Intrinsic Effect of Pyridine-N-Position on Structural Properties of Cu-Based Low-Dimensional Coordination Frameworks


Abstract
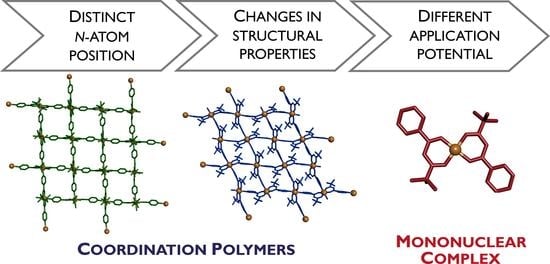
:
1. Introduction
2. Results and Discussion
2.1. Synthesis and Analysis
2.2. X-ray Structure Determinations
2.2.1. Ligand [HL3][NO3]
2.2.2. Network N1
2.2.3. Network N2
2.2.4. Complex C1
2.3. Powder X-ray Diffraction (PXRD) Analyses
2.4. SEM Analysis
2.5. Framework Thermal Stability
2.6. Gas Sorption Studies
3. Materials and Methods
3.1. General Methods
3.2. X-ray Crystallography
3.3. General Preparation of L–L3
3.4. Synthesis of [Cu(L1 or 2)2]n–N1 and N2
3.5. Synthesis of Cu(L3)2–C1
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| calcd. | calculated |
| CCD | Charge-coupled Device |
| XRD | X-Ray diffraction |
| PXRD | powder X-Ray diffraction |
| TGA | thermogravimetric analysis |
| TOF-MS | time-of-flight mass spectrometry |
| SEM | scanning electron microscopy |
| LFD | large field detector |
| THF | tetrahydrofuran |
| MeOH | methanol |
| NaH | sodium hydride |
| BET | Brunauer–Emmett–Teller isotherm |
| CPs | coordination polymers |
References
- Gorczyński, A.; Harrowfield, J.M.; Patroniak, V.; Stefankiewicz, A.R. Quaterpyridines as Scaffolds for Functional Metallosupramolecular Materials. Chem. Rev. 2016, 116, 14620–14674. [Google Scholar] [CrossRef]
- Northrop, B.H.; Yang, H.-B.; Stang, P.J. Coordination-driven self-assembly of functionalized supramolecular metallacycles. Chem. Commun. 2008, 5896–5908. [Google Scholar] [CrossRef] [PubMed]
- Bocian, A.; Drożdż, W.; Szymańska, M.; Lewandowski, J.; Fik-Jaskółka, M.; Gorczyński, A.; Patroniak, V.; Stefankiewicz, A.R. Complex-decorated surfactant-encapsulated clusters (CD-SECs) as novel multidynamic hybrid materials. Nanoscale 2020, 12, 4743–4750. [Google Scholar] [CrossRef]
- Markiewicz, G.; Walczak, A.; Perlitius, F.; Piasecka, M.; Harrowfield, J.M.; Stefankiewicz, A.R. Photoswitchable transition metal complexes with azobenzene-functionalized imine-based ligands: Structural and kinetic analysis. Dalton Trans. 2018, 47, 14254–14262. [Google Scholar] [CrossRef] [PubMed]
- Drożdż, W.; Walczak, A.; Bessin, Y.; Gervais, V.; Cao, X.-Y.; Lehn, J.-M.; Ulrich, S.; Stefankiewicz, A.R. Multivalent Metallosupramolecular Assemblies as Effective DNA Binding Agents. Chem. Eur. J. 2018, 24, 10802–10811. [Google Scholar] [CrossRef] [PubMed]
- Brzechwa-Chodzyńska, A.; Zieliński, M.; Gilski, M.; Harrowfield, J.M.; Stefankiewicz, A.R. Dynamer and Metallodynamer Interconversion: An Alternative View to Metal Ion Complexation. Inorg. Chem. 2020, 59, 8552–8561. [Google Scholar] [CrossRef] [PubMed]
- Kitagawa, S.; Kitaura, R.; Noro, S.-I. Functional Porous Coordination Polymers. Angew. Chem. Int. Ed. 2004, 43, 2334–2375. [Google Scholar] [CrossRef] [PubMed]
- Janiak, C. Engineering coordination polymers towards applications. Dalton Trans. 2003, 2781–2804. [Google Scholar] [CrossRef]
- Duan, J.; Jin, W.; Kitagawa, S. Water-resistant porous coordination polymers for gas separation. Coord. Chem. Rev. 2017, 332, 48–74. [Google Scholar] [CrossRef] [Green Version]
- Uemura, T.; Yanai, N.; Kitagawa, S. Polymerization reactions in porous coordination polymers. Chem. Soc. Rev. 2009, 38, 1228–1236. [Google Scholar] [CrossRef] [Green Version]
- Agarwal, R.A.; Gupta, N.K. CO2 sorption behavior of imidazole, benzimidazole and benzoic acid based coordination polymers. Coord. Chem. Rev. 2017, 332, 100–121. [Google Scholar] [CrossRef]
- Toma, H.E.; Rocha, R.C. Linkage Isomerization Reactions. Croatica Chemica Acta 2001, 74, 499–528. [Google Scholar]
- Burmeister, J.L. Linkage isomerism in metal complexes. Coord. Chem. Rev. 1968, 3, 225–245. [Google Scholar] [CrossRef]
- Lee, S.-L.; Hu, F.-L.; Shang, X.-J.; Shi, Y.-X.; Tan, A.L.; Mizera, J.; Clegg, J.K.; Zhang, W.-H.; Young, D.J.; Lang, J.-P. Efficient ring-opening polymerization (ROP) of ε-caprolactone catalysed by isomeric pyridyl β-diketonate iron(III) complexes. New J. Chem. 2017, 41, 14457–14465. [Google Scholar] [CrossRef]
- Wu, H.-B.; Wang, Q.-M. Construction of Heterometallic Cages with Tripodal Metalloligands. Angew. Chem. Int. Ed. 2009, 48, 7343–7345. [Google Scholar] [CrossRef] [Green Version]
- Burrows, A.D.; Mahon, M.F.; Renouf, C.L.; Richardson, C.; Warren, A.J.; Warren, J.E. Dipyridyl β-diketonate complexes and their use as metalloligands in the formation of mixed-metal coordination networks. Dalton Trans. 2012, 41, 4153–4163. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hou, G.-G.; Liu, Y.; Liu, Q.-K.; Ma, J.-P.; Dong, Y.-B. NbO lattice MOFs based on octahedral M(II) and ditopic pyridyl substituted diketonate ligands: Structure, encapsulation and guest-driven luminescent property. Chem. Commun. 2011, 47, 10731–10733. [Google Scholar] [CrossRef]
- Liu, F.; Zhou, Y. Polymeric [Eu(L)3(H2O)] (HL = 1-(pyridin-4-yl)butane-1,3-dione and Dimeric [Eu2(L)6(H2O)2] (HL = 1,3-di(pyridin-3-yl)propane-1,3-dione) as selective and sensitive chemosensors for Hg(II) ion. Inorg. Chem. Commun. 2010, 13, 1410–1413. [Google Scholar] [CrossRef]
- Wang, M.; Vajpayee, V.; Shanmugaraju, S.; Zheng, Y.-R.; Zhao, Z.; Kim, H.; Mukherjee, P.S.; Chi, K.-W.; Stang, P.J. Coordination-Driven Self-Assembly of M3L2 Trigonal Cages from Preorganized Metalloligands Incorporating Octahedral Metal Centers and Fluorescent Detection of Nitroaromatics. Inorg. Chem. 2011, 50, 1506–1512. [Google Scholar] [CrossRef] [Green Version]
- Carlucci, L.; Ciani, G.; Proserpio, D.M.; Visconti, M. The novel metalloligand [Fe(bppd)3] (bppd = 1,3-bis(4-pyridyl)-1,3-propanedionate) for the crystal engineering of heterometallic coordination networks with different silver salts. Anionic control of the structures. CrystEngComm 2011, 13, 5891–5902. [Google Scholar] [CrossRef]
