The Lysine Specific Demethylase-1 Negatively Regulates the COL9A1 Gene in Human Articular Chondrocytes

 , and
, and
Abstract
:
1. Introduction
2. Results
2.1. LSD1 Labeling in OA Cartilage
2.2. LSD1 Depletion Leads to the Up-Regulation of the Expression of the Cartilage-Specific Gene COL9A1 in HACs
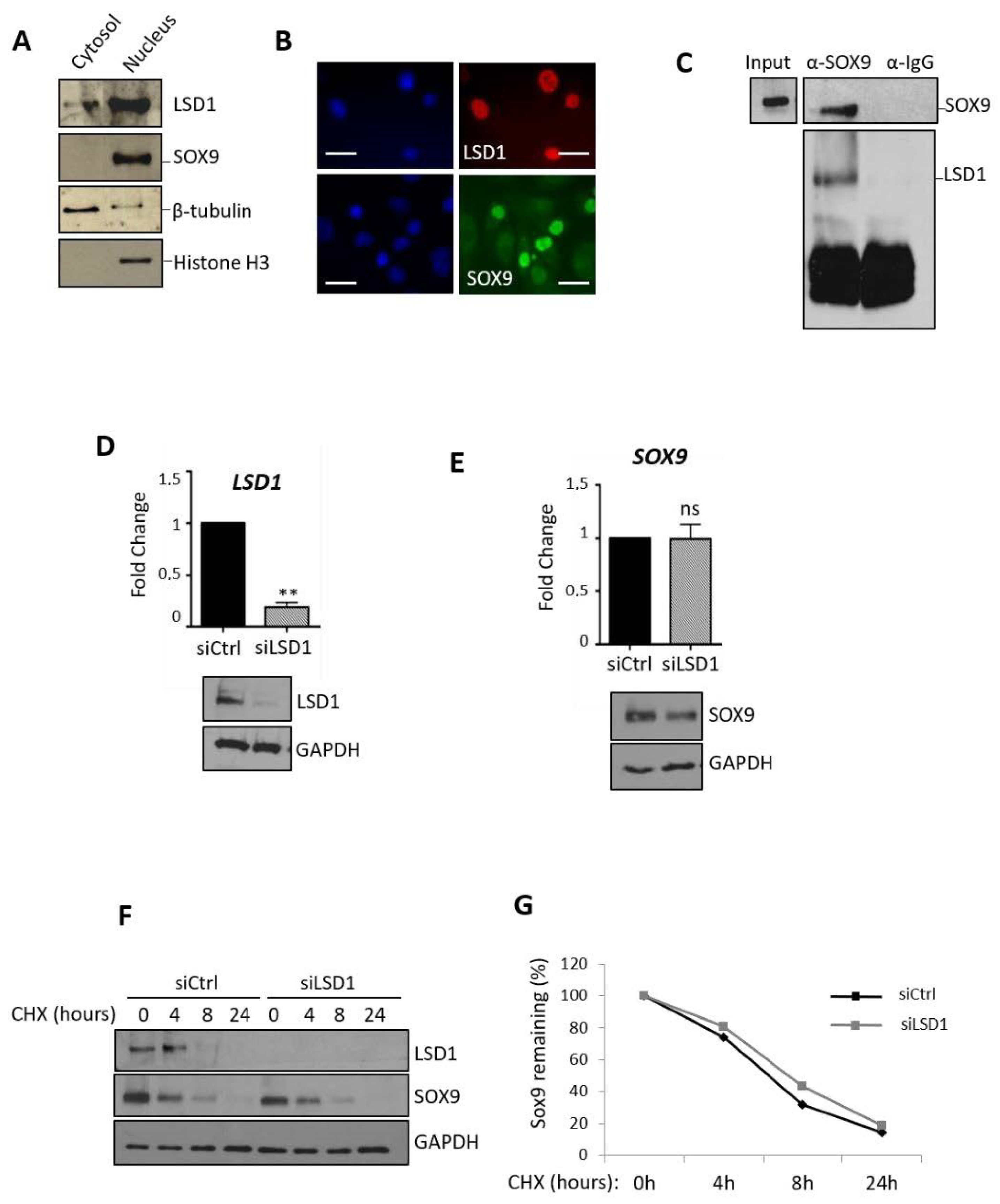
2.3. LSD1 Does not Affect the Level of Expression of SOX9 or Its Stability but Associates with SOX9 Protein
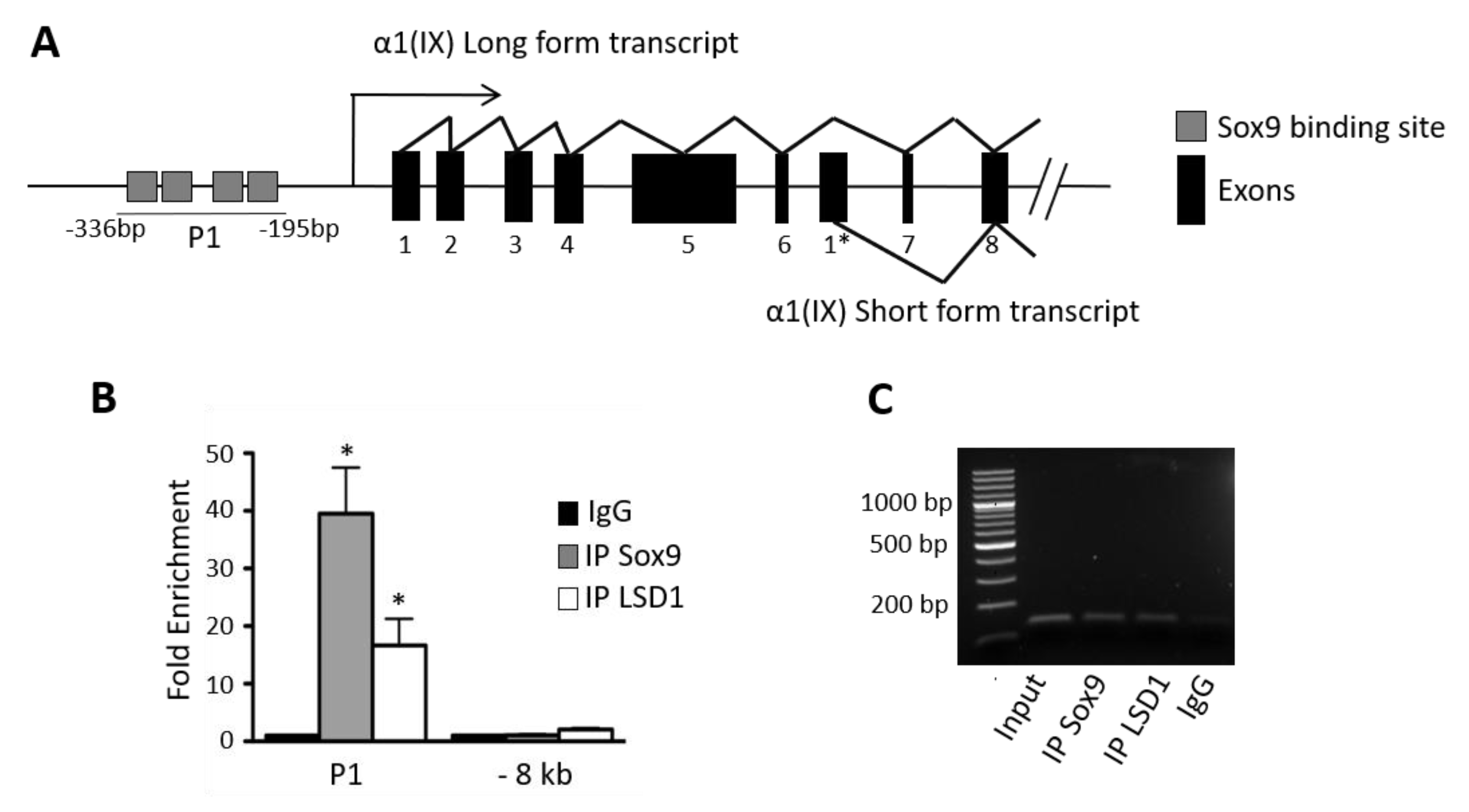
2.4. LSD1 Is Recruited onto the SOX9- Binding Region of the COL9A1 Promoter
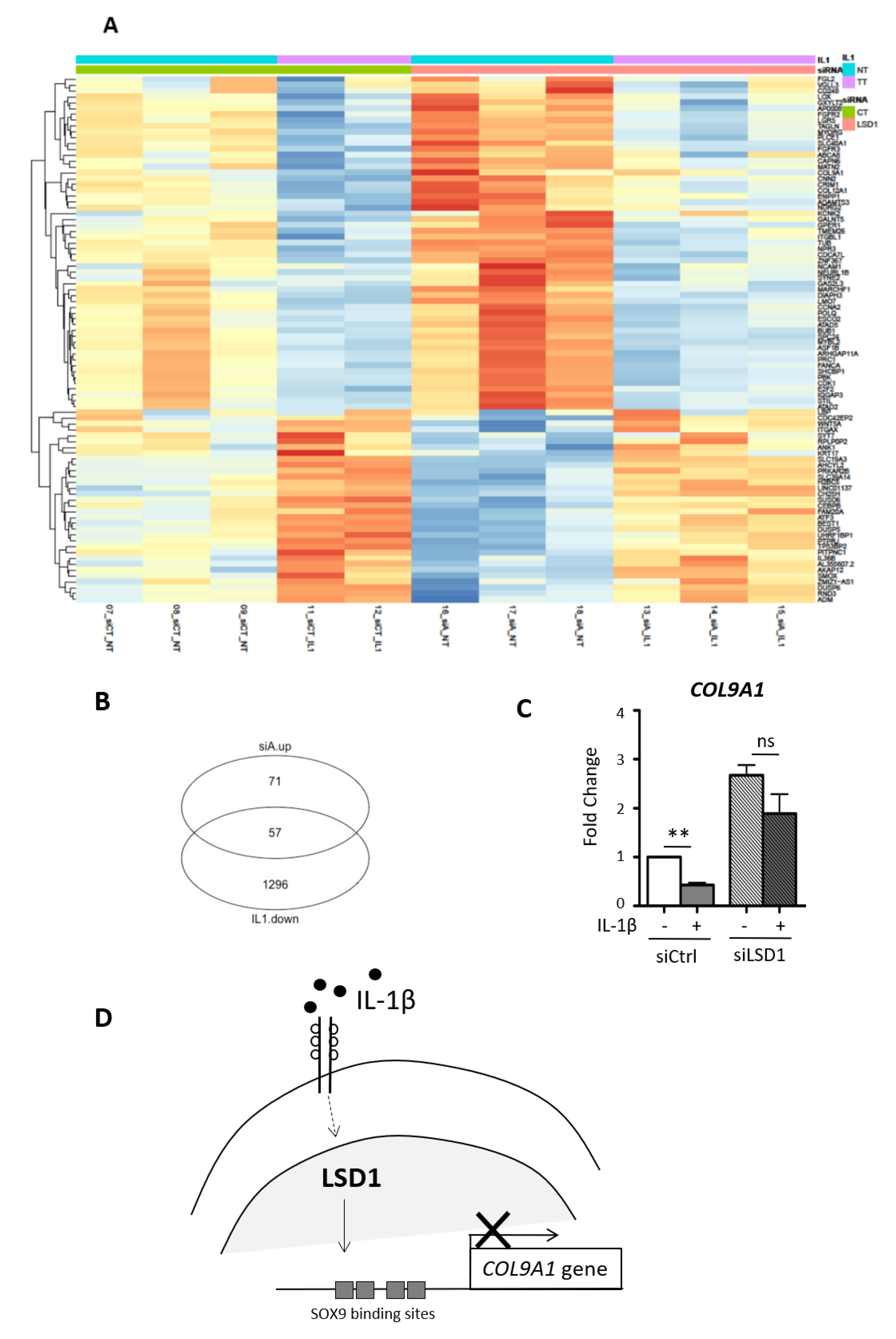
2.5. The Down-Regulation of the COL9A1 Gene Expression by the Inflammatory Cytokine IL1-β Is Blocked When LSD1 Is Depleted
3. Discussion
4. Materials and Methods
4.1. Cartilage Samples
4.2. Chondrocyte Isolation and Primary Monolayer Culture
4.3. Western Blot
4.4. Immunofluorescence
4.5. Transient Transfection
4.6. Subcellular Fractionation
4.7. Co-Immunoprecipitation
4.8. Real-Time PCR Analysis
4.9. RNA Sequencing and Bioinformatic Analysis
4.10. Chromatin Immunoprecipitation (ChIP) Assay
4.11. Immunohistochemistry
4.12. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Sulzbacher, I. Osteoarthritis: Histology and pathogenesis. Wien. Med. Wochenschr. 2012, 163, 212–219. [Google Scholar] [CrossRef] [PubMed]
- Aigner, T.; Kurz, B.; Fukui, N.; Sandell, L. Roles of chondrocytes in the pathogenesis of osteoarthritis. Curr. Opin. Rheumatol. 2002, 14, 578–584. [Google Scholar] [CrossRef] [PubMed]
- Eyre, D. The collagens of articular cartilage. Semin. Arthritis Rheum. 1991, 21, 2–11. [Google Scholar] [CrossRef]
- Eyre, D.R.; Weis, M.A.; Wu, J.-J. Articular cartilage collagen: An irreplaceable framework? Eur. Cells Mater. 2006, 12, 57–63. [Google Scholar] [CrossRef]
