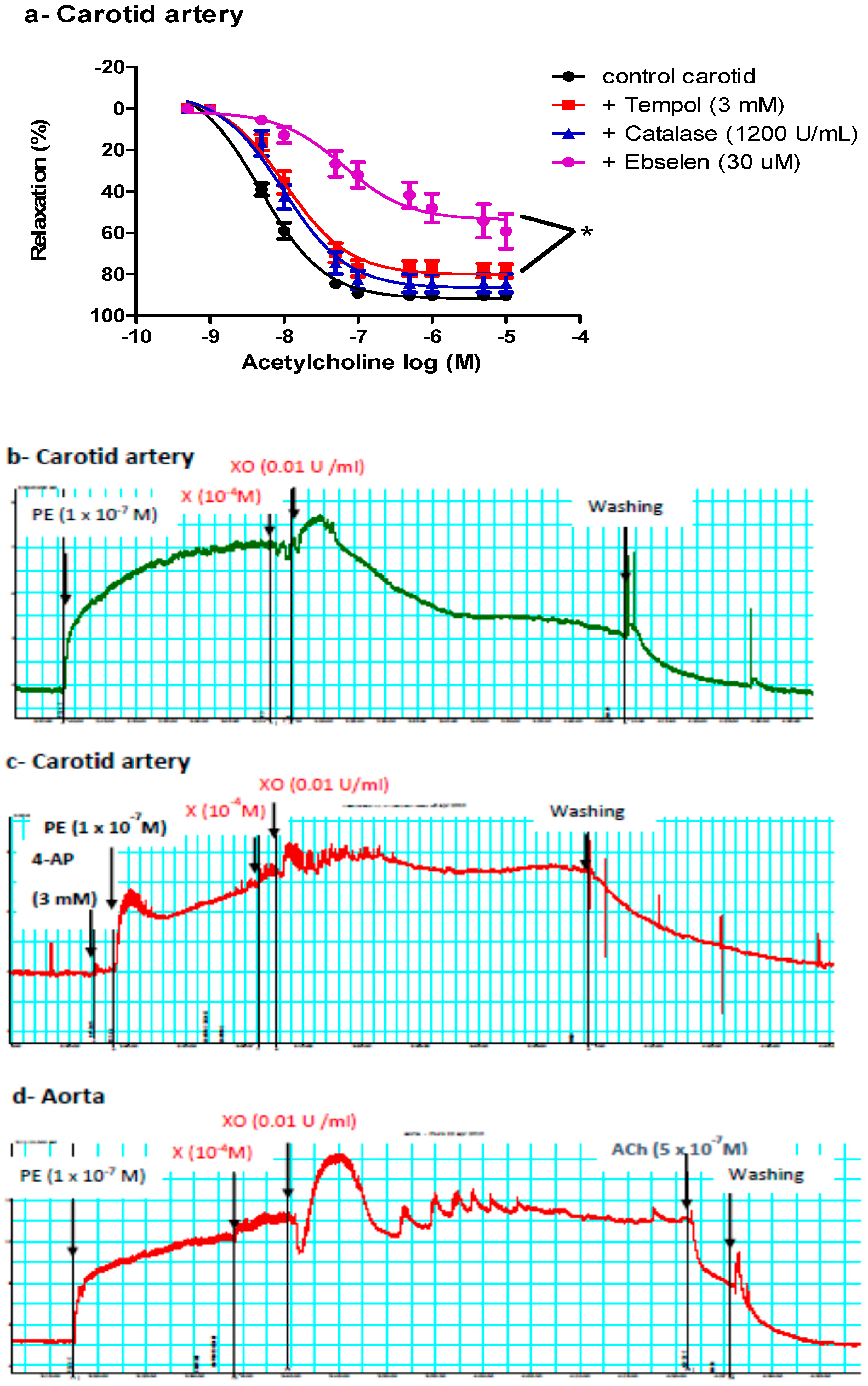
2.4.1. Effects of Tempol, Catalase, or Ebselen on ACh-Induced Vasodilatation
2.4.2. Effect of Exogenous O2− or ONOO− on Pre-Constricted Aortae and Carotid Arteries
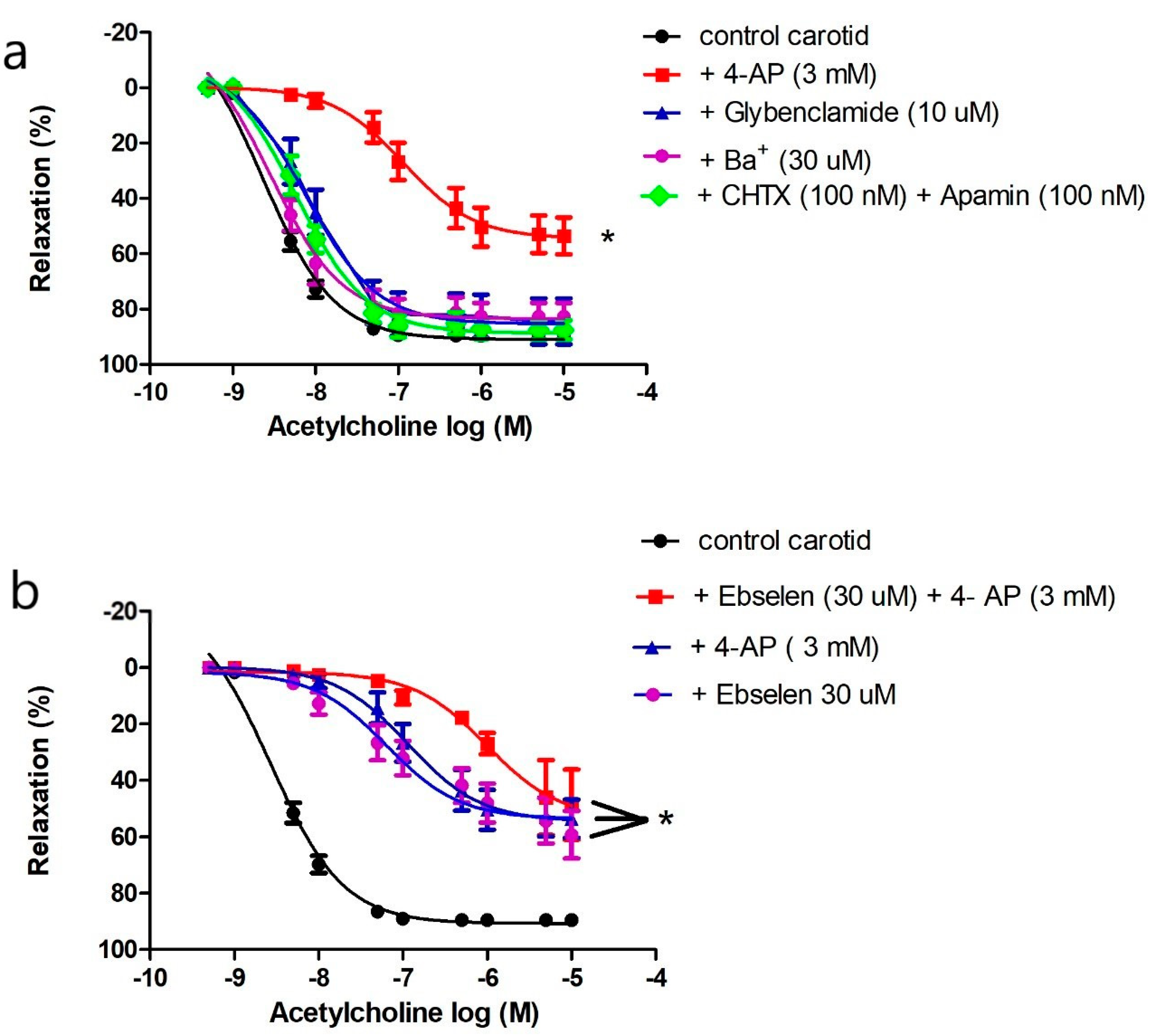
2.4.3. Effects of Potassium Channel Inhibitors on Endothelium-Dependent Vasodilatation in the Carotid Artery
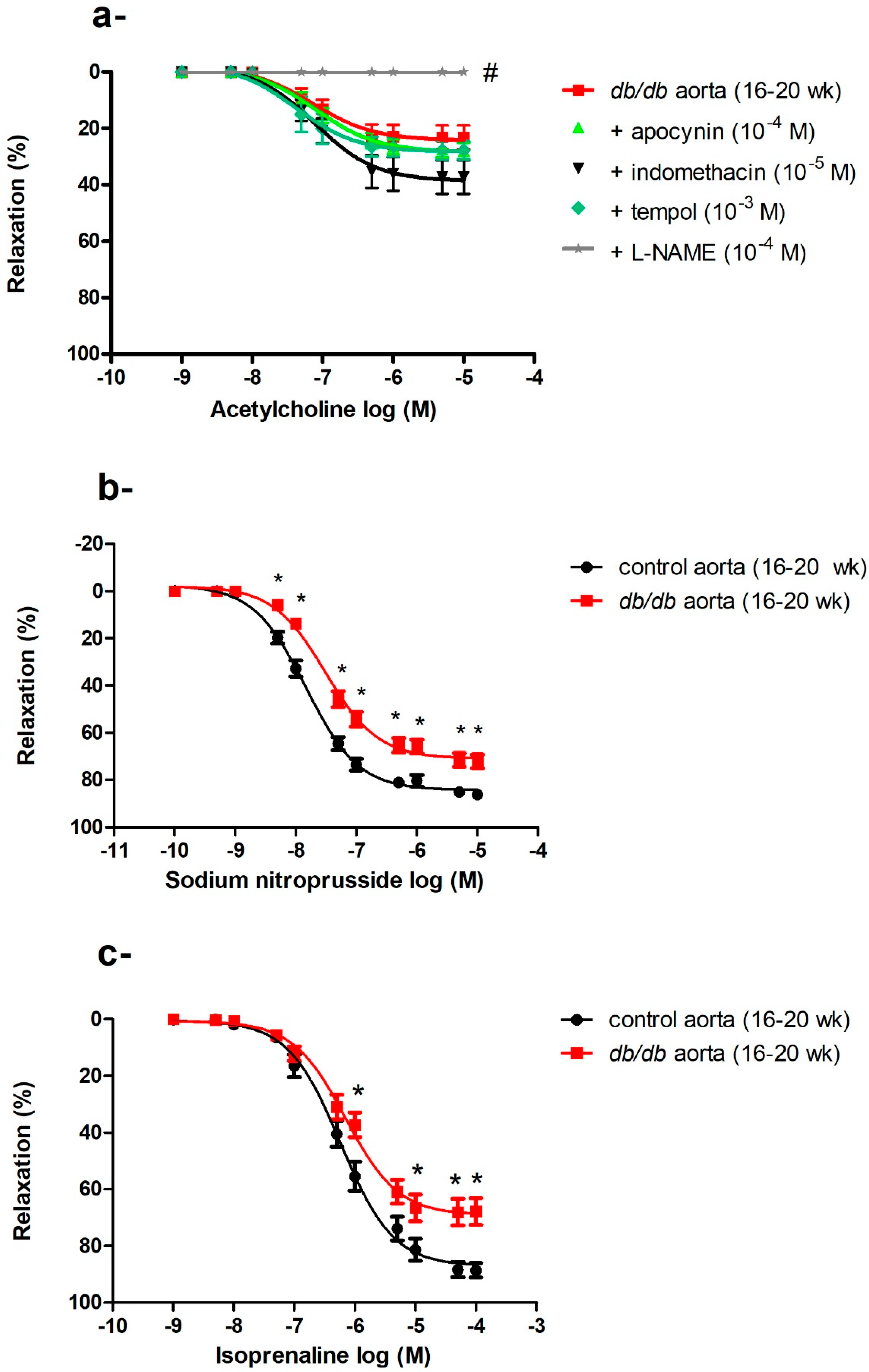
2.5. Generalized Vascular Dysfunction in the Aortae of db/db Mice
2.5.1. Effects of Indomethacin, Apocynin, L-NAME, and Tempol on Impaired ACh-Induced Vasodilatation in the Aortae of db/db Mice
2.5.2. Impaired Endothelium-Independent Vasodilatation in the Aortae of db/db Mice
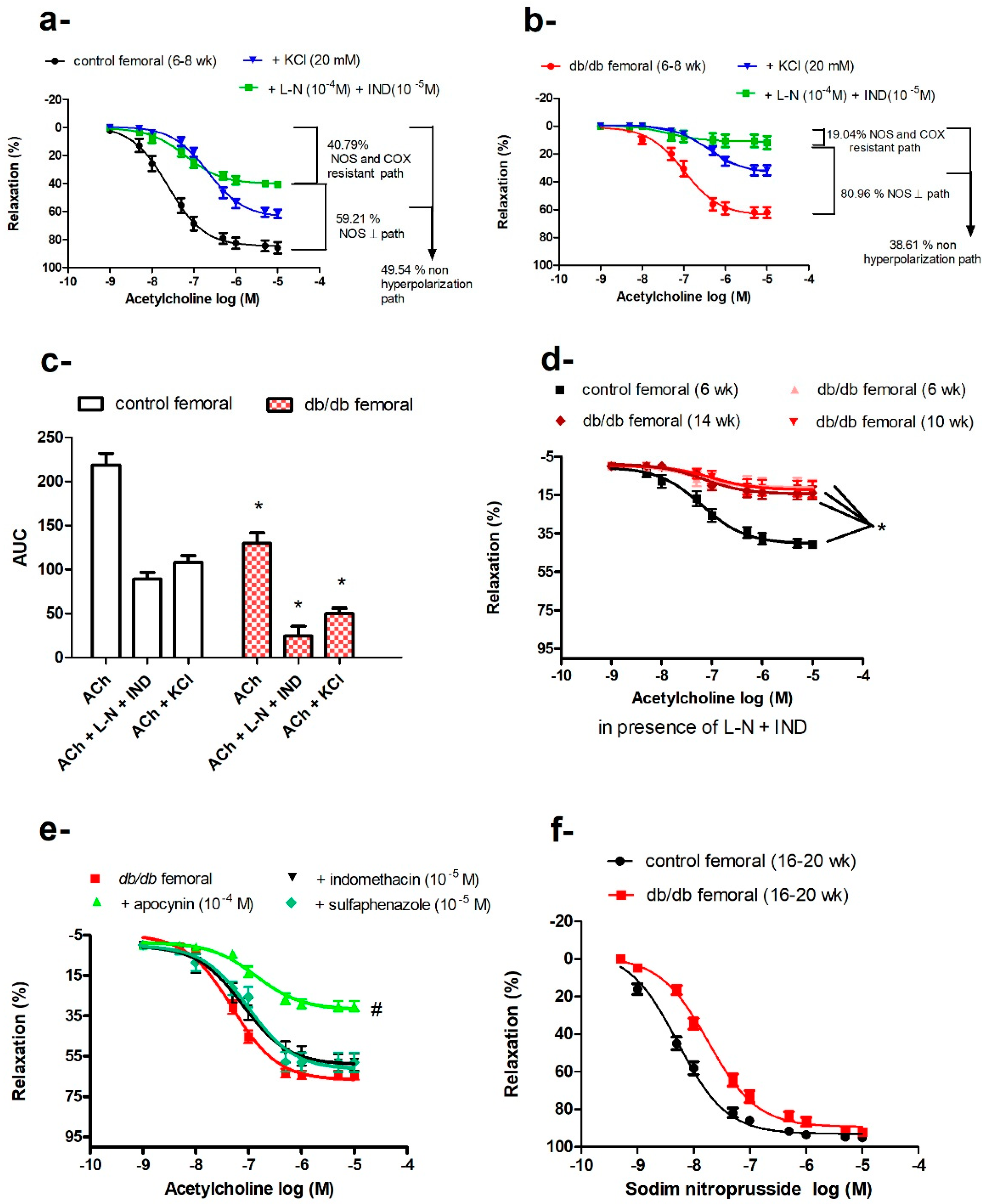
2.6. Impaired Vasodilatation in Femoral Arteries of db/db Mice
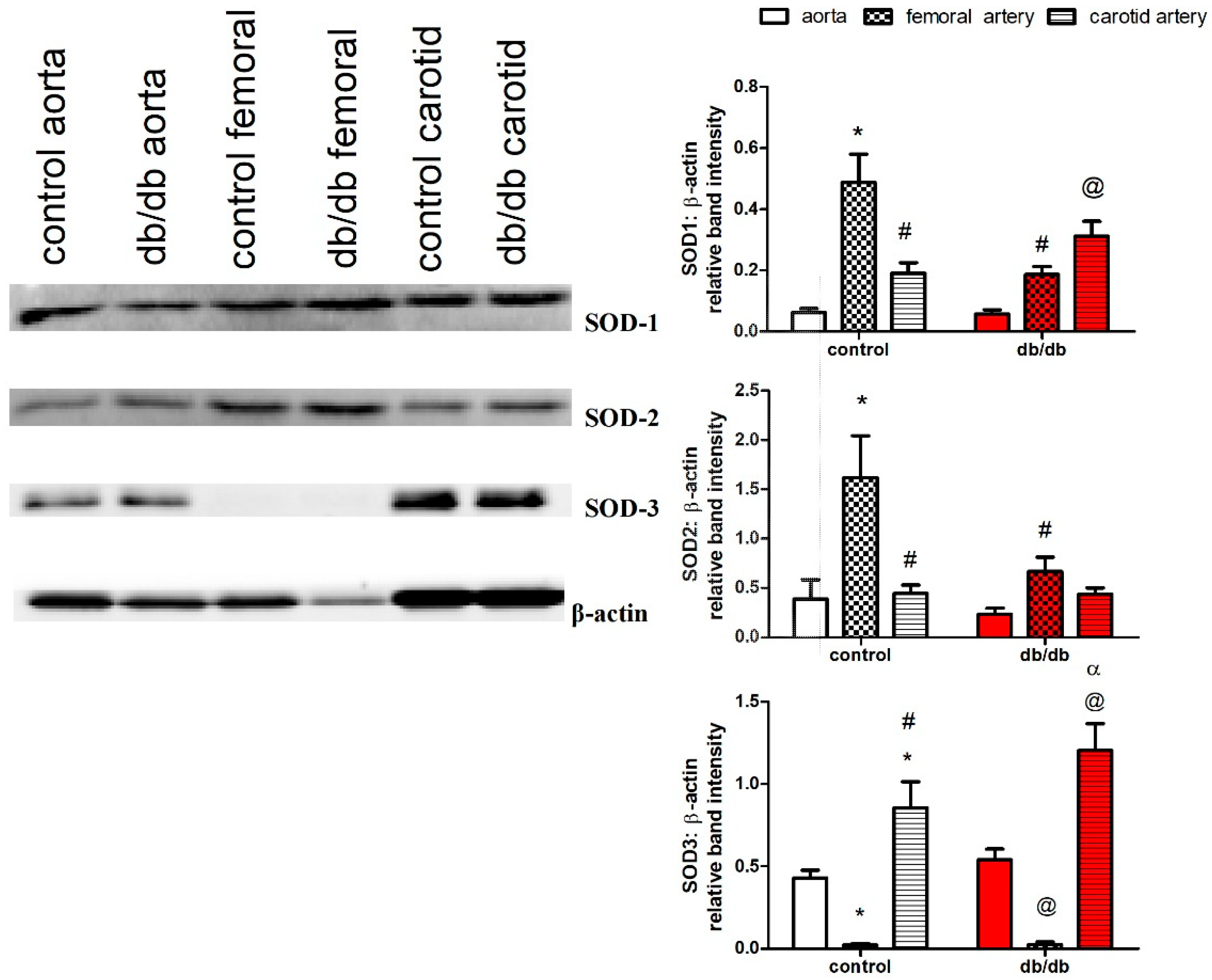
2.7. Protein Expression of SOD-1, SOD-2, and SOD-3 in the Aortae, as well as Carotid and Femoral Arteries, of db/db and Control Mice
3. Discussion
3.1. Aortae
3.2. Carotid Artery
3.3. Femoral Artery
4. Materials and Methods
4.1. Animals
4.2. Intraperitoneal Glucose Tolerance Test
4.3. Plasma and Tissue Sample Collection
4.4. Assessment of Endothelium-Dependent and-Independent Vasodilatation
4.5. Western Blot
4.6. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Bigagli, E.; Lodovici, M. Circulating Oxidative Stress Biomarkers in Clinical Studies on Type 2 Diabetes and Its Complications. Oxidative Med. Cell. Longev. 2019, 2019, 5953685. [Google Scholar] [CrossRef]
- Brunner, H.; Cockcroft, J.R.; Deanfield, J.; Donald, A.; Ferrannini, E.; Halcox, J.; Kiowski, W.; Lüscher, T.F.; Mancia, G.; Natali, A.; et al. Endothelial function and dysfunction. Part II: Association with cardiovascular risk factors and diseases. A statement by the Working Group on Endothelins and Endothelial Factors of the European Society of Hypertension. J. Hypertens. 2005, 23, 233–246. [Google Scholar] [CrossRef]
- Shi, Y.; Vanhoutte, P.M. Macro- and microvascular endothelial dysfunction in diabetes. J. Diabetes 2017, 9, 434–449. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McCarron, J.G.; Wilson, C.; Heathcote, H.R.; Zhang, X.; Buckley, C.; Lee, M.D. Heterogeneity and emergent behaviour in the vascular endothelium. Curr. Opin. Pharmacol. 2019, 45, 23–32. [Google Scholar] [CrossRef] [PubMed]
- Lerman, A.; Zeiher, A.M. Endothelial function: Cardiac events. Circulation 2005, 111, 363–368. [Google Scholar] [CrossRef] [PubMed]
- Stehouwer, C.D.A.; Gall, M.A.; Twisk, J.W.R.; Knudsen, E.; Emeis, J.J.; Parving, H.H. Increased urinary albumin excretion, endothelial dysfunction, and chronic low-grade inflammation in type 2 diabetes: Progressive, interrelated, and independently associated with risk of death. Diabetes 2002, 51, 1157–1165. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nitenberg, A.; Cosson, E.; Pham, I. Postprandial endothelial dysfunction: Role of glucose, lipids and insulin. Diabetes Metab. 2006, 32, 2S28–2S33. [Google Scholar] [CrossRef]
