The Role of Thiocyanate in Modulating Myeloperoxidase Activity during Disease

 , ,
, , 

Abstract
:Table of Contents
- Introduction
- 1.1.
- Sources, Secretion and Elimination of SCN−
- 1.1.1.
- Exogenous and Endogenous Sources of SCN−
- 1.1.2.
- Secretion and Elimination of SCN−
- 1.2.
- Role of MPO in SCN− Biochemistry
- 1.3.
- Halides and the Formation of MPO-mediated Oxidants
- SCN− in Diseases
- 2.1.
- Positive Effect of SCN− in Disease Outcome
- 2.1.1.
- Cardiovascular Disease
- 2.1.2.
- Respiratory Disease
- Respiratory Viral Infections
- 2.2.
- Negative Effect of SCN− in Disease Outcome
- 2.2.1.
- Smoking and Respiratory Infections
- 2.2.2.
- Autoimmune Rheumatic Diseases
- 2.2.3.
- Gastrointestinal Disease
- Conclusions
1. Introduction
1.1. Sources, Secretion and Elimination of SCN−
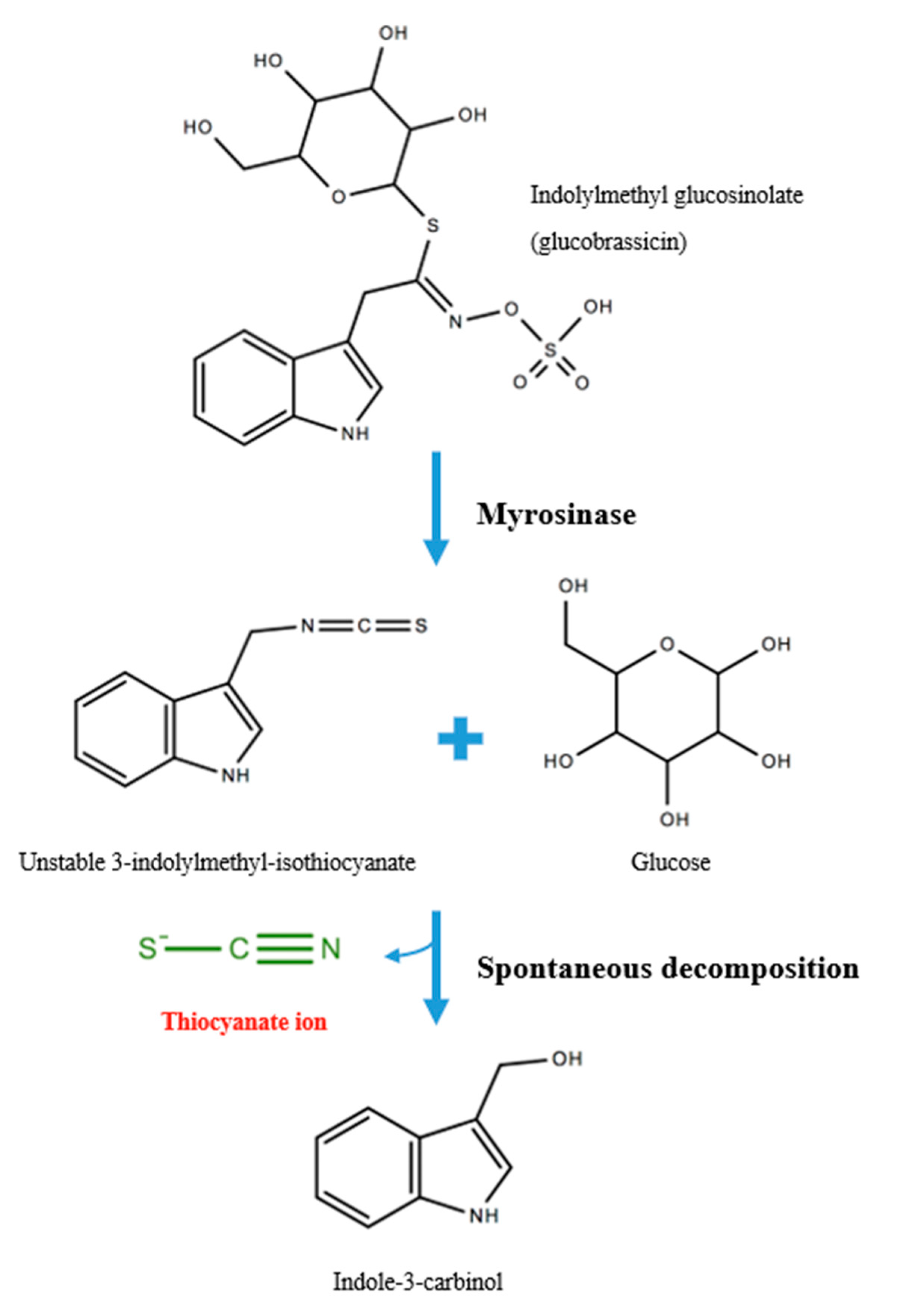
1.1.1. Exogenous and Endogenous Sources of SCN−
1.1.2. Secretion and Elimination of SCN−
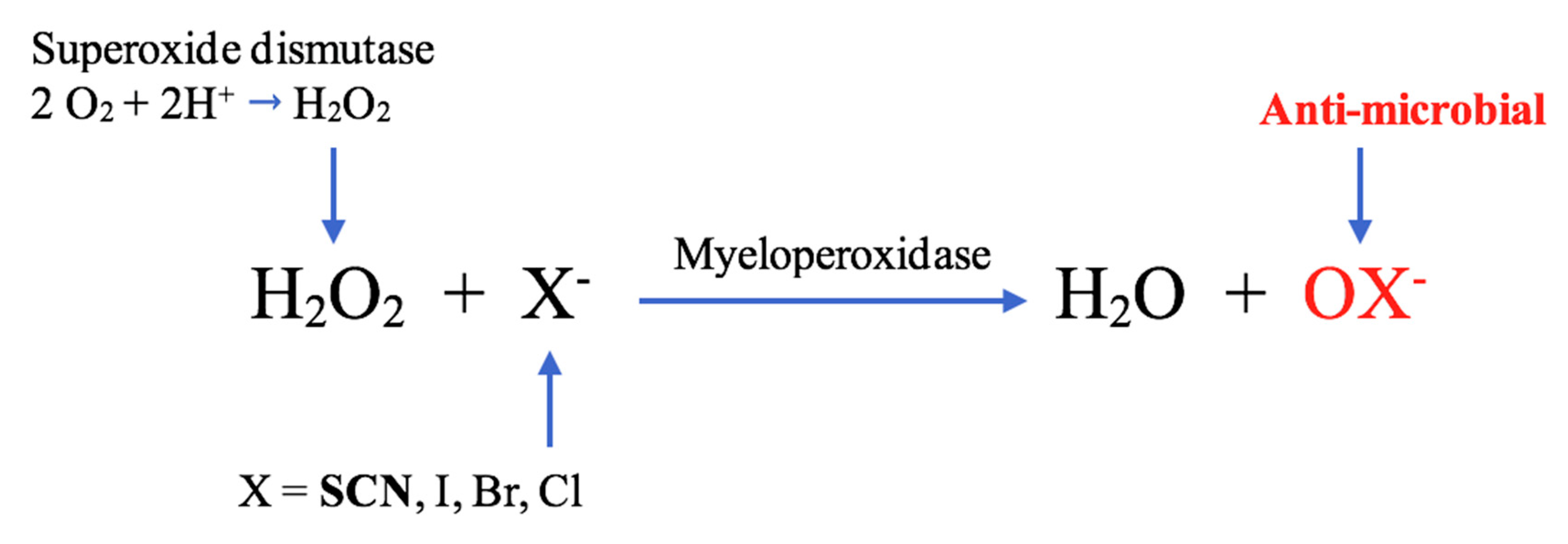
1.2. Role of MPO in SCN− Biochemistry
1.3. Halides and the Formation of MPO-mediated Oxidants
2. SCN− in Diseases
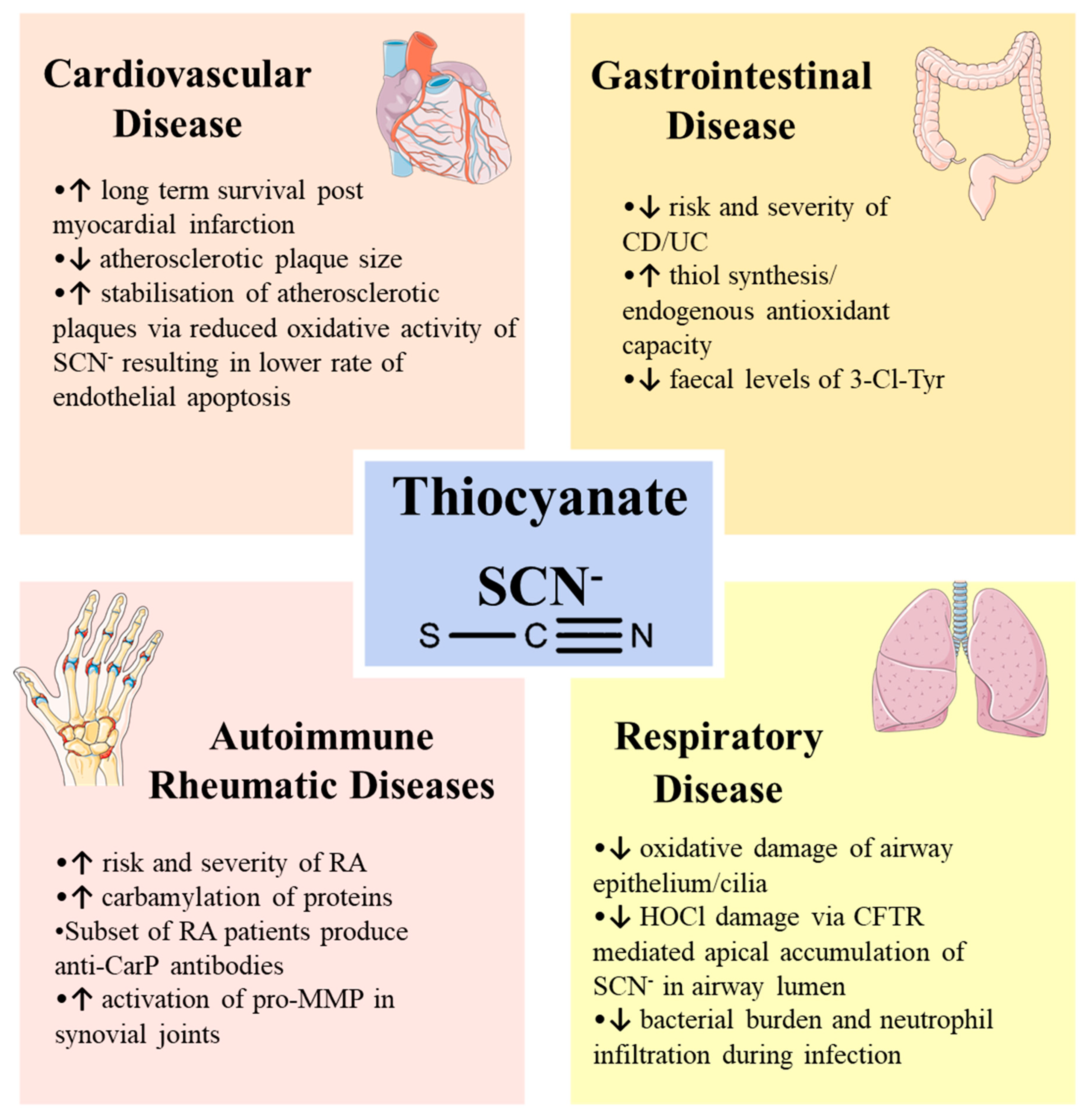
2.1. Positive Effect of SCN− in Disease Outcome
2.1.1. Cardiovascular Disease
2.1.2. Respiratory Disease
Respiratory Viral Infections
2.2. Negative Effect of SCN− in Disease Outcome
2.2.1. Smoking and Respiratory Infections
2.2.2. Autoimmune Rheumatic Diseases
2.2.3. Gastrointestinal Disease
3. Conclusions
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| 3-Cl-Tyr | 3-chlorotyrosine |
| ApoE | Apolipoprotein E Knockout |
| βENaC | Beta Epithelial Sodium Channel |
| CF | Cystic Fibrosis |
| CFTR | Cystic Fibrosis Transmembrane Conductance Regulator |
| CN− | Cyanide Ion |
| DSS | Dextran Sodium Sulfate |
| GSH | Glutathione |
| H2O2 | Hydrogen Peroxide |
| HOBr | Hypobromous Acid |
| HOCl | Hypochlorous Acid |
| HOSCN | Hypothiocyanous Acid |
| IBD | Inflammatory Bowel Disease |
| IL-4 | Interleukin-4 |
| LPO | Lactoperoxidase |
| MMP | Matrix Metalloproteinase |
| MPO | Myeloperoxidase |
| NADPH | Nicotinamide Adenine Dinucleotide Phosphate |
| NaSCN | Sodium Thiocyanate |
| OCN− | Cyanate Ion |
| OSCN− | Hypothiocyanite Ion |
| RA | Rheumatoid Arthritis |
| ROS | Reactive Oxygen Species |
| S2O32− | Thiosulfate Ion |
| SARS-CoV-2 | Severe Acute Respiratory Syndrome Coronavirus 2 |
| SCN− | Thiocyanate Ion |
| UC | Ulcerative Colitis |
References
- Thomas, E.L. Lactoperoxidase-catalyzed oxidation of thiocyanate: Equilibria between oxidized forms of thiocyanate. Biochemistry 1981, 20, 3273–3280. [Google Scholar] [CrossRef] [PubMed]
- Das, D.; De, P.K.; Banerjee, R.K. Thiocyanate, a plausible physiological electron donor of gastric peroxidase. Biochem. J. 1995, 305 Pt 1, 59–64. [Google Scholar] [CrossRef]
- Fragoso, M.A.; Fernandez, V.; Forteza, R.; Randell, S.H.; Salathe, M.; Conner, G.E. Transcellular thiocyanate transport by human airway epithelia. J. Physiol. 2004, 561, 183–194. [Google Scholar] [CrossRef] [PubMed]
- Schultz, C.P.; Ahmed, M.K.; Dawes, C.; Mantsch, H.H. Thiocyanate levels in human saliva: Quantitation by Fourier transform infrared spectroscopy. Anal. Biochem. 1996, 240, 7–12. [Google Scholar] [CrossRef] [PubMed]
- Minarowski, L.; Sands, D.; Minarowska, A.; Karwowska, A.; Sulewska, A.; Gacko, M.; Chyczewska, E. Thiocyanate concentration in saliva of cystic fibrosis patients. Folia Histochem. Cytobiol. 2008, 46, 245–246. [Google Scholar] [CrossRef] [Green Version]
- Madiyal, A.; Ajila, V.; Babu, S.G.; Hegde, S.; Kumari, S.; Madi, M.; Achalli, S.; Alva, P.; Ullal, H. Status of thiocyanate levels in the serum and saliva of non-smokers, ex-smokers and smokers. Afr. Health Sci. 2018, 18, 727–736. [Google Scholar] [CrossRef] [Green Version]
- Leung, A.M.; Lamar, A.; He, X.; Braverman, L.E.; Pearce, E.N. Iodine status and thyroid function of Boston-area vegetarians and vegans. J. Clin. Endocrinol. Metab. 2011, 96, E1303–E1307. [Google Scholar] [CrossRef] [Green Version]
- Han, H.; Kwon, H. Estimated dietary intake of thiocyanate from Brassicaceae family in Korean diet. J. Toxicol. Envrion. Health A 2009, 72, 1380–1387. [Google Scholar] [CrossRef]
- McGregor, D.I. Thiocyanate ion, a hydrolysis product of glucosinolates from rape and mustard seed. Can. J. Plant Sci. 1978, 58, 795–800. [Google Scholar] [CrossRef]
- Buratti, M.; Xaiz, D.; Caravelliand, G.; Colombi, A. Validation of urinary thiocyanate as a biomarker of tobacco smoking. Biomarkers 1997, 2, 81–85. [Google Scholar] [CrossRef]
- Lenney, W.; Gilchrist, F.J. Pseudomonas aeruginosa and cyanide production. Eur. Respir. J. 2011, 37, 482. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nagahara, N.; Okazaki, T.; Nishino, T. Cytosolic mercaptopyruvate sulfurtransferase is evolutionarily related to mitochondrial rhodanese. Striking similarity in active site amino acid sequence and the increase in the mercaptopyruvate sulfurtransferase activity of rhodanese by site-directed mutagenesis. J. Biol. Chem. 1995, 270, 16230–16235. [Google Scholar] [CrossRef] [Green Version]
- Wrobel, M.; Jurkowska, H.; Sliwa, L.; Srebro, Z. Sulfurtransferases and cyanide detoxification in mouse liver, kidney, and brain. Toxicol. Mech. Methods 2004, 14, 331–337. [Google Scholar] [CrossRef] [PubMed]
- Weuffen, W.; Franzke, C.; Thurkow, B. The alimentary ingestion, analysis and biological significance of thiocyanate. Nahrung 1984, 28, 341–355. [Google Scholar] [CrossRef]
- Chung, J.; Wood, J.L. Oxidation of Thiocyanate to Cyanide Catalyzed by Hemoglobin. J. Biol. Chem. 1971, 246, 555–560. [Google Scholar] [PubMed]
- Wells, D.G.; Langman, M.J.; Wilson, J. Thiocyanate metabolism in human vitamin B12 deficiency. Br. Med. J. 1972, 4, 588–590. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Broderick, K.E.; Potluri, P.; Zhuang, S.; Scheffler, I.E.; Sharma, V.S.; Pilz, R.B.; Boss, G.R. Cyanide detoxification by the cobalamin precursor cobinamide. Exp. Biol. Med. 2006, 231, 641–649. [Google Scholar] [CrossRef]
- Thomson, E.; Brennan, S.; Senthilmohan, R.; Gangell, C.L.; Chapman, A.L.; Sly, P.D.; Kettle, A.J.; Balding, E.; Berry, L.J.; Carlin, J.B.; et al. Identifying peroxidases and their oxidants in the early pathology of cystic fibrosis. Free Radic. Biol. Med. 2010, 49, 1354–1360. [Google Scholar] [CrossRef]
- Chandler, J.D.; Day, B.J. Biochemical mechanisms and therapeutic potential of pseudohalide thiocyanate in human health. Free Radic. Res. 2015, 49, 695–710. [Google Scholar] [CrossRef] [Green Version]
- Wijkstrom-Frei, C.; El-Chemaly, S.; Ali-Rachedi, R.; Gerson, C.; Cobas, M.A.; Forteza, R.; Salathe, M.; Conner, G.E. Lactoperoxidase and human airway host defense. Am. J. Respir. Cell Mol. Biol. 2003, 29, 206–212. [Google Scholar] [CrossRef]
- Ashby, M. Hypothiocyanite. Adv. Inorg. Chem. 2012, 64, 263–303. [Google Scholar] [CrossRef]
- Fletcher, K.; Honour, A.J.; Rowlands, E.N. Studies on the concentration of radioiodide and thiocyanate by slices of the salivary gland. Biochem. J. 1956, 63, 194–199. [Google Scholar] [CrossRef] [PubMed]
- Tenovuo, J.; Pruitt, K.M.; Thomas, E.L. Peroxidase antimicrobial system of human saliva: Hypothiocyanite levels in resting and stimulated saliva. J. Dent. Res. 1982, 61, 982–985. [Google Scholar] [CrossRef] [PubMed]
- Pedemonte, N.; Caci, E.; Sondo, E.; Caputo, A.; Rhoden, K.; Pfeffer, U.; Di Candia, M.; Bandettini, R.; Ravazzolo, R.; Zegarra-Moran, O.; et al. Thiocyanate transport in resting and IL-4-stimulated human bronchial epithelial cells: Role of pendrin and anion channels. J. Immunol. 2007, 178, 5144–5153. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moskwa, P.; Lorentzen, D.; Excoffon, K.J.; Zabner, J.; McCray, P.B., Jr.; Nauseef, W.M.; Dupuy, C.; Banfi, B. A novel host defense system of airways is defective in cystic fibrosis. Am. J. Respir. Crit. Care Med. 2007, 175, 174–183. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gould, N.S.; Gauthier, S.; Kariya, C.T.; Min, E.; Huang, J.; Brian, D.J. Hypertonic saline increases lung epithelial lining fluid glutathione and thiocyanate: Two protective CFTR-dependent thiols against oxidative injury. Respir. Res. 2010, 11, 119. [Google Scholar] [CrossRef] [Green Version]
- Pettigrew, A.R.; Fell, G.S. Simplified colorimetric determination of thiocyanate in biological fluids, and its application to investigation of the toxic amblyopias. Clin. Chem. 1972, 18, 996–1000. [Google Scholar] [CrossRef]
- Tenovuo, J.; Makinen, K.K. Concentration of thiocyanate and ionizable iodine in saliva of smokers and nonsmokers. J. Dent. Res. 1976, 55, 661–663. [Google Scholar] [CrossRef]
- Junge, B. Changes in serum thiocyanate concentration on stopping smoking. Br. Med. J. 1985, 291, 22. [Google Scholar] [CrossRef] [Green Version]
- Kalburgi, C.V.; Naik, K.L.; Kokatnur, M.V.; Warad, S. Estimation and correlation of salivary thiocyanate levels in healthy and different forms of tobacco users having chronic periodontitis: A cross-sectional biochemical study. Contemp. Clin. Dent. 2014, 5, 182–186. [Google Scholar] [CrossRef]
- Schulz, V.; Bonn, R.; Kindler, J. Kinetics of elimination of thiocyanate in 7 healthy subjects and in 8 subjects with renal failure. Klin. Wochenschr. 1979, 57, 243–247. [Google Scholar] [CrossRef] [PubMed]
- van Haeringen, N.J.; Ensink, F.T.; Glasius, E. The peroxidase-thiocyanate-hydrogenperoxide system in tear fluid and saliva of different species. Exp. Eye Res. 1979, 28, 343–347. [Google Scholar] [CrossRef]
- Lorentzen, D.; Durairaj, L.; Pezzulo, A.A.; Nakano, Y.; Launspach, J.; Stoltz, D.A.; Zamba, G.; McCray, P.B., Jr.; Zabner, J.; Welsh, M.J.; et al. Concentration of the antibacterial precursor thiocyanate in cystic fibrosis airway secretions. Free Radic. Biol. Med. 2011, 50, 1144–1150. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wokes, F.; Wedgwood, P.; Wyatt, J. Thiocyanates in milk and other biological fluids. Biochem. J. 1952, 50, xix–xx. [Google Scholar]
- Kirk, A.B.; Dyke, J.V.; Martin, C.F.; Dasgupta, P.K. Temporal patterns in perchlorate, thiocyanate, and iodide excretion in human milk. Environ. Health Perspect. 2007, 115, 182–186. [Google Scholar] [CrossRef]
- Fiedler, T.J.; Davey, C.A.; Fenna, R.E. X-ray crystal structure and characterization of halide-binding sites of human myeloperoxidase at 1.8 A resolution. J. Biol. Chem. 2000, 275, 11964–11971. [Google Scholar] [CrossRef] [Green Version]
- Zeng, J.; Fenna, R.E. X-ray crystal structure of canine myeloperoxidase at 3 Å resolution. J. Mol. Biol. 1992, 226, 185–207. [Google Scholar] [CrossRef]
- Dolphin, D.; Forman, A.; Borg, D.C.; Fajer, J.; Felton, R.H. Compounds I of catalase and horse radish peroxidase: Pi-cation radicals. Proc. Natl. Acad. Sci. USA 1971, 68, 614–618. [Google Scholar] [CrossRef] [Green Version]
- Vlasova, I.I. Peroxidase Activity of Human Hemoproteins: Keeping the Fire under Control. Molecules 2018, 23, 2561. [Google Scholar] [CrossRef] [Green Version]
- Kettle, A.J.; Anderson, R.F.; Hampton, M.B.; Winterbourn, C.C. Reactions of superoxide with myeloperoxidase. Biochemistry 2007, 46, 4888–4897. [Google Scholar] [CrossRef]
- Chandler, J.D.; Day, B.J. Thiocyanate: A potentially useful therapeutic agent with host defense and antioxidant properties. Biochem. Pharmacol. 2012, 84, 1381–1387. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baldridge, C.W.; Gerard, R.W. The extra respiration of phagocytosis. Am. J. Physiol. Leg. Content 1932, 103, 235–236. [Google Scholar] [CrossRef]
- Cross, A.R.; Segal, A.W. The NADPH oxidase of professional phagocytes--prototype of the NOX electron transport chain systems. Biochim. Biophys. Acta 2004, 1657, 1–22. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morgan, P.E.; Laura, R.P.; Maki, R.A.; Reynolds, W.F.; Davies, M.J. Thiocyanate supplementation decreases atherosclerotic plaque in mice expressing human myeloperoxidase. Free Radic. Res. 2015, 49, 743–749. [Google Scholar] [CrossRef] [Green Version]
- Pattison, D.I.; Davies, M.J.; Hawkins, C.L. Reactions and reactivity of myeloperoxidase-derived oxidants: Differential biological effects of hypochlorous and hypothiocyanous acids. Free Radic. Res. 2012, 46, 975–995. [Google Scholar] [CrossRef]
- Furtmüller, P.G.; Burner, U.; Obinger, C. Reaction of Myeloperoxidase Compound I with Chloride, Bromide, Iodide, and Thiocyanate. Biochemistry 1998, 37, 17923–17930. [Google Scholar] [CrossRef]
- Davies, M.J.; Hawkins, C.L.; Pattison, D.I.; Rees, M.D. Mammalian heme peroxidases: From molecular mechanisms to health implications. Antioxid. Redox Signal. 2008, 10, 1199–1234. [Google Scholar] [CrossRef]
- Pattison, D.I.; Davies, M.J. Absolute rate constants for the reaction of hypochlorous acid with protein side chains and peptide bonds. Chem. Res. Toxicol. 2001, 14, 1453–1464. [Google Scholar] [CrossRef]
- Skaff, O.; Pattison, D.I.; Davies, M.J. Hypothiocyanous acid reactivity with low-molecular-mass and protein thiols: Absolute rate constants and assessment of biological relevance. Biochem. J. 2009, 422, 111–117. [Google Scholar] [CrossRef] [Green Version]
- Gorman, W.F.; Messinger, E.; Herman, M. Toxicity of thiocyanates used in treatment of hypertension. Ann. Intern. Med. 1949, 30, 1054–1059. [Google Scholar] [CrossRef]
- Ruddell, W.S.; Bone, E.S.; Hill, M.J.; Blendis, L.M.; Walters, C.L. Gastric-juice nitrite. A risk factor for cancer in the hypochlorhydric stomach? Lancet 1976, 2, 1037–1039. [Google Scholar] [CrossRef]
- Heliovaara, M.; Karvonen, M.J.; Punsar, S.; Rautanen, Y.; Haapakoski, J. Serum thiocyanate concentration and cigarette smoking in relation to overall mortality and to deaths from coronary heart disease and lung cancer. J. Chronic. Dis. 1981, 34, 305–311. [Google Scholar] [CrossRef]
- Lijinsky, W.; Kovatch, R.M. Chronic toxicity tests of sodium thiocyanate with sodium nitrite in F344 rats. Toxicol. Ind. Health 1989, 5, 25–29. [Google Scholar] [CrossRef] [PubMed]
- Shiue, I. Urinary thiocyanate concentrations are associated with adult cancer and lung problems: US NHANES, 2009–2012. Env. Sci. Pollut. Res. Int. 2015, 22, 5952–5960. [Google Scholar] [CrossRef]
- World Health Organization. Global Health Estimates 2016: Disease Burden by Cause, Age, Sex, by Country and by Region, 2000–2016; World Health Organization: Geneva, Switzerland, 2018. [Google Scholar]
- Pasceri, V.; Cheng, J.S.; Willerson, J.T.; Yeh, E.T. Modulation of C-reactive protein-mediated monocyte chemoattractant protein-1 induction in human endothelial cells by anti-atherosclerosis drugs. Circulation 2001, 103, 2531–2534. [Google Scholar] [CrossRef] [Green Version]
- Ronald, J.A.; Chen, J.W.; Chen, Y.; Hamilton, A.M.; Rodriguez, E.; Reynolds, F.; Hegele, R.A.; Rogers, K.A.; Querol, M.; Bogdanov, A.; et al. Enzyme-sensitive magnetic resonance imaging targeting myeloperoxidase identifies active inflammation in experimental rabbit atherosclerotic plaques. Circulation 2009, 120, 592–599. [Google Scholar] [CrossRef] [Green Version]
- Heslop, C.L.; Frohlich, J.J.; Hill, J.S. Myeloperoxidase and C-reactive protein have combined utility for long-term prediction of cardiovascular mortality after coronary angiography. J. Am. Coll. Cardiol. 2010, 55, 1102–1109. [Google Scholar] [CrossRef] [Green Version]
- Exner, M.; Hermann, M.; Hofbauer, R.; Hartmann, B.; Kapiotis, S.; Gmeiner, B. Thiocyanate catalyzes myeloperoxidase-initiated lipid oxidation in LDL. Free Radic. Biol. Med. 2004, 37, 146–155. [Google Scholar] [CrossRef]
- Hadfield, K.A.; Pattison, D.I.; Brown, B.E.; Hou, L.; Rye, K.A.; Davies, M.J.; Hawkins, C.L. Myeloperoxidase-derived oxidants modify apolipoprotein A-I and generate dysfunctional high-density lipoproteins: Comparison of hypothiocyanous acid (HOSCN) with hypochlorous acid (HOCl). Biochem. J. 2013, 449, 531–542. [Google Scholar] [CrossRef] [Green Version]
- Abdo, A.I.; Rayner, B.S.; van Reyk, D.M.; Hawkins, C.L. Low-density lipoprotein modified by myeloperoxidase oxidants induces endothelial dysfunction. Redox. Biol. 2017, 13, 623–632. [Google Scholar] [CrossRef]
- Marsche, G.; Hammer, A.; Oskolkova, O.; Kozarsky, K.F.; Sattler, W.; Malle, E. Hypochlorite-modified high density lipoprotein, a high affinity ligand to scavenger receptor class B, type I, impairs high density lipoprotein-dependent selective lipid uptake and reverse cholesterol transport. J. Biol. Chem. 2002, 277, 32172–32179. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marsche, G.; Zimmermann, R.; Horiuchi, S.; Tandon, N.N.; Sattler, W.; Malle, E. Class B scavenger receptors CD36 and SR-BI are receptors for hypochlorite-modified low density lipoprotein. J. Biol. Chem. 2003, 278, 47562–47570. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marsche, G.; Heller, R.; Fauler, G.; Kovacevic, A.; Nuszkowski, A.; Graier, W.; Sattler, W.; Malle, E. 2-Chlorohexadecanal Derived From Hypochlorite-Modified High-Density Lipoprotein–Associated Plasmalogen Is a Natural Inhibitor of Endothelial Nitric Oxide Biosynthesis. Arterioscler. Thromb. Vasc. Biol. 2004, 24, 2302–2306. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vita, J.A.; Brennan, M.L.; Gokce, N.; Mann, S.A.; Goormastic, M.; Shishehbor, M.H.; Penn, M.S.; Keaney, J.F., Jr.; Hazen, S.L. Serum myeloperoxidase levels independently predict endothelial dysfunction in humans. Circulation 2004, 110, 1134–1139. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tiyerili, V.; Camara, B.; Becher, M.U.; Schrickel, J.W.; Lütjohann, D.; Mollenhauer, M.; Baldus, S.; Nickenig, G.; Andrié, R.P. Neutrophil-derived myeloperoxidase promotes atherogenesis and neointima formation in mice. Int. J. Cardiol. 2016, 204, 29–36. [Google Scholar] [CrossRef]
- Roth Flach, R.J.; Su, C.; Bollinger, E.; Cortes, C.; Robertson, A.W.; Opsahl, A.C.; Coskran, T.M.; Maresca, K.P.; Keliher, E.J.; Yates, P.D.; et al. Myeloperoxidase inhibition in mice alters atherosclerotic lesion composition. PLoS ONE 2019, 14, e0214150. [Google Scholar] [CrossRef] [Green Version]
- Brennan, M.L.; Anderson, M.M.; Shih, D.M.; Qu, X.D.; Wang, X.; Mehta, A.C.; Lim, L.L.; Shi, W.; Hazen, S.L.; Jacob, J.S.; et al. Increased atherosclerosis in myeloperoxidase-deficient mice. J. Clin. Investig. 2001, 107, 419–430. [Google Scholar] [CrossRef] [Green Version]
- Zoellner, H. Dental infection and vascular disease. Semin. Thromb. Hemost. 2011, 37, 181–192. [Google Scholar] [CrossRef]
- Nedoboy, P.E.; Morgan, P.E.; Mocatta, T.J.; Richards, A.M.; Winterbourn, C.C.; Davies, M.J. High plasma thiocyanate levels are associated with enhanced myeloperoxidase-induced thiol oxidation and long-term survival in subjects following a first myocardial infarction. Free Radic. Res. 2014, 48, 1256–1266. [Google Scholar] [CrossRef]
- Wang, Z.; Nicholls, S.J.; Rodriguez, E.R.; Kummu, O.; Horkko, S.; Barnard, J.; Reynolds, W.F.; Topol, E.J.; DiDonato, J.A.; Hazen, S.L. Protein carbamylation links inflammation, smoking, uremia and atherogenesis. Nat. Med. 2007, 13, 1176–1184. [Google Scholar] [CrossRef]
- Chandler, J.D.; Nichols, D.P.; Nick, J.A.; Hondal, R.J.; Day, B.J. Selective metabolism of hypothiocyanous acid by mammalian thioredoxin reductase promotes lung innate immunity and antioxidant defense. J. Biol. Chem. 2013, 288, 18421–18428. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zietzer, A.; Niepmann, S.T.; Camara, B.; Lenart, M.A.; Jansen, F.; Becher, M.U.; Andrie, R.; Nickenig, G.; Tiyerili, V. Sodium thiocyanate treatment attenuates atherosclerotic plaque formation and improves endothelial regeneration in mice. PLoS ONE 2019, 14, e0214476. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.; Szep, S.; Lu, Z. The antioxidant role of thiocyanate in the pathogenesis of cystic fibrosis and other inflammation-related diseases. Proc. Natl. Acad. Sci. USA 2009, 106, 20515–20519. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kantar, A.; Oggiano, N.; Giorgi, P.L.; Braga, P.C.; Fiorini, R. Polymorphonuclear leukocyte-generated oxygen metabolites decrease beat frequency of human respiratory cilia. Lung 1994, 172, 215–222. [Google Scholar] [CrossRef] [PubMed]
- Worlitzsch, D.; Herberth, G.; Ulrich, M.; Doring, G. Catalase, myeloperoxidase and hydrogen peroxide in cystic fibrosis. Eur. