Developing Electron Microscopy Tools for Profiling Plasma Lipoproteins Using Methyl Cellulose Embedment, Machine Learning and Immunodetection of Apolipoprotein B and Apolipoprotein(a)

 , ,
, ,  and
and 
Abstract
:1. Introduction
2. Results
2.1. Contrasting and Optimization of Methylcellulose (MC) Film Thickness
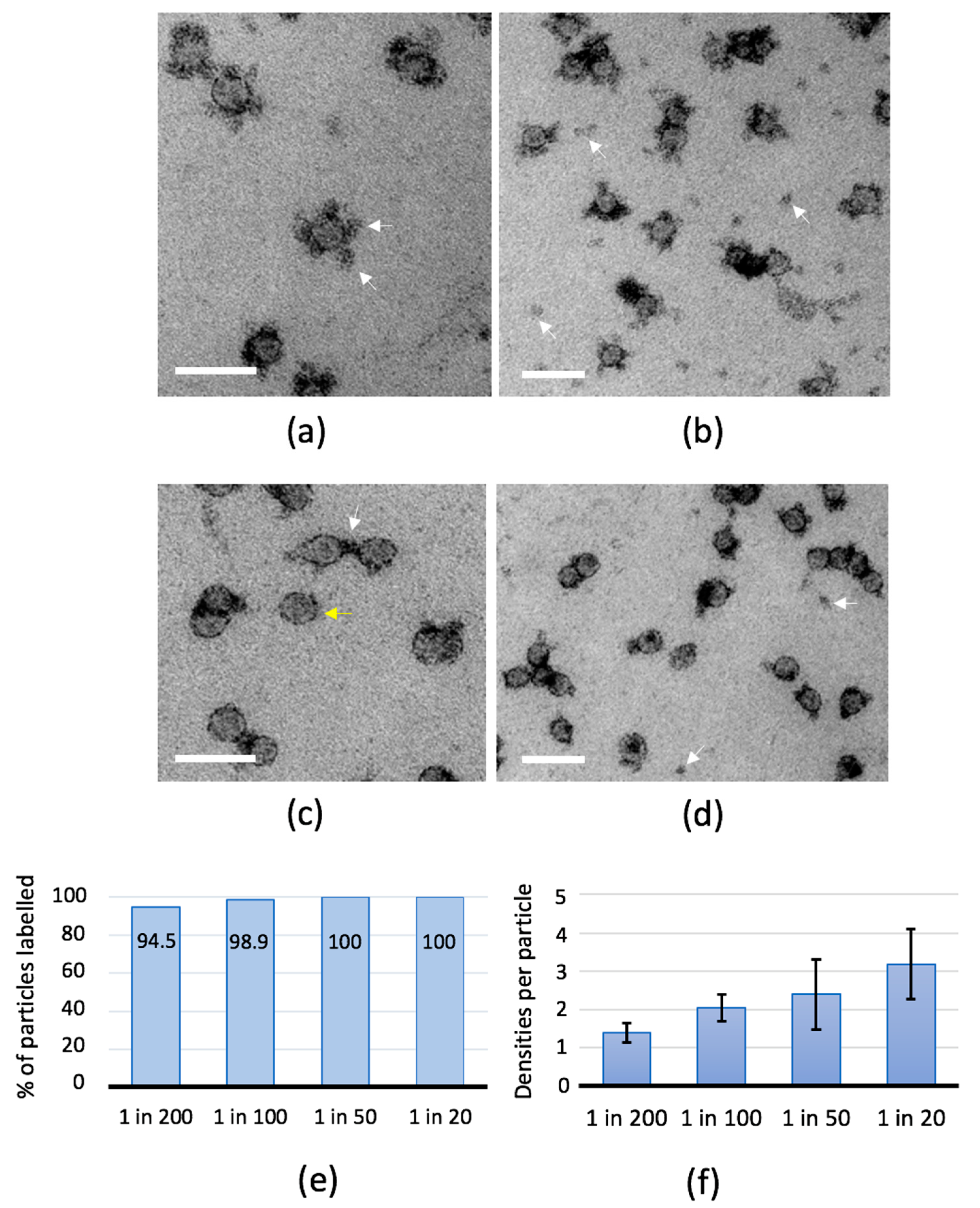
2.2. Antibody-Binding Studies
2.3. Optimizing Visualisation of Lipoprotein Particles from Human Plasma
2.4. Lp(a) Particles Identified Using Anti-Apolipoprotein(a) Antibodies
2.5. Deep Learning Approach to Identifying Lipoproteins
3. Discussion
4. Materials and Methods
4.1. Materials and Chemicals
4.2. Contrasting
4.3. Antibody-Binding Experiments
4.4. Gel Filtration
4.5. Mask R-CNN
4.6. Mask R-CNN Architecture and Implementation
4.7. Transfer Learning
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| ApoB | apolipoprotein B |
| BSA | 0.1% bovine serum albumin in PBS |
| Cryo-EM | cryo-electron microscopy |
| CNN | convolutional neural network |
| CV | coefficient of variation |
| CVD | cardiovascular risk |
| EM | electron microscopy |
| FPN | feature pyramid network |
| FSG | 0.5% fish skin gelatin in PBS |
| GPU | graphic processing unit |
| HDL | high-density lipoprotein |
| IDL | intermediate-density lipoprotein |
| KS | Kolmogorov-Smirnov |
| LDL | low-density lipoprotein |
| LP | lipoprotein particle |
| Lp(a) | lipoprotein (a) |
| mAP | median average precision |
| MC | methyl cellulose |
| NMR | nuclear magnetic resonance |
| PBS | phosphate buffered saline |
| R-CNN | region-based convolutional neural network |
| ResNet-50 | residual neural network that is 50 layers deep |
| RLP | remnant lipoprotein |
| RPN | region proposed network |
| SD | standard deviation |
| sdLDL | small dense LDL |
| STA | sodium silicotungstate |
| UA | uranyl acetate |
| VLDL | very low-density lipoprotein |
References
- Feingold, K.R.; Grunfeld, C. Introduction to Lipids and Lipoproteins. In Endotext; Feingold, K.R., Anawalt, B., Boyce, A., Eds.; MDText.com, Inc.: South Dartmouth, MA, USA, 2000. [Google Scholar]
- Karimi, N.; Cvjetkovic, A.; Jang, S.C.; Crescitelli, R.; Hosseinpour Feizi, M.A.; Nieuwland, R.; Lötvall, J.; Lässer, C. Detailed Analysis of the Plasma Extracellular Vesicle Proteome after Separation from Lipoproteins. Cell. Mol. Life Sci. 2018, 75, 2873–2886. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goldstein, J.L.; Brown, M.S. A Century of Cholesterol and Coronaries: From Plaques to Genes to Statins. Cell 2015, 161, 161–172. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lipid Modification Therapy for Preventing Cardiovascular Disease NICE. Available online: http://pathways.nice.org.uk/pathways/cardiovascular-disease-prevention (accessed on 7 August 2020).