- O’Driscoll, G.; Green, D.; Maiorana, A.; Stanton, K.; Colreavy, F.; Taylor, R. Improvement in endothelial function by angiotensin-converting enzyme inhibition in non-insulin-dependent diabetes mellitus. J. Am. Coll. Cardiol. 1999, 33, 1506–1511. [Google Scholar] [CrossRef]
- De Vriese, A.S.; Verbeuren, T.J.; Van De Voorde, J.; Lameire, N.H.; Vanhoutte, P.M. Endothelial dysfunction in diabetes. Br. J. Pharmacol. 2000, 130, 963–974. [Google Scholar] [CrossRef] [Green Version]
- Bagi, Z.; Koller, A.; Kaley, G. Superoxide-NO interaction decreases flow- and agonist-induced dilations of coronary arterioles in Type 2 diabetes mellitus. Am. J. Physiol. Heart Circ. Physiol. 2003, 285, H1404–H1410. [Google Scholar] [CrossRef] [Green Version]
- Granstam, E.; Granstam, S.-O. Involvement of Nitric Oxide in the Regulation of Regional Hemodynamics in Streptozotocin-Diabetic Rats. Physiol. Res. 2003, 52, 159–169. [Google Scholar]
- Fortes, Z.B.; Lerne, J.G.; Scivoletto, R. Vascular reactivity in diabetes mellitus: Role of the endothelial cell. Br. J. Pharmacol. 1983, 79, 771–781. [Google Scholar] [CrossRef] [Green Version]
- Szerafin, T.; Erdei, N.; Fülöp, T.; Pasztor, E.T.; Édes, I.; Koller, A.; Bagi, Z. Increased cyclooxygenase-2 expression and prostaglandin-mediated dilation in coronary arterioles of patients with diabetes mellitus. Circ. Res. 2006, 99. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Skyrme-Jones, R.A.; Berry, K.L.; O’Brien, R.C.; Meredith, I.T. Basal and exercise-induced skeletal muscle blood flow is augmented in type I diabetes mellitus. Clin. Sci. 2000, 98, 111–120. [Google Scholar] [CrossRef]
- Andrew, R.; Skyrme-Jones, P.; O’brien, R.C.; Meredith, I.T. Vasodilator prostanoids, but not nitric oxide, may account for skeletal muscle hyperaemia in Type I diabetes mellitus. Clin. Sci. 2000, 99, 383–392. [Google Scholar]
- Yamamoto, E.; Nakamura, T.; Kataoka, K.; Tokutomi, Y.; Dong, Y.F.; Fukuda, M.; Nako, H.; Yasuda, O.; Ogawa, H.; Kim-Mitsuyama, S. Nifedipine prevents vascular endothelial dysfunction in a mouse model of obesity and type 2 diabetes, by improving eNOS dysfunction and dephosphorylation. Biochem. Biophys. Res. Commun. 2010, 403, 258–263. [Google Scholar] [CrossRef] [PubMed]
- Lagaud, G.J.; Masih-Khan, E.; Kai, S.; van Breemen, C.; Dubé, G.P. Influence of type II diabetes on arterial tone and endothelial function in murine mesenteric resistance arteries. J. Vasc. Res. 2001, 38, 578–589. [Google Scholar] [CrossRef]
- Belmadani, S.; Palen, D.I.; Gonzalez-Villalobos, R.A.; Boulares, H.A.; Matrougui, K. Elevated epidermal growth factor receptor phosphorylation induces resistance artery dysfunction in diabetic db/db mice. Diabetes 2008, 57, 1629–1637. [Google Scholar] [CrossRef] [Green Version]
- Park, Y.; Capobianco, S.; Gao, X.; Falck, J.R.; Dellsperger, K.C.; Zhang, C. Role of EDHF in type 2 diabetes-induced endothelial dysfunction. Am. J. Physiol. Heart Circ. Physiol. 2008, 295, 1982–1988. [Google Scholar] [CrossRef] [Green Version]
- Bagi, Z.; Erdei, N.; Toth, A.; Li, W.; Hintze, T.H.; Koller, A.; Kaley, G. Type 2 diabetic mice have increased arteriolar tone and blood pressure: Enhanced release of COX-2-derived constrictor prostaglandins. Arterioscler. Thromb. Vasc. Biol. 2005, 25, 1610–1616. [Google Scholar] [CrossRef] [Green Version]
- Thorin, E.; Shreeve, S.M. Heterogeneity of vascular endothelial cells in normal and disease states. Pharmacol. Ther. 1998, 78, 155–166. [Google Scholar] [CrossRef]
- Ding, H.; Triggle, C.R. Endothelial dysfunction in diabetes: Multiple targets for treatment. Pflugers Arch. Eur. J. Physiol. 2010, 459, 977–994. [Google Scholar] [CrossRef] [PubMed]
- Basu, S. F2-isoprostanes in human health and diseases: From molecular mechanisms to clinical implications. Antioxid. Redox Signal. 2008, 10, 1405–1434. [Google Scholar] [CrossRef] [PubMed]
- Shi, Y.; Vanhoutte, P.M. Oxidative stress and COX cause hyper-responsiveness in vascular smooth muscle of the femoral artery from diabetic rats. Br. J. Pharmacol. 2008, 154, 639–651. [Google Scholar] [CrossRef] [Green Version]
- Sallam, N.; Fisher, A.; Golbidi, S.; Laher, I. Weight and inflammation are the major determinants of vascular dysfunction in the aortae of db/db mice. Naunyn. Schmiedebergs Arch. Pharmacol. 2011, 383, 483–492. [Google Scholar] [CrossRef] [PubMed]
- Matsumoto, T.; Kobayashi, T.; Wachi, H.; Seyama, Y.; Kamata, K. Vascular NAD(P)H oxidase mediates endothelial dysfunction in basilar arteries from Otsuka Long-Evans Tokushima Fatty (OLETF) rats. Atherosclerosis 2007, 192, 15–24. [Google Scholar] [CrossRef]
- Félétou, M.; Vanhoutte, P.M. Endothelial dysfunction: A multifaceted disorder (The Wiggers Award Lecture). Am. J. Physiol. Heart Circ. Physiol. 2006, 291, H985–H1002. [Google Scholar] [CrossRef]
- Szewczyk, A.; Jarmuszkiewicz, W.; Koziel, A.; Sobieraj, I.; Nobik, W.; Lukasiak, A.; Skup, A.; Bednarczyk, P.; Drabarek, B.; Dymkowska, D.; et al. Mitochondrial mechanisms of endothelial dysfunction. Pharmacol. Rep. 2015, 67, 704–710. [Google Scholar] [CrossRef]
- Gómez-Moreno, D.; Adrover, J.M.; Hidalgo, A. Neutrophils as effectors of vascular inflammation. Eur. J. Clin. Investig. 2018, 48, e12940. [Google Scholar] [CrossRef] [Green Version]
- Trebak, M.; Ginnan, R.; Singer, H.A.; Jourd’heuil, D. Interplay between calcium and reactive oxygen/nitrogen species: An essential paradigm for vascular smooth muscle signaling. Antioxid. Redox Signal. 2010, 12, 657–674. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.-X.; Zheng, Y.-M. ROS-dependent signaling mechanisms for hypoxic Ca(2+) responses in pulmonary artery myocytes. Antioxid. Redox Signal. 2010, 12, 611–623. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gutterman, D.D.; Miura, H.; Liu, Y. Redox modulation of vascular tone: Focus of potassium channel mechanisms of dilation. Arterioscler. Thromb. Vasc. Biol. 2005, 25, 671–678. [Google Scholar] [CrossRef] [PubMed]
- Chatterjee, S.; Fisher, A.B. Mechanotransduction in the endothelium: Role of membrane proteins and reactive oxygen species in sensing, transduction, and transmission of the signal with altered blood flow. Antioxid. Redox Signal. 2014, 20, 899–913. [Google Scholar] [CrossRef] [PubMed]
- Veit, F.; Pak, O.; Brandes, R.P.; Weissmann, N. Hypoxia-dependent reactive oxygen species signaling in the pulmonary circulation: Focus on ion Channels. Antioxid. Redox Signal. 2015, 22, 537–552. [Google Scholar] [CrossRef] [Green Version]
- Dunham-Snary, K.J.; Hong, Z.G.; Xiong, P.Y.; Del Paggio, J.C.; Herr, J.E.; Johri, A.M.; Archer, S.L. A mitochondrial redox oxygen sensor in the pulmonary vasculature and ductus arteriosus. Pflugers Arch. Eur. J. Physiol. 2016, 468, 43–58. [Google Scholar] [CrossRef] [Green Version]
- Luo, Z.; Chen, Y.; Chen, S.; Welch, W.J.; Andresen, B.T.; Jose, P.A.; Wilcox, C.S. Comparison of inhibitors of superoxide generation in vascular smooth muscle cells. Br. J. Pharmacol. 2009, 157, 935–943. [Google Scholar] [CrossRef] [Green Version]
- Harden, T.K. Agonist-induced desensitization of the beta-adrenergic receptor-linked adenylate cyclase. Pharmacol. Rev. 1983, 35, 5–32. [Google Scholar] [PubMed]
- Miike, T.; Kunishiro, K.; Kanda, M.; Azukizawa, S.; Kurahashi, K.; Shirahase, H. Impairment of endothelium-dependent ACh-induced relaxation in aorta of diabetic db/db mice--possible dysfunction of receptor and/or receptor-G protein coupling. Naunyn. Schmiedeberg’s Arch. Pharmacol. 2008, 377, 401–410. [Google Scholar] [CrossRef] [PubMed]
- San Martín, A.; Du, P.; Dikalova, A.; Lassègue, B.; Aleman, M.; Góngora, M.C.; Brown, K.; Joseph, G.; Harrison, D.G.; Taylor, W.R.; et al. Reactive oxygen species-selective regulation of aortic inflammatory gene expression in Type 2 diabetes. Am. J. Physiol. Heart Circ. Physiol. 2007, 292, H2073–H2082. [Google Scholar] [CrossRef] [PubMed]
- Viswanad, B.; Srinivasan, K.; Kaul, C.L.; Ramarao, P. Effect of tempol on altered angiotensin II and acetylcholine-mediated vascular responses in thoracic aorta isolated from rats with insulin resistance. Pharmacol. Res. 2006, 53, 209–215. [Google Scholar] [CrossRef] [PubMed]
- Erdös, B.; Miller, A.W.; Busija, D.W. Alterations in KATP and KCa channel function in cerebral arteries of insulin-resistant rats. Am. J. Physiol.-Heart Circ. Physiol. 2002, 283, H2472–H2477. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Caballero, A.E.; Arora, S.; Saouaf, R.; Lim, S.C.; Smakowski, P.; Park, J.Y.; King, G.L.; LoGerfo, F.W.; Horton, E.S.; Veves, A. Microvascular and macrovascular reactivity is reduced in subjects at risk for type 2 diabetes. Diabetes 1999, 48, 1856–1862. [Google Scholar] [CrossRef] [PubMed]