- Olsen, R.; Olsen, B.R. Collagen IX. Int. J. Biochem. Cell Biol. 1997, 29, 555–558. [Google Scholar] [CrossRef]
- Holden, P.; Meadows, R.S.; Chapman, K.L.; Grant, M.E.; Kadler, K.E.; Briggs, M.D. Cartilage Oligomeric Matrix Protein Interacts with Type IX Collagen, and Disruptions to These Interactions Identify a Pathogenetic Mechanism in a Bone Dysplasia Family. J. Boil. Chem. 2000, 276, 6046–6055. [Google Scholar] [CrossRef] [Green Version]
- Heilig, J.; Dietmar, H.F.; Brachvogel, B.; Paulsson, M.; Zaucke, F.; Niehoff, A. Collagen IX deficiency leads to premature vascularization and ossification of murine femoral heads through an imbalance of pro- and antiangiogenic factors. Osteoarthr. Cartil. 2020, 28, 988–999. [Google Scholar] [CrossRef]
- Blumbach, K.; DeGroot, J.; Paulsson, M.; Van Osch, G.J.V.M.; Zaucke, F.; Bastiaansen-Jenniskens, Y.M. Combined role of type IX collagen and cartilage oligomeric matrix protein in cartilage matrix assembly: Cartilage oligomeric matrix protein counteracts type IX collagen–induced limitation of cartilage collagen fibril growth in mouse chondrocyte cultures. Arthritis Rheum. 2009, 60, 3676–3685. [Google Scholar] [CrossRef]
- Dreier, R. Hypertrophic differentiation of chondrocytes in osteoarthritis: The developmental aspect of degenerative joint disorders. Arthritis Res. 2010, 12, 216. [Google Scholar] [CrossRef] [Green Version]
- Fasslert, R.; Schnegelsberg, P.N.J.; Dausmant, J.; Shinya, T.Y.; Mccarthy, M.T.; Olse, B.R.; Jaenisch, R. Mice lacking al (IX) collagen develop noninflammatory degenerative joint disease. Proc. Natl. Acad. Sci. USA 1994, 91, 5070–5074. [Google Scholar] [CrossRef] [Green Version]
- Hu, K.; Xu, L.; Cao, L.; Flahiff, C.M.; Brussiau, J.; Ho, K.; Setton, L.A.; Youn, I.; Guilak, F.; Olsen, B.R.; et al. Pathogenesis of osteoarthritis-like changes in the joints of mice deficient in type IX collagen. Arthritis Rheum. 2006, 54, 2891–2900. [Google Scholar] [CrossRef] [PubMed]
- Akiyama, H.; Chaboissier, M.-C.; Martin, J.F.; Schedl, A.; De Crombrugghe, B. The transcription factor Sox9 has essential roles in successive steps of the chondrocyte differentiation pathway and is required for expression of Sox5 and Sox6. Genes Dev. 2002, 16, 2813–2828. [Google Scholar] [CrossRef] [Green Version]
- Haag, J.; Gebhard, P.M.; Aigner, T. SOX Gene Expression in Human Osteoarthritic Cartilage. Pathobiology 2008, 75, 195–199. [Google Scholar] [CrossRef]
- Tchetina, E.V.; Squires, G.; Poole, A.R. Increased type II collagen degradation and very early focal cartilage degeneration is associated with upregulation of chondrocyte differentiation related genes in early human articular cartilage lesions. J. Rheumatol. 2005, 32, 876–886. [Google Scholar]
- Bridgewater, L.C.; Lefebvre, V.; De Crombrugghe, B. Chondrocyte-specific enhancer elements in the Col11a2 gene resemble the Col2a1 tissue-specific enhancer. J. Boil. Chem. 1998, 273, 14998–15006. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Genzer, M.A.; Bridgewater, L.C. A Col9a1 enhancer element activated by two interdependent SOX9 dimers. Nucleic Acids Res. 2007, 35, 1178–1186. [Google Scholar] [CrossRef]
- Lefebvre, V.; Huang, W.; Harley, V.R.; Goodfellow, P.N.; De Crombrugghe, B. SOX9 is a potent activator of the chondrocyte-specific enhancer of the pro alpha1(II) collagen gene. Mol. Cell. Boil. 1997, 17, 2336–2346. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sekiya, I.; Tsuji, K.; Koopman, P.; Watanabe, H.; Yamada, Y.; Shinomiya, K.; Nifuji, A.; Noda, M. SOX9 Enhances Aggrecan Gene Promoter/Enhancer Activity and Is Up-regulated by Retinoic Acid in a Cartilage-derived Cell Line, TC6. J. Boil. Chem. 2000, 275, 10738–10744. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ikeda, T.; Kamekura, S.; Mabuchi, A.; Kou, I.; Seki, S.; Takato, T.; Nakamura, K.; Kawaguchi, H.; Ikegawa, S.; Chung, U.-I. The combination of SOX5, SOX6, and SOX9 (the SOX trio) provides signals sufficient for induction of permanent cartilage. Arthritis Rheum. 2004, 50, 3561–3573. [Google Scholar] [CrossRef] [PubMed]
- Hata, K.; Takashima, R.; Amano, K.; Ono, K.; Nakanishi, M.; Yoshida, M.; Wakabayashi, M.; Matsuda, A.; Maeda, Y.; Suzuki, Y.; et al. Arid5b facilitates chondrogenesis by recruiting the histone demethylase Phf2 to Sox9-regulated genes. Nat. Commun. 2013, 4, 2850. [Google Scholar] [CrossRef] [Green Version]
- Morera, L.; Lübbert, M.; Jung, M. Targeting histone methyltransferases and demethylases in clinical trials for cancer therapy. Clin. Epigenetics 2016, 8, 57. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kouzarides, T. Chromatin Modifications and Their Function. Cell 2007, 128, 693–705. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bannister, A.J.; Kouzarides, T. Regulation of chromatin by histone modifications. Cell Res. 2011, 21, 381–395. [Google Scholar] [CrossRef] [PubMed]