- Zhang, Y.; Chen, B.; Fronczek, F.R.; Maverick, A.W. A Nanoporous Ag−Fe Mixed-Metal−Organic Framework Exhibiting Single-Crystal-to-Single-Crystal Transformations upon Guest Exchange. Inorg. Chem. 2008, 47, 4433–4435. [Google Scholar] [CrossRef] [PubMed]
- Andrews, P.C.; Deacon, G.B.; Frank, R.; Fraser, B.H.; Junk, P.C.; MacLellan, J.G.; Massi, M.; Moubaraki, B.; Murray, K.S.; Silberstein, M. Formation of HoIII Trinuclear Clusters and GdIII Monodimensional Polymers Induced by ortho and para Regioisomers of Pyridyl-Functionalised β-Diketones: Synthesis, Structure, and Magnetic Properties. Eur. J. Inorg. Chem. 2009, 2009, 744–751. [Google Scholar] [CrossRef]
- Chen, B.; Fronczek, F.R.; Maverick, A.W. Porous Cu−Cd Mixed-Metal−Organic Frameworks Constructed from Cu(Pyac)2 {Bis[3-(4-pyridyl)pentane-2,4-dionato]copper(II)}. Inorg. Chem. 2004, 43, 8209–8211. [Google Scholar] [CrossRef] [PubMed]
- Walczak, A.; Stefankiewicz, A.R. pH-Induced Linkage Isomerism of Pd(II) Complexes: A Pathway to Air- and Water-Stable Suzuki–Miyaura-Reaction Catalysts. Inorg. Chem. 2018, 57, 471–477. [Google Scholar] [CrossRef] [PubMed]
- Walczak, A.; Stachowiak, H.; Kurpik, G.; Kaźmierczak, J.; Hreczycho, G.; Stefankiewicz, A.R. High catalytic activity and selectivity in hydrosilylation of new Pt(II) metallosupramolecular complexes based on ambidentate ligands. J. Catal. 2019, 373, 139–146. [Google Scholar] [CrossRef]
- Abdine, R.A.A.; Kurpik, G.; Walczak, A.; Aeash, S.A.A.; Stefankiewicz, A.R.; Monnier, F.; Taillefer, M. Mild temperature amination of aryl iodides and aryl bromides with aqueous ammonia in the presence of CuBr and pyridyldiketone ligands. J. Catal. 2019, 376, 119–122. [Google Scholar] [CrossRef]
- Lee, J.; Kovalevsky, A.Y.; Novozhilova, I.V.; Bagley, K.A.; Coppens, P.; Richter-Addo, G.B. Single- and Double-Linkage Isomerism in a Six-Coordinate Iron Porphyrin Containing Nitrosyl and Nitro Ligands. J. Am. Chem. Soc. 2004, 126, 7180–7181. [Google Scholar] [CrossRef]
- Van Rijt, S.H.; Hebden, A.J.; Amaresekera, T.; Deeth, R.J.; Clarkson, G.J.; Parsons, S.; McGowan, P.C.; Sadler, P.J. Amide Linkage Isomerism As an Activity Switch for Organometallic Osmium and Ruthenium Anticancer Complexes. J. Med. Chem. 2009, 52, 7753–7764. [Google Scholar] [CrossRef]
- Coronado, E.; Giménez-López, M.C.; Levchenko, G.; Romero, F.M.; García-Baonza, V.; Milner, A.; Paz-Pasternak, M. Pressure-Tuning of Magnetism and Linkage Isomerism in Iron(II) Hexacyanochromate. J. Am. Chem. Soc. 2005, 127, 4580–4581. [Google Scholar] [CrossRef]
- Shatruk, M.; Dragulescu-Andrasi, A.; Chambers, K.E.; Stoian, S.A.; Bominaar, E.L.; Achim, C.; Dunbar, K.R. Properties of Prussian Blue Materials Manifested in Molecular Complexes: Observation of Cyanide Linkage Isomerism and Spin-Crossover Behavior in Pentanuclear Cyanide Clusters. J. Am. Chem. Soc. 2007, 129, 6104–6116. [Google Scholar] [CrossRef]
- Coronado, E.; Giménez-López, M.C.; Korzeniak, T.; Levchenko, G.; Romero, F.M.; Segura, A.; García-Baonza, V.; Cezar, J.C.; de Groot, F.M.F.; Milner, A.; et al. Pressure-Induced Magnetic Switching and Linkage Isomerism in K0.4Fe4[Cr(CN)6]2.8·16H2O: X-ray Absorption and Magnetic Circular Dichroism Studies. J. Am. Chem. Soc. 2008, 130, 15519–15532. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Castellano, M.; Ruiz-García, R.; Cano, J.; Ferrando-Soria, J.; Pardo, E.; Fortea-Pérez, F.R.; Stiriba, S.-E.; Barros, W.P.; Stumpf, H.O.; Cañadillas-Delgado, L.; et al. Metallosupramolecular approach toward multifunctional magnetic devices for molecular spintronics. Coord. Chem. Rev. 2015, 303, 110–138. [Google Scholar] [CrossRef]
- Domasevitch, K.V.; Vreshch, V.D.; Lysenko, A.B.; Krautscheid, H. Two-dimensional square-grid frameworks formed by self-associating copper(II) complexes with 1-(3-pyridyl)- and 1-(4-pyridyl)-substituted butane-1,3-diones. Acta Crystallographica C 2006, 62, m443–m447. [Google Scholar] [CrossRef] [PubMed]
- Dudek, M.; Clegg, J.K.; Glasson, C.R.K.; Kelly, N.; Gloe, K.; Gloe, K.; Kelling, A.; Buschmann, H.-J.; Jolliffe, K.A.; Lindoy, L.F.; et al. Interaction of Copper(II) with Ditopic Pyridyl-β-diketone Ligands: Dimeric, Framework, and Metallogel Structures. Cryst. Growth Des. 2011, 11, 1697–1704. [Google Scholar] [CrossRef]
- Kołodziejski, M.; Walczak, A.; Hnatejko, Z.; Harrowfield, J.; Stefankiewicz, A.R. Unsymmetrical bidentate ligands as a basis for construction of ambidentate ligands for functional materials: Properties of 4,4-dimethyl-1-phenylpentane-1,3-dionate. Polyhedron 2017, 137, 270–277. [Google Scholar] [CrossRef]
- Gilli, P.; Pretto, L.; Bertolasi, V.; Gilli, G. Predicting Hydrogen-Bond Strengths from Acid−Base Molecular Properties. The pKa Slide Rule: Toward the Solution of a Long-Lasting Problem. Acc. Chem. Res. 2009, 42, 33–44. [Google Scholar] [CrossRef]
- Gimenez-Marques, M.; Calvo Galve, N.; Palomino, M.; Valencia, S.; Rey, F.; Sastre, G.; Vitorica-Yrezabal, I.J.; Jimenez-Ruiz, M.; Rodriguez-Velamazan, J.A.; Gonzalez, M.A.; et al. Gas confinement in compartmentalized coordination polymers for highly selective sorption. Chem. Sci. 2017, 8, 3109–3120. [Google Scholar] [CrossRef] [Green Version]
- Brock, A.J.; Whittaker, J.J.; Powell, J.A.; Pfrunder, M.C.; Grosjean, A.; Parsons, S.; McMurtrie, J.C.; Clegg, J.K. Elastically Flexible Crystals have Disparate Mechanisms of Molecular Movement Induced by Strain and Heat. Angew. Chem. Int. Ed. 2018, 57, 11325–11328. [Google Scholar] [CrossRef]
- Worthy, A.; Grosjean, A.; Pfrunder, M.C.; Xu, Y.; Yan, C.; Edwards, G.; Clegg, J.K.; McMurtrie, J.C. Atomic resolution of structural changes in elastic crystals of copper(II) acetylacetonate. Nat. Chem. 2018, 10, 65–69. [Google Scholar] [CrossRef]
- Gould, J.A.; Athwal, H.S.; Blake, A.J.; Lewis, W.; Hubberstey, P.; Champness, N.R.; Schröder, M. Gas adsorption and structural diversity in a family of Cu(II) pyridyl-isophthalate metal–organic framework materials. Philos. T. R. Soc. A 2017, 375, 20160334. [Google Scholar] [CrossRef] [Green Version]
- Liu, Y.; Zhao, Y.; Liu, Z.-Q.; Liu, X.-H.; Zhang, X.-D.; Sun, W.-Y. Coordination polymers with salicylaldehyde ligands: Structural diversity, selective sorption and luminescence sensing properties. CrystEngComm 2020, 22, 304–310. [Google Scholar] [CrossRef]
- Zeng, M.-H.; Hu, S.; Chen, Q.; Xie, G.; Shuai, Q.; Gao, S.-L.; Tang, L.-Y. Apical Ligand Substitution, Shape Recognition, Vapor-Adsorption Phenomenon, and Microcalorimetry for a Pillared Bilayer Porous Framework That Shrinks or Expands in Crystal-to-Crystal Manners upon Change in the Cobalt(II) Coordination Environment. Inorg. Chem. 2009, 48, 7070–7079. [Google Scholar] [CrossRef] [PubMed]
- Kanoo, P.; Mostafa, G.; Matsuda, R.; Kitagawa, S.; Kumar Maji, T. A pillared-bilayer porous coordination polymer with a 1D channel and a 2D interlayer space, showing unique gas and vapor sorption. Chem. Commun. 2011, 47, 8106–8108. [Google Scholar] [CrossRef] [PubMed]
- Zeng, M.-H.; Feng, X.-L.; Chen, X.-M. Crystal-to-crystal transformations of a microporous metal–organic laminated framework triggered by guest exchange, dehydration and readsorption. Dalton Trans. 2004, 2217–2223. [Google Scholar] [CrossRef] [PubMed]
- Dolomanov, O.V.; Bourhis, L.J.; Gildea, R.J.; Howard, J.A.K.; Puschmann, H. OLEX2: A complete structure solution, refinement and analysis program. J. Appl. Crystallogr. 2009, 42, 339–341. [Google Scholar] [CrossRef]
- Sheldrick, G.M. SHELXT – Integrated space-group and crystal-structure determination. Acta Crystallographica A 2015, 71, 3–8. [Google Scholar] [CrossRef] [Green Version]
- Sheldrick, G.M. Crystal structure refinement with SHELXL. Acta Crystallographica C 2015, 71, 3–8. [Google Scholar] [CrossRef]
- Agilent. CrysAlis PRO; Agilent Technologies Ltd and O: Yarnton, UK, 2014. [Google Scholar]
- Oxford Diffraction. CrysAlis RED; Oxford Diffraction Ltd: Oxfordshire, UK, 2006. [Google Scholar]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Parameters | N1–[Cu(L1)2]n | N2–[Cu(L2)2]n | tC1–Cu(L3)2 | mC1–Cu(L3)2 | [HL3][NO3] |
|---|---|---|---|---|---|
| CCDC code | 1979395 | 1979396 | 2009855 | 1979398 | 1979397 |
| Empirical formula | C24H28CuN2O4 | C24H28CuN2O4 | C24H28CuN2O4 | C24H28CuN2O4 | C12H16N2O5 |
| Formula weight | 472.02 | 472.02 | 472.02 | 472.02 | 268.27 |
| Temperature/K | 130.6(3) | 293(2) | 293(2) | 133(2) | 293(2) |
| Crystal system | monoclinic | monoclinic | triclinic | Monoclinic | Monoclinic |
| Space group | P21/n | P21/n | P-1 | P21/n | P21/c |
| a/Å | 9.67360(10) | 11.5058(3) | 6.3111(8) | 13.1273(6) | 11.3313(4) |
| b/Å | 12.46690(10) | 9.3195(2) | 9.4952(11) | 6.0457(2) | 10.0225(4) |
| c/Å | 9.73330(10) | 12.3363(3) | 10.5041(13) | 14.2591(5) | 11.9033(5) |
| α/° | 90 | 90 | 94.356(10) | 90 | 90 |
| β/° | 90.0270(10) | 108.256(3) | 106.293(11) | 92.322(3) | 91.277(3) |
| γ/° | 90 | 90 | 98.971(10) | 90 | 90 |
| Volume/Å3 | 1173.83(2) | 1256.22(6) | 592.01(13) | 1130.73(8) | 1351.50(9) |
| Z | 2 | 2 | 1 | 2 | 4 |
| ρcalcg/cm3 | 1.335 | 1.248 | 1.324 | 1.386 | 1.318 |
| μ/mm-1 | 1.569 | 0.898 | 0.953 | 1.629 | 0.103 |
| F(000) | 494.0 | 494.0 | 247.0 | 494.0 | 568.0 |
| Crystal size/mm3 | 0.62 × 0.36 × 0.34 | 0.15 × 0.1 × 0.05 | 0.3 × 0.05 × 0.03 | 0.2 × 0.1 × 0.02 | 0.2 × 0.15 × 0.1 |
| Radiation | Cu Kα (λ = 1.54184) | MoKα (λ = 0.71073) | MoKα (λ = 0.71073) | CuKα (λ = 1.54184) | MoKα (λ = 0.71073) |
| 2Θ range for data collection/° | 11.578 to 152.524 | 8.218 to 53.446 | 8.528 to 50.042 | 8.978 to 152.556 | 6.46 to 59.074 |
| Index ranges | −10 ≤ h ≤ 12, −15 ≤ k ≤ 15, −12 ≤ l ≤ 10 | −14 ≤ h ≤ 14, −11 ≤ k ≤ 11, −15 ≤ l ≤ 15 | −7 ≤ h ≤ 7, −11 ≤ k ≤ 11, −6 ≤ l ≤ 12 | −15 ≤ h ≤ 16, −7 ≤ k ≤ 7, −17 ≤ l ≤ 12 | −15 ≤ h ≤ 15, −13 ≤ k ≤ 13, −14 ≤ l ≤ 14 |
| Reflections collected | 21,676 | 56,535 | 2094 | 4407 | 50,829 |
| Independent reflections | 2428 (Rint = 0.0320, Rsigma = 0.0123) | 2662 (Rint = 0.0444, Rsigma = 0.0160) | 2094 (Rint = merged, Rsigma = 0.0956) | 2299 (Rint = 0.0209, Rsigma = 0.0254) | 3209 (Rint = 0.0762, Rsigma = 0.0317) |
| Data/restraints/parameters | 2428/0/188 | 2662/0/166 | 2094/0/146 | 2299/0/176 | 3209/0/214 |
| Goodness-of-fit on F2 | 1.083 | 1.038 | 1.096 | 1.074 | 1.098 |
| Final R indexes [I ≥ 2σ (I)] | R1 = 0.0321, wR2 = 0.0860 | R1 = 0.0359, wR2 = 0.0928 | R1 = 0.0701, wR2 = 0.1488 | R1 = 0.0460, wR2 = 0.1288 | R1 = 0.0752, wR2 = 0.1715 |
| Final R indexes (all data) | R1 = 0.0325, wR2 = 0.0862 | R1 = 0.0448, wR2 = 0.0976 | R1 = 0.1134, wR2 = 0.1698 | R1 = 0.0517, wR2 = 0.1357 | R1 = 0.1101, wR2 = 0.1906 |
| Largest diff. peak/hole/e Å-3 | 0.32/-0.38 | 0.37/-0.29 | 0.37/-0.49 | 0.92/-0.55 | 0.61/-0.17 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Walczak, A.; Kurpik, G.; Stefankiewicz, A.R. Intrinsic Effect of Pyridine-N-Position on Structural Properties of Cu-Based Low-Dimensional Coordination Frameworks. Int. J. Mol. Sci. 2020, 21, 6171. https://doi.org/10.3390/ijms21176171
Walczak A, Kurpik G, Stefankiewicz AR. Intrinsic Effect of Pyridine-N-Position on Structural Properties of Cu-Based Low-Dimensional Coordination Frameworks. International Journal of Molecular Sciences. 2020; 21(17):6171. https://doi.org/10.3390/ijms21176171
Chicago/Turabian StyleWalczak, Anna, Gracjan Kurpik, and Artur R. Stefankiewicz. 2020. "Intrinsic Effect of Pyridine-N-Position on Structural Properties of Cu-Based Low-Dimensional Coordination Frameworks" International Journal of Molecular Sciences 21, no. 17: 6171. https://doi.org/10.3390/ijms21176171
APA StyleWalczak, A., Kurpik, G., & Stefankiewicz, A. R. (2020). Intrinsic Effect of Pyridine-N-Position on Structural Properties of Cu-Based Low-Dimensional Coordination Frameworks. International Journal of Molecular Sciences, 21(17), 6171. https://doi.org/10.3390/ijms21176171