- Andersen, K.; Pedersen, B.K. The role of inflammation in vascular insulin resistance with focus on IL-6. Horm. Metab. Res. 2008, 40, 635–639. [Google Scholar] [CrossRef]
- Wasserman, D.H.; Wang, T.J.; Brown, N.J. The vasculature in prediabetes. Circ. Res. 2018, 122, 1135–1150. [Google Scholar] [CrossRef] [PubMed]
- Ormazabal, V.; Nair, S.; Elfeky, O.; Aguayo, C.; Salomon, C.; Zuñiga, F.A. Association between insulin resistance and the development of cardiovascular disease. Cardiovasc. Diabetol. 2018, 17. [Google Scholar] [CrossRef]
- Katakami, N. Mechanism of development of atherosclerosis and cardiovascular disease in diabetes mellitus. J. Atheroscler. Thromb. 2018, 25, 27–39. [Google Scholar] [CrossRef] [Green Version]
- Iantorno, M.; Campia, U.; Di Daniele, N.; Nistico, S.P.; Forleo, G.B.; Cardillo, C.; Tesauro, M. Obesity, inflammation and endothelial dysfunction. J. Biol. Regul. Homeost. Agents 2014, 28, 169–176. [Google Scholar]
- Pannirselvam, M.; Ding, H.; Anderson, T.J.; Triggle, C.R. Pharmacological characteristics of endothelium-derived hyperpolarizing factor-mediated relaxation of small mesenteric arteries from db/db mice. Eur. J. Pharmacol. 2006, 551, 98–107. [Google Scholar] [CrossRef]
- Guzik, T.J.; Touyz, R.M. Oxidative Stress, Inflammation, and Vascular Aging in Hypertension. Hypertension 2017, 70, 660–667. [Google Scholar] [CrossRef]
- Okon, E.B.; Chung, A.W.Y.; Zhang, H.; Laher, I.; Van Breemen, C. Hyperglycemia and hyperlipidemia are associated with endothelial dysfunction during the development of type 2 diabetes. Can. J. Physiol. Pharmacol. 2007, 85, 562–567. [Google Scholar] [CrossRef]
- Woodman, R.J.; Chew, G.T.; Watts, G.F. Mechanisms, significance and treatment of vascular dysfunction in type 2 diabetes mellitus: Focus on lipid-regulating therapy. Drugs 2005, 65, 31–74. [Google Scholar] [CrossRef] [PubMed]
- Tschudi, M.R.; Barton, M.; Bersinger, N.A.; Moreau, P.; Cosentino, F.; Noll, G.; Malinski, T.; Lüscher, T.F. Effect of age on kinetics of nitric oxide release in rat aorta and pulmonary artery. J. Clin. Investig. 1996, 98, 899–905. [Google Scholar] [CrossRef]
- Gendron, M.È.; Théorêt, J.F.; Mamarbachi, A.M.; Drouin, A.; Nguyen, A.; Bolduc, V.; Thorin-Trescases, N.; Merhi, Y.; Thorin, E. Late chronic catechin antioxidant treatment is deleterious to the endothelial function in aging mice with established atherosclerosis. Am. J. Physiol. Heart Circ. Physiol. 2010, 298. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Majithiya, J.B.; Balaraman, R. Time-dependent changes in antioxidant enzymes and vascular reactivity of aorta in streptozotocin-induced diabetic rats treated with curcumin. J. Cardiovasc. Pharmacol. 2005, 46, 697–705. [Google Scholar] [CrossRef] [PubMed]
- Collins, R.; Armitage, J.; Parish, S.; Sleight, P.; Peto, R. MRC/BHF Heart Protection Study of antioxidant vitamin supplementation in 20 536 high-risk individuals: A randomised placebo-controlled trial. Lancet 2002, 360, 23–33. [Google Scholar] [CrossRef]
- Rapola, J.M.; Virtamo, J.; Haukka, J.K.; Heinonen, O.P.; Albanes, D.; Taylor, P.R.; Huttunen, J.K. Effect of vitamin E and beta carotene on the incidence of angina pectoris. A randomized, double-blind, controlled trial. JAMA 1996, 275, 693–698. [Google Scholar] [CrossRef]
- Daiber, A.; Chlopicki, S. Revisiting pharmacology of oxidative stress and endothelial dysfunction in cardiovascular disease: Evidence for redox-based therapies. Free Radic. Biol. Med. 2020, 157, 15–37. [Google Scholar] [CrossRef]
- Kowaluk, E.A.; Seth, P.; Fung, H.L. Metabolic activation of sodium nitroprusside to nitric oxide in vascular smooth muscle. J. Pharmacol. Exp. Ther. 1992, 262, 916–922. [Google Scholar]
- Matsushita, M.; Tanaka, Y.; Koike, K. Studies on the mechanisms underlying beta-adrenoceptor-mediated relaxation of rat abdominal aorta. J. Smooth Muscle Res. 2006, 42, 217–225. [Google Scholar] [CrossRef] [Green Version]
- Chai, Q.; Liu, Z.; Chen, L. Effects of streptozotocin-induced diabetes on Kv channels in rat small coronary smooth muscle cells. Chin. J. Physiol. 2005, 48, 57–63. [Google Scholar]
- Pelligrino, D.A.; Koenig, H.M.; Wang, Q.; Albrecht, R.F. Protein kinase C suppresses receptor-mediated pial arteriolar relaxation in the diabetic rat. Neuroreport 1994, 5, 417–420. [Google Scholar] [CrossRef]
- Bubolz, A.H.; Li, H.; Wu, Q.; Liu, Y. Enhanced oxidative stress impairs cAMP-mediated dilation by reducing Kv channel function in small coronary arteries of diabetic rats. Am. J. Physiol. Heart Circ. Physiol. 2005, 289, H1873–H1880. [Google Scholar] [CrossRef] [PubMed]
- Chai, Q.; Xu, X.; Jia, Q.; Dong, Q.; Liu, Z.; Zhang, W.; Chen, L. Molecular basis of dysfunctional K V channels in small coronary artery smooth muscle cells of streptozotocin-induced diabetic rats. Chin. J. Physiol. 2007, 50, 171–177. [Google Scholar] [PubMed]
- Jin, N.; Packer, C.S.; Rhoades, R.A. Reactive oxygen-mediated contraction in pulmonary arterial smooth muscle: Cellular mechanisms. Can. J. Physiol. Pharmacol. 1991, 69, 383–388. [Google Scholar] [CrossRef] [PubMed]
- Waypa, G.B.; Schumacker, P.T. Hypoxia-induced changes in pulmonary and systemic vascular resistance: Where is the O2 sensor? Respir. Physiol. Neurobiol. 2010, 174, 201–211. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bhattacharya, I.; Mundy, A.L.; Widmer, C.C.; Kretz, M.; Barton, M. Regional heterogeneity of functional changes in conduit arteries after high-fat diet. Obesity 2008, 16, 743–748. [Google Scholar] [CrossRef]
- Shi, Y.; Ku, D.D.; Man, R.Y.; Vanhoutte, P.M. Augmented EDHF-mediated relaxations attenuate endothelial dysfunction in femoral and mesenteric, but not in carotid arteries from type I diabetic rats. J. Pharmacol. Exp. Ther. 2006. [Google Scholar] [CrossRef]
- Bagi, Z.; Koller, A.; Kaley, G. PPARgamma activation, by reducing oxidative stress, increases NO bioavailability in coronary arterioles of mice with Type 2 diabetes. Am. J. Physiol.-Heart Circ. Physiol. 2004, 286, H742–H748. [Google Scholar] [CrossRef] [Green Version]
- Pagano, P.J.; Griswold, M.C.; Najibi, S.; Marklund, S.L.; Cohen, R.A. Resistance of endothelium-dependent relaxation to elevation of O(-)(2) levels in rabbit carotid artery. Am. J. Physiol. 1999, 277, H2109–H2114. [Google Scholar] [CrossRef] [Green Version]
- Liaudet, L.; Vassalli, G.; Pacher, P. Role of peroxynitrite in the redox regulation of cell signal transduction pathways. Front. Biosci. 2009, 14, 4809–4814. [Google Scholar] [CrossRef] [Green Version]
- Beckman, J.S. Oxidative damage and tyrosine nitration from peroxynitrite. Chem. Res. Toxicol. 1996, 9, 836–844. [Google Scholar] [CrossRef] [PubMed]
- Wei, E.P.; Kontos, H.A.; Beckman, J.S. Mechanisms of cerebral vasodilation by superoxide, hydrogen peroxide, and peroxynitrite. Am. J. Physiol. 1996, 271, H1262–H1266. [Google Scholar] [CrossRef] [PubMed]
- Hong, K.W.; Rhim, B.Y.; Lee, W.S.; Jeong, B.R.; Kim, C.D.; Shin, Y.W. Release of superoxide-dependent relaxing factor(s) from endothelial cells. Am. J. Physiol. Heart Circ. Physiol. 1989, 257. [Google Scholar] [CrossRef] [PubMed]
- Benkusky, N.A. Attenuation of vascular relaxation after development of tachyphylaxis to peroxynitrite in vivo. Am. J. Physiol. Heart Circ. Physiol. 1998, 275. [Google Scholar] [CrossRef]
- Nossaman, B.D.; Dabisch, P.A.; Liles, J.T.; Baber, S.R.; Champion, H.C.; Kaye, A.D.; Feng, C.-J.; Anwar, M.; Bivalacqua, T.J.; Santiago, J.A.; et al. Peroxynitrite does not impair pulmonary and systemic vascular responses. J. Appl. Physiol. 2004, 96, 455–462. [Google Scholar] [CrossRef]
- Villa, L.M.; Salas, E.; Darley-Usmar, V.M.; Radomski, M.W.; Moncada, S. Peroxynitrite induces both vasodilatation and impaired vascular relaxation in the isolated perfused rat heart. Proc. Natl. Acad. Sci. USA 1994, 91, 12383–12387. [Google Scholar] [CrossRef] [Green Version]
- Wu, M.; Pritchard, K.A.; Kaminski, P.M.; Fayngersh, R.P.; Hintze, T.H.; Wolin, M.S. Involvement of nitric oxide and nitrosothiols in relaxation of pulmonary arteries to peroxynitrite. Am. J. Physiol. 1994, 266, H2108–H2113. [Google Scholar] [CrossRef]
- Richardson, R.S.; Donato, A.J.; Uberoi, A.; Wray, D.W.; Lawrenson, L.; Nishiyama, S.; Bailey, D.M. Exercise-induced brachial artery vasodilation: Role of free radicals. Am. J. Physiol.-Heart Circ. Physiol. 2007, 292, H1516–H1522. [Google Scholar] [CrossRef]
- Dabisch, P.A.; Liles, J.T.; Baber, S.R.; Golwala, N.H.; Murthy, S.N.; Kadowitz, P.J. Analysis of L-NAME-dependent and -resistant responses to acetylcholine in the rat. Am. J. Physiol.-Heart Circ. Physiol. 2008, 294, H688–H698. [Google Scholar] [CrossRef] [Green Version]
- Ohashi, M.; Faraci, F.; Heistad, D. Peroxynitrite hyperpolarizes smooth muscle and relaxes internal carotid artery in rabbit via ATP-sensitive K+ channels. Am. J. Physiol. Heart Circ. Physiol. 2005, 289, H2244–H2250. [Google Scholar] [CrossRef]
- Lewis, S.J.; Hoque, A.; Walton, T.M.; Kooy, N.W. Potential role of nitration and oxidation reactions in the effects of peroxynitrite on the function of β-adrenoceptor sub-types in the rat. Eur. J. Pharmacol. 2005, 518, 187–194. [Google Scholar] [CrossRef] [PubMed]
- Hill, C.E.; Phillips, J.K.; Sandow, S.L. Heterogeneous control of blood flow amongst different vascular beds. Med. Res. Rev. 2001, 21, 1–60. [Google Scholar] [CrossRef]
- Goto, K.; Kitazono, T. Endothelium-dependent hyperpolarization (EDH) in diabetes: Mechanistic insights and therapeutic implications. Int. J. Mol. Sci. 2019, 20, 3737. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pannirselvam, M.; Verma, S.; Anderson, T.J.; Triggle, C.R. Cellular basis of endothelial dysfunction in small mesenteric arteries from spontaneously diabetic (db/db -/-) mice: Role of decreased tetrahydrobiopterin bioavailability. Br. J. Pharmacol. 2002, 136, 255–263. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lin, D.; Takemoto, D.J. Oxidative activation of protein kinase Cγ through the C1 domain: Effects on gap junctions. J. Biol. Chem. 2005, 280, 13682–13693. [Google Scholar] [CrossRef] [Green Version]
- Mulvany, M.J.; Halpern, W. Contractile properties of small arterial resistance vessels in spontaneously hypertensive and normotensive rats. Circ. Res. 1977, 41, 19–26. [Google Scholar] [CrossRef] [Green Version]







© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sallam, N.A.; Laher, I. Redox Signaling and Regional Heterogeneity of Endothelial Dysfunction in db/db Mice. Int. J. Mol. Sci. 2020, 21, 6147. https://doi.org/10.3390/ijms21176147
Sallam NA, Laher I. Redox Signaling and Regional Heterogeneity of Endothelial Dysfunction in db/db Mice. International Journal of Molecular Sciences. 2020; 21(17):6147. https://doi.org/10.3390/ijms21176147
Chicago/Turabian StyleSallam, Nada A., and Ismail Laher. 2020. "Redox Signaling and Regional Heterogeneity of Endothelial Dysfunction in db/db Mice" International Journal of Molecular Sciences 21, no. 17: 6147. https://doi.org/10.3390/ijms21176147
APA StyleSallam, N. A., & Laher, I. (2020). Redox Signaling and Regional Heterogeneity of Endothelial Dysfunction in db/db Mice. International Journal of Molecular Sciences, 21(17), 6147. https://doi.org/10.3390/ijms21176147