- Maiques-Diaz, A.; Somervaille, T.C.P. LSD1: Biologic roles and therapeutic targeting. Epigenomics 2016, 8, 1103–1116. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- El Mansouri, F.E.; Nebbaki, S.-S.; Kapoor, M.; Afif, H.; Martel-Pelletier, J.; Pelletier, J.-P.; Benderdour, M.; Fahmi, H. Lysine-specific demethylase 1-mediated demethylation of histone H3 lysine 9 contributes to interleukin 1β-induced microsomal prostaglandin E synthase 1 expression in human osteoarthritic chondrocytes. Arthritis Res. Ther. 2014, 16, R113. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, J.; Scully, K.; Zhu, X.; Cai, L.; Zhang, J.; Prefontaine, G.G.; Krones, A.; Ohgi, K.A.; Zhu, P.; Garcia-Bassets, I.; et al. Opposing LSD1 complexes function in developmental gene activation and repression programmes. Nature 2007, 446, 882–887. [Google Scholar] [CrossRef] [PubMed]
- Zhang, P.; Jimenez, S.A.; Stokes, D.G. Regulation of HumanCOL9A1Gene Expression. J. Boil. Chem. 2002, 278, 117–123. [Google Scholar] [CrossRef] [Green Version]
- Rice, S.J.; Beier, F.; Young, D.A.; Loughlin, J. Interplay between genetics and epigenetics in osteoarthritis. Nat. Rev. Rheumatol. 2020, 16, 268–281. [Google Scholar] [CrossRef]
- Castillo-Aguilera, O.; Depreux, P.; Halby, L.; Arimondo, P.B.; Goossens, L. DNA Methylation Targeting: The DNMT/HMT Crosstalk Challenge. Biomolecules 2017, 7, 3. [Google Scholar] [CrossRef]
- Imagawa, K.; De Andres, M.C.; Hashimoto, K.; Itoi, E.; Otero, M.; Roach, H.I.; Goldring, M.B.; Oreffo, R.O.C. Association of Reduced Type IX Collagen Gene Expression in Human Osteoarthritic Chondrocytes With Epigenetic Silencing by DNA Hypermethylation. Arthritis Rheumatol. 2014, 66, 3040–3051. [Google Scholar] [CrossRef] [Green Version]
- Zhang, M.; Lu, Q.; Egan, B.; Zhong, X.; Brandt, K.D.; Wang, J. Epigenetically mediated spontaneous reduction of NFAT1 expression causes imbalanced metabolic activities of articular chondrocytes in aged mice. Osteoarthr. Cartil. 2016, 24, 1274–1283. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yoneyama, M.; Tochio, N.; Umehara, T.; Koshiba, S.; Inoue, M.; Yabuki, T.; Aoki, M.; Seki, E.; Matsuda, T.; Watanabe, S.; et al. Structural and Functional Differences of SWIRM Domain Subtypes. J. Mol. Boil. 2007, 369, 222–238. [Google Scholar] [CrossRef] [PubMed]
- Yang, M.; Gocke, C.B.; Luo, X.; Borek, D.; Tomchick, D.R.; Machius, M.; Otwinowski, Z.; Yu, H. Structural Basis for CoREST-Dependent Demethylation of Nucleosomes by the Human LSD1 Histone Demethylase. Mol. Cell 2006, 23, 377–387. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.; Zhou, B.P. Epigenetic regulation of LSD1 during mammary carcinogenesis. Mol. Cell. Oncol. 2014, 1, e963426. [Google Scholar] [CrossRef] [PubMed]
- Rodova, M.; Lu, Q.; Li, Y.; Woodbury, B.G.; Crist, J.D.; Gardner, B.M.; Yost, J.G.; Zhong, X.B.; Anderson, H.C.; Wang, J. Nfat1 regulates adult articular chondrocyte function through its age-dependent expression mediated by epigenetic histone methylation. J. Bone Miner. Res. 2011, 26, 1974–1986.29. [Google Scholar] [CrossRef] [Green Version]
- Nicholson, T.B.; Chen, T. LSD1 demethylates histone and non-histone proteins. Epigenetics 2009, 4, 129–132. [Google Scholar] [CrossRef] [Green Version]
- Ito, T.; Yadav, N.; Lee, J.; Furumatsu, T.; Yamashita, S.; Yoshida, K.; Taniguchi, N.; Hashimoto, M.; Tsuchiya, M.; Ozaki, T.; et al. Arginine methyltransferase CARM1/PRMT4 regulates endochondral ossification. BMC Dev. Biol. 2009, 9, 47. [Google Scholar] [CrossRef] [Green Version]
- Huang, W.; Zhou, X.; Lefebvre, V.; De Crombrugghe, B. Phosphorylation of SOX9 by Cyclic AMP-Dependent Protein Kinase A Enhances SOX9’s Ability To Transactivate aCol2a1 Chondrocyte-Specific Enhancer. Mol. Cell. Boil. 2000, 20, 4149–4158. [Google Scholar] [CrossRef] [Green Version]
- Walport, L.J.; Hopkinson, R.J.; Chowdhury, R.; Schiller, R.; Ge, W.; Kawamura, A.; Schofield, C.J. Arginine demethylation is catalysed by a subset of JmjC histone lysine demethylases. Nat. Commun. 2016, 7, 11974. [Google Scholar] [CrossRef]
- Diab, M. The role of type IX collagen in osteoarthritis and rheumatoid arthritis. Orthop. Rev. 1993, 22, 165–170. [Google Scholar]
- Kamper, M.; Hamann, N.; Prein, C.; Clausen-Schaumann, H.; Farkas, Z.; Aszodi, A.; Niehoff, A.; Paulsson, M.; Zaucke, F. Early changes in morphology, bone mineral density and matrix composition of vertebrae lead to disc degeneration in aged collagen IX -/- mice. Matrix Biol. 2016, 49, 132–143. [Google Scholar] [CrossRef]
- Ge, X.; Ma, X.; Meng, J.; Zhang, C.; Ma, K.; Zhou, C. Role of Wnt-5A in interleukin-1β-induced matrix metalloproteinase expression in rabbit temporomandibular joint condylar chondrocytes. Arthritis Rheum. 2009, 60, 2714–2722. [Google Scholar] [CrossRef] [PubMed]
- Yang, C.-Y.; Chanalaris, A.; Troeberg, L. ADAMTS and ADAM metalloproteinases in osteoarthritis - looking beyond the ’usual suspects’. Osteoarthr. Cartil. 2017, 25, 1000–1009. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Choi, M.-C.; Jo, J.; Park, J.; Kang, H.K.; Park, Y. NFk-B Signaling Pathways in Osteoarthritic Cartilage Destruction. Cells 2019, 8, 734. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Andrews, S. FastQC A Quality Control Application for FastQ Files. Available online: https://www.bioinformatics.babraham.ac.uk/projects/fastqc/ (accessed on 30 August 2020).
- Ewels, P.; Magnusson, M.; Lundin, S.; Käller, M. MultiQC: Summarize analysis results for multiple tools and samples in a single report. Bioinformatics 2016, 32, 3047–3048. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, D.; Langmead, B.; Salzberg, S.L. HISAT: A fast spliced aligner with low memory requirements. Nat. Methods 2015, 12, 357–360. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liao, Y.; Smyth, G.K.; Shi, W. featureCounts: An efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics 2013, 30, 923–930. [Google Scholar] [CrossRef] [Green Version]
- Love, M.L.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Boil. 2014, 15, 31. [Google Scholar] [CrossRef] [Green Version]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Application | Gene | Forward Primer | Reverse Primer |
|---|---|---|---|
| qPCR | LSD1 | 5′-TGAGAAAATCCACGCTGGCA -3′ | 5′-TCCTCCCTGTGCTCTAGGTC-3′ |
| SOX9 | 5′-ACGCCGAGCTCAGCAAGA-3′ | 5′-CACGAACGGCCGCTTCT-3′ | |
| COL2A1 | 5′-TCCATGTTGCAGAAAACCTTCA-3′ | 5′-GGAAGAGTGGAGACTACTGGATTGAC-3′ | |
| COL9A1 | 5′-ACGGTTTGCCTGGAGCTAT-3′ | 5′-ACCGTCTCGGCCATTTCT-3′ | |
| COL11A2 | 5′-CCTGACCCACTGA GTATGTTCAT-3′ | 5′-TTGCAGGATCAGGGAAAGTGA-3′ | |
| ACAN | 5′-TCGAGGACAGCGAGGCC-3′ | 5′-TCGAGGGTGTAGCGTGTAGAGA-3′ | |
| RPL30 | 5′-CCTAAGGCAGGAAGATGGGGTG-3′ | 5′-AGTCTGCTTGTACCCCAGGA-3′ | |
| ChIP | COL9A1 (P1) | 5′-CCTCCCAGTGGGCACATTTT-3′ | 5′-TCCAGGCAAGGTGCTATAGGAACA-3′ |
| −8 kb | 5’-AGCTGTTTGACTGGTTCACCA-3’ | 5’-TACCTCGCAACAATCCAGCA-3’ |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Durand, A.-L.; Dufour, A.; Aubert-Foucher, E.; Oger-Desfeux, C.; Pasdeloup, M.; Lustig, S.; Servien, E.; Vaz, G.; Perrier-Groult, E.; Mallein-Gerin, F.; et al. The Lysine Specific Demethylase-1 Negatively Regulates the COL9A1 Gene in Human Articular Chondrocytes. Int. J. Mol. Sci. 2020, 21, 6322. https://doi.org/10.3390/ijms21176322
Durand A-L, Dufour A, Aubert-Foucher E, Oger-Desfeux C, Pasdeloup M, Lustig S, Servien E, Vaz G, Perrier-Groult E, Mallein-Gerin F, et al. The Lysine Specific Demethylase-1 Negatively Regulates the COL9A1 Gene in Human Articular Chondrocytes. International Journal of Molecular Sciences. 2020; 21(17):6322. https://doi.org/10.3390/ijms21176322
Chicago/Turabian StyleDurand, Anne-Laure, Alexandre Dufour, Elisabeth Aubert-Foucher, Christine Oger-Desfeux, Marielle Pasdeloup, Sebastien Lustig, Elvire Servien, Gualter Vaz, Emeline Perrier-Groult, Frederic Mallein-Gerin, and et al. 2020. "The Lysine Specific Demethylase-1 Negatively Regulates the COL9A1 Gene in Human Articular Chondrocytes" International Journal of Molecular Sciences 21, no. 17: 6322. https://doi.org/10.3390/ijms21176322
APA StyleDurand, A. -L., Dufour, A., Aubert-Foucher, E., Oger-Desfeux, C., Pasdeloup, M., Lustig, S., Servien, E., Vaz, G., Perrier-Groult, E., Mallein-Gerin, F., & Lafont, J. E. (2020). The Lysine Specific Demethylase-1 Negatively Regulates the COL9A1 Gene in Human Articular Chondrocytes. International Journal of Molecular Sciences, 21(17), 6322. https://doi.org/10.3390/ijms21176322