- Schaefer, E.J.; Tsunoda, F.; Diffenderfer, M.; Polisecki, E.; Thai, N.; Asztalos, B. The Measurement of Lipids, Lipoproteins, Apolipoproteins, Fatty Acids, and Sterols, and Next Generation Sequencing for the Diagnosis and Treatment of Lipid Disorders. Endotext 2000. [Google Scholar]
- Hayashi, T.; Koba, S.; Ito, Y.; Hirano, T. Method for Estimating High sdLDL-C by Measuring Triglyceride and Apolipoprotein B Levels. Lipids Health Dis. 2017, 16, 21. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Duran, E.K.; Aday, A.W.; Cook, N.; Buring, J.E.; Ridker, P.M.; Pradhan, A.D. Triglyceride-Rich Lipoprotein Cholesterol, Small Dense LDL Cholesterol, and Incident Cardiovascular Disease. J. Am. Coll. Cardiol. 2020, 75, 2122. [Google Scholar] [CrossRef]
- Willeit, P.; Ridker, P.M.; Nestel, P.J.; Simes, J.; Tonkin, A.M.; Pedersen, T.R.; Schwartz, G.G.; Olsson, A.G.; Colhoun, H.M.; Kronenberg, F.; et al. Baseline and On-Statin Treatment Lipoprotein(a) Levels for Prediction of Cardiovascular Events: Individual Patient-Data Meta-Analysis of Statin Outcome Trials. Lancet 2018, 392, 1311–1320. [Google Scholar] [CrossRef] [Green Version]
- Tsimikas, S. Lipoprotein(a): Novel Target and Emergence of Novel Therapies to Lower Cardiovascular Disease Risk. Curr. Opin. Endocrinol. Diabetes Obes. 2016, 23, 157–164. [Google Scholar] [CrossRef] [Green Version]
- Ivanova, E.A.; Myasoedova, V.A.; Melnichenko, A.A.; Grechko, A.V.; Orekhov, A.N. Small Dense Low-Density Lipoprotein as Biomarker for Atherosclerotic Diseases. Oxid. Med. Cell. Longev. 2017, 2017, 1–10. [Google Scholar] [CrossRef]
- Sines, J.; Rothnagel, R.; van Heel, M.; Gaubatz, J.W.; Morrisett, J.D.; Chiu, W. Electron Cryomicroscopy and Digital Image Processing of Lipoprotein(a). Chem. Phys. Lipids 1994, 67, 81–89. [Google Scholar] [CrossRef]
- Nordestgaard, B.G.; Chapman, M.J.; Ray, K.; Borén, J.; Andreotti, F.; Watts, G.F.; Ginsberg, H.; Amarenco, P.; Catapano, A.L.; Descamps, O.S.; et al. Lipoprotein(a) as a Cardiovascular Risk Factor: Current Status. Eur. Hear. J. 2010, 31, 2844–2853. [Google Scholar] [CrossRef]
- Kamstrup, P.R.; Benn, M.; Tybjærg-Hansen, A.; Nordestgaard, B.G. Extreme Lipoprotein(a) Levels and Risk of Myocardial Infarction in the General Population: The Copenhagen City Heart Study. Circulation 2008, 117, 176–184. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kamstrup, P.R.; Nordestgaard, B.G. Lipoprotein(a) Should Be Taken Much More Seriously. Biomark. Med. 2009, 3, 439–441. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Clarke, R.; Peden, J.F.; Hopewell, J.C.; Kyriakou, T.; Goel, A.; Heath, S.C.; Parish, S.; Barlera, S.; Franzosi, M.G.; Rust, S.; et al. Genetic Variants Associated with Lp(a) Lipoprotein Level and Coronary Disease. N. Engl. J. Med. 2009, 361, 2518–2528. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McCormick, S.P.A. Lipoprotein(a): Biology and Clinical Importance. Clin. Biochem. Rev. 2004, 25, 69–80. [Google Scholar]
- Marcovina, S.M.; Albers, J.J. Lipoprotein (a) Measurements for Clinical Application. J. Lipid Res. 2016, 57, 526–537. [Google Scholar] [CrossRef] [Green Version]
- Wilson, D.P.; Jacobson, T.A.; Jones, P.H.; Koschinsky, M.L.; McNeal, C.J.; Nordestgaard, B.G.; Orringer, C.E. Use of Lipoprotein(a) in Clinical Practice: A Biomarker Whose Time Has Come. A Scientific Statement from the National Lipid Association. J. Clin. Lipidol. 2019, 13, 374–392. [Google Scholar] [CrossRef]
- Kim, J.Y.; Park, J.H.; Jeong, S.W.; Schellingerhout, D.; Park, J.E.; Lee, D.K.; Choi, W.J.; Chae, S.L.; Kim, D.E. High Levels of Remnant Lipoprotein Cholesterol is a Risk Factor for Large Artery Atherosclerotic Stroke. J. Clin. Neurol. 2011, 7, 203–209. [Google Scholar] [CrossRef]
- Masuoka, H.; Kamei, S.; Wagayama, H.; Ozaki, M.; Kawasaki, A.; Tanaka, T.; Kitamura, M.; Katoh, S.; Shintani, U.; Misaki, M.; et al. Association of Remnant-Like Particle Cholesterol with Coronary Artery Disease in Patients with Normal Total Cholesterol Levels. Am. Heart J. 2000, 139, 305–310. [Google Scholar] [CrossRef]
- May, H.T.; Muhlestein, J.B.; Ma, Y.; López, J.A.G.; Coll, B.; Nelson, J. Effects of Evolocumab on the ApoA1 Remnant Ratio: A Pooled Analysis of Phase 3 Studies. Cardiol. Ther. 2019, 8, 91–102. [Google Scholar] [CrossRef] [Green Version]
- Welsh, C.; Celis-Morales, C.A.; Brown, R.; Mackay, D.F.; Lewsey, J.; Mark, P.B.; Gray, S.R.; Ferguson, L.D.; Anderson, J.J.; Lyall, D.M.; et al. Comparison of Conventional Lipoprotein Tests and Apolipoproteins in the Prediction of Cardiovascular Disease. Circulation 2019, 140, 542–552. [Google Scholar] [CrossRef]
- Allaire, J.; Vors, C.; Couture, P.; Lamarche, B. LDL Particle Number and Size and Cardiovascular Risk: Anything New Under The Sun? Curr. Opin. Lipidol. 2017, 28, 261–266. [Google Scholar] [CrossRef] [PubMed]
- Freedman, D.S.; Otvos, J.D.; Jeyarajah, E.J.; Shalaurova, I.; Cupples, A.; Parise, H.; D’Agostino, R.B.; Wilson, P.W.F.; Schaefer, E.J. Sex and Age Differences in Lipoprotein Subclasses Measured by Nuclear Magnetic Resonance Spectroscopy: The Framingham study. Clin. Chem. 2004, 50, 1189–1200. [Google Scholar] [CrossRef] [PubMed]
- Hole, P.; Sillence, K.; Hannell, C.; Manus Maguire, C.; Roesslein, M.; Suarez, G.; Capracotta, S.; Magdolenova, Z.; Horev-Azaria, L.; Dybowska, A.; et al. Interlaboratory Comparison of Size Measurements on Nanoparticles Using Nanoparticle Tracking Analysis (NTA). J. Nanopart. Res. 2013, 15, 2101. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Momen-Heravi, F.; Balaj, L.; Alian, S.; Tigges, J.; Toxavidis, V.; Ericsson, M.; Distel, R.J.; Ivanov, A.R.; Skog, J.; Kuo, W.P. Alternative Methods for Characterization of Extracellular Vesicles. Front. Physiol. 2012, 3, 354. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Manus Maguire, C.; Rösslein, M.; Wick, P.; Prina-Mello, A. Characterisation of Particles in Solution—A Perspective on Light Scattering and Comparative Technologies. Sci. Technol. Adv. Mater. 2018, 19, 732–735. [Google Scholar] [CrossRef] [PubMed]
- Witte, D.R.; Taskinen, M.R.; Perttunen-Nio, H.; Van Tol, A.; Livingstone, S.; Colhoun, H.M. Study of Agreement between Ldl Size as Measured by Nuclear Magnetic Resonance and Gradient Gel Electrophoresis. J. Lipid Res. 2004, 45, 1069–1075. [Google Scholar] [CrossRef] [Green Version]
- Otvos, J.D.; Jeyarajah, E.J.; Bennett, D.W.; Krauss, R.M. Development of a Proton Nuclear Magnetic Resonance Spectroscopic Method for Determining Plasma Lipoprotein Concentrations and Subspecies Distributions from a Single, Rapid Measurement. Clin. Chem. 1992, 38, 1632–1638. [Google Scholar] [CrossRef]
- Soo, C.Y.; Song, Y.; Zheng, Y.; Campbell, E.C.; Riches, A.C.; Gunn-Moore, F.; Powis, S.J. Nanoparticle Tracking Analysis Monitors Microvesicle and Exosome Secretion from Immune Cells. Immunology 2012, 136, 192–197. [Google Scholar] [CrossRef] [Green Version]
- Welton, J.L.; Webber, J.P.; Botos, L.-A.; Jones, M.; Clayton, A. Ready-Made Chromatography Columns for Extracellular Vesicle Isolation from Plasma. J. Extracell. Vesicles 2015, 4, 27269. [Google Scholar] [CrossRef]
- Sódar, B.W.; Kittel, Á.; Pálóczi, K.; Vukman, K.V.; Osteikoetxea, X.; Szabó-Taylor, K.; Németh, A.; Sperlágh, B.; Baranyai, T.; Giricz, Z.; et al. Low-Density Lipoprotein Mimics Blood Plasma-Derived Exosomes and Microvesicles During Isolation and Detection. Sci. Rep. 2016, 6, 24316. [Google Scholar] [CrossRef] [Green Version]
- Caulfield, M.P.; Li, S.; Lee, G.; Blanche, P.J.; Salameh, W.A.; Benner, W.H.; Reitz, R.E.; Krauss, R.M. Direct Determination of Lipoprotein Particle Sizes and Concentrations by Ion Mobility Analysis. Clin. Chem. 2008, 54, 1307–1316. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hacker, C.; Asadi, J.; Pliotas, C.; Ferguson, S.; Sherry, L.; Marius, P.; Tello, J.; Jackson, D.; Naismith, J.; Lucocq, J.M. Nanoparticle Suspensions Enclosed in Methylcellulose: A New Approach For Quantifying Nanoparticles in Transmission Electron Microscopy. Sci. Rep. 2016, 6, 1–13. [Google Scholar] [CrossRef]
- Griffiths, G.; McDowall, A.; Back, R.; Dubochet, J. On the preparation of cryosections for immunocytochemistry. J. Ultrasructure Res. 1984, 89, 65–78. [Google Scholar] [CrossRef]
- Asadi, J.; Ferguson, S.; Raja, H.; Hacker, C.; Marius, P.; Ward, R.; Pliotas, C.; Naismith, J.; Lucocq, J. Enhanced Imaging of Lipid Rich Nanoparticles Embedded in Methylcellulose Films for Transmission Electron Microscopy Using Mixtures of Heavy Metals. Micron 2017, 99, 40–48. [Google Scholar] [CrossRef] [Green Version]
- Coronado-Gray, A.; Van Antwerpen, R. Lipid Composition Influences the Shape of Human Low Density Lipoprotein in Vitreous Ice. Lipids 2005, 40, 495–500. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van Rik, A.; Chen, G.C.; Pullinger, C.R.; Kane, J.P.; LaBelle, M.; Krauss, R.M.; Luna-Chavez, C.; Forte, T.M.; Gilkey, J.C. Cryo-Electron Microscopy of Low Density Lipoprotein and Reconstituted Discoidal High Density Lipoprotein: Imaging of the Apolipoprotein Moiety. J. Lipid Res. 1997, 38, 659–669. [Google Scholar]
- Wang, R.; Pokhariya, H.; McKenna, S.J.; Lucocq, J. Recognition of Immunogold Markers in Electron Micrographs. J. Struct. Biol. 2011, 176, 151–158. [Google Scholar] [CrossRef]
- Tong, H.; Zhang, L.; Kaspar, A.; Rames, M.J.; Huang, L.; Woodnutt, G.; Ren, G. Peptide-Conjugation Induced Conformational Changes in Human Igg1 Observed by Optimized Negative-Staining and Individual-Particle Electron Tomography. Sci. Rep. 2013, 3, 1089. [Google Scholar] [CrossRef]
- Yu, Y.; Kuang, Y.-L.; Lei, D.; Zhai, X.; Zhang, M.; Krauss, R.M.; Ren, G. Polyhedral 3D Structure of Human Plasma Very Low Density Lipoproteins by Individual Particle Cryo-Electron Tomography 1. J. Lipid Res. 2016, 57, 1879–1888. [Google Scholar] [CrossRef] [Green Version]
- Xu, R.; Fitts, A.; Li, X.; Fernandes, J.; Pochampally, R.; Mao, J.; Liu, Y.M. Quantification of Small Extracellular Vesicles by Size Exclusion Chromatography with Fluorescence Detection. Anal. Chem. 2016, 88, 10390–10394. [Google Scholar] [CrossRef] [Green Version]
- Böing, A.N.; van der Pol, E.; Grootemaat, A.E.; Coumans, F.A.W.; Sturk, A.; Nieuwland, R. Single-Step Isolation of Extracellular Vesicles by Size-Exclusion Chromatography. J. Extracell. Vesicles 2014, 3, 23430. [Google Scholar] [CrossRef] [PubMed]
- Kontush, A. HDL Particle Number and Size as Predictors of Cardiovascular Disease. Front. Pharmacol. 2015, 6, 218. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rajman, I.; Eacho, P.I.; Chowienczyk, P.J.; Ritter, J.M. LDL Particle Size: An Important Drug Target? Br. J. Clin. Pharmacol. 1999, 48, 125–133. [Google Scholar] [CrossRef] [PubMed]
- He, K.; Gkioxari, G.; Dollar, P.; Girshick, R. Mask R-CNN. In Proceedings of the IEEE International Conference on Computer Vision (ICCV), Venice, Italy, 22–29 October 2017. [Google Scholar] [CrossRef]
- Ren, S.; He, K.; Girshick, R.; Sun, J. Faster R-CNN: Towards Real-Time Object Detection with Region Proposal Networks. IEEE Trans. Pattern Anal. Mach. Intell. 2017, 39, 1137–1149. [Google Scholar] [CrossRef] [Green Version]
- Rodriguez-Garcia, E.; Ruiz-Nava, J.; Santamaria-Fernandez, S.; Fernandez-Garcia, J.C.; Vargas-Candela, A.; Yahyaoui, R.; Tinahones, F.J.; Bernal-Lopez, M.R.; Gomez-Huelgas, R. Characterization of Lipid Profile by Nuclear Magnetic Resonance Spectroscopy (1 H NMR) of Metabolically Healthy Obese Women after Weight Loss with Mediterranean Diet and Physical Exercise. Medicine 2017, 96, e7040. [Google Scholar] [CrossRef]
- Mallol, R.; Amigó, N.; Rodríguez, M.A.; Heras, M.; Vinaixa, M.; Plana, N.; Rock, E.; Ribalta, J.; Yanes, O.; Masana, L.; et al. Liposcale: A Novel Advanced Lipoprotein Test Based on 2d Diffusion-Ordered 1H NMR Spectroscopy. J. Lipid Res. 2015, 56, 737–746. [Google Scholar] [CrossRef] [Green Version]
- Takov, K.; Yellon, D.M.; Davidson, S.M. Comparison of Small Extracellular Vesicles Isolated from Plasma by Ultracentrifugation or Size-Exclusion Chromatography: Yield, Purity and Functional Potential. J. Extracell. Vesicles 2019, 8, 1560809. [Google Scholar] [CrossRef]
- Munroe, W.H.; Phillips, M.L.; Schumaker, V.N. Excessive Centrifugal Fields Damage High Density Lipoprotein. J. Lipid Res. 2015, 56, 1172–1181. [Google Scholar] [CrossRef] [Green Version]
- Chatterton, J.E.; Phillips, M.L.; Curtiss, L.K.; Milne, R.; Fruchart, J.C.; Schumaker, V.N. Immunoelectron Microscopy of Low Density Lipoproteins Yields a Ribbon and Bow Model for the Conformation of Apolipoprotein B on the Lipoprotein Surface. J. Lipid Res. 1995, 36, 2027–2037. [Google Scholar]
- Chiu, Y.-J.; Cai, W.; Lee, T.; Kraimer, J.; Lo, Y.-H. Quantitative Analysis of Exosome Secretion Rates of Single Cells. Bio-protocol 2017, 7, e2143. [Google Scholar] [CrossRef] [Green Version]
- O’Neal, D.; Grieve, G.; Rae, D. Factors Influencing Lp-Particle Size as Determined by Gradient Gel Electrophoresis. J. Lipid Res. 1996, 37, 1655–1663. [Google Scholar]
- Vuola, A.O.; Akram, S.U.; Kannala, J. Mask-RCNN and U-Net Ensembled for Nuclei Segmentation. In Proceedings of the IEEE 16th International Symposium on Biomedical Imaging, Venice, Italy, 8–11 April 2019. [Google Scholar] [CrossRef] [Green Version]
- Eggert, C.; Brehm, S.; Winschel, A.; Zecha, D.; Lienhart, R. A Closer Look: Small Object Detection in Faster R-CNN. In Proceedings of the IEEE International Conference on Multimedia and Expo (ICME) Expo, Hong Kong, China, 10–14 July 2017. [Google Scholar] [CrossRef]
- Lucocq, J. Can Data Provenance Go the Full Monty? Trends Cell Biol. 2012, 22, 229–230. [Google Scholar] [CrossRef]
- Sterio, D.C. The Unbiased Estimation of Number and Sizes of Arbitrary Particles Using the Disector. J. Microsc. 1984, 134, 127–136. [Google Scholar] [CrossRef]
- Ronneberger, O.; Fischer, P.; Brox, T. U-Net: Convolutional Networks for Biomedical Image Segmentation; Springer: Cham, Germany, 2015. [Google Scholar] [CrossRef] [Green Version]
- Falk, T.; Mai, D.; Bensch, R.; Çiçek, Ö.; Abdulkadir, A.; Marrakchi, Y.; Böhm, A.; Deubner, J.; Jäckel, Z.; Seiwald, K.; et al. U-Net: Deep Learning for Cell Counting, Detection, and Morphometry. Nat. Methods 2019, 16, 67–70. [Google Scholar] [CrossRef]
- Moen, E.; Bannon, D.; Kudo, T.; Graf, W.; Covert, M.; Van Valen, D. Deep Learning for Cellular Image Analysis. Nat. Methods 2019, 16, 1233–1246. [Google Scholar] [CrossRef]
- Van Valen, D.A.; Kudo, T.; Lane, K.M.; Macklin, D.N.; Quach, N.T.; DeFelice, M.M.; Maayan, I.; Tanouchi, Y.; Ashley, E.A.; Covert, M.W. Deep Learning Automates the Quantitative Analysis of Individual Cells in Live-Cell Imaging Experiments. PLoS Comput. Biol. 2016, 12, e1005177. [Google Scholar] [CrossRef] [Green Version]
- Caicedo, J.C.; Roth, J.; Goodman, A.; Becker, T.; Karhohs, K.W.; Broisin, M.; Molnar, C.; McQuin, C.; Singh, S.; Theis, F.J.; et al. Evaluation of Deep Learning Strategies for Nucleus Segmentation in Fluorescence Images. Cytom. Part A 2019, 95, 952–965. [Google Scholar] [CrossRef] [Green Version]
- Hu, Z.; Fang, W.; Gou, T.; Wu, W.; Hu, J.; Zhou, S.; Mu, Y. A Novel Method Based on a Mask R-CNN Model for Processing dPCR Images. Anal. Methods 2019, 11, 3410–3418. [Google Scholar] [CrossRef]
- Girshick, R.; Donahue, J.; Darrell, T.; Malik, J. Rich Feature Hierarchies for Accurate Object Detection and Semantic Segmentation. In Proceedings of the IEEE Conference on Computer Vision and Pattern Recognition, Columbus, OH, USA, 24–27 June 2014. [Google Scholar] [CrossRef] [Green Version]
- Girshick, R. Fast R-CNN. In Proceedings of the IEEE International Conference on Computer Vision, Santiago, Chile, 7–13 December 2015. [Google Scholar] [CrossRef]
- Waleed, A. Mask R-CNN for object Detection and Instance Segmentation on Keras and TensorFlow. Available online: https://github.com/matterport/Mask_RCNN (accessed on 7 August 2020).
- Lin, T.Y.; Dollár, P.; Girshick, R.; He, K.; Hariharan, B.; Belongie, S. Feature Pyramid Networks for Object Detection. In Proceedings of the 30th IEEE Conference on Computer Vision and Pattern Recognition, Honolulu, HI, USA, 21–26 July 2017. [Google Scholar] [CrossRef] [Green Version]
- Johnson, J.W. Automatic Nucleus Segmentation with Mask-RCNN. Adv. Intell. Syst. Comput. 2020, 944, 399–407. [Google Scholar] [CrossRef]












{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| RPN Anchor Scales (Pixels) | Augmentation Included Gaussian Noise & Sharpen? | Detection Rate (%) | False Detects per 100 | mAP | Number of Overlapping Pairs (Ground Truth) |
|---|---|---|---|---|---|
| {16,32,64,128,256} | Yes | 84.0 | 7.7 | 0.60 | 42 (63) |
| {32,64,128,256,512} | No | 83.7 | 8.5 | 0.61 | 38 (63) |
| Sample | RPN Anchor Scales (Pixels) | Augmentation Included Gaussian Noise & Sharpen? | Detection Rate (%) | False Detects per 100 | Number of Overlapping Pairs (Ground Truth) |
|---|---|---|---|---|---|
| 1 | {16,32,64,128,256} | Yes | 99.7 | 4.1 | 889 (895) |
| {32,64,128,256,512} | No | 99.3 | 9.4 | 892 (895) | |
| 2 | {16,32,64,128,256} | Yes | 99.9 | 0.6 | 1404 (1412) |
| {32,64,128,256,512} | No | 99.9 | 2.8 | 1411 (1412) |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Giesecke, Y.; Soete, S.; MacKinnon, K.; Tsiaras, T.; Ward, M.; Althobaiti, M.; Suveges, T.; Lucocq, J.E.; McKenna, S.J.; Lucocq, J.M. Developing Electron Microscopy Tools for Profiling Plasma Lipoproteins Using Methyl Cellulose Embedment, Machine Learning and Immunodetection of Apolipoprotein B and Apolipoprotein(a). Int. J. Mol. Sci. 2020, 21, 6373. https://doi.org/10.3390/ijms21176373
Giesecke Y, Soete S, MacKinnon K, Tsiaras T, Ward M, Althobaiti M, Suveges T, Lucocq JE, McKenna SJ, Lucocq JM. Developing Electron Microscopy Tools for Profiling Plasma Lipoproteins Using Methyl Cellulose Embedment, Machine Learning and Immunodetection of Apolipoprotein B and Apolipoprotein(a). International Journal of Molecular Sciences. 2020; 21(17):6373. https://doi.org/10.3390/ijms21176373
Chicago/Turabian StyleGiesecke, Yvonne, Samuel Soete, Katarzyna MacKinnon, Thanasis Tsiaras, Madeline Ward, Mohammed Althobaiti, Tamas Suveges, James E. Lucocq, Stephen J. McKenna, and John M. Lucocq. 2020. "Developing Electron Microscopy Tools for Profiling Plasma Lipoproteins Using Methyl Cellulose Embedment, Machine Learning and Immunodetection of Apolipoprotein B and Apolipoprotein(a)" International Journal of Molecular Sciences 21, no. 17: 6373. https://doi.org/10.3390/ijms21176373
APA StyleGiesecke, Y., Soete, S., MacKinnon, K., Tsiaras, T., Ward, M., Althobaiti, M., Suveges, T., Lucocq, J. E., McKenna, S. J., & Lucocq, J. M. (2020). Developing Electron Microscopy Tools for Profiling Plasma Lipoproteins Using Methyl Cellulose Embedment, Machine Learning and Immunodetection of Apolipoprotein B and Apolipoprotein(a). International Journal of Molecular Sciences, 21(17), 6373. https://doi.org/10.3390/ijms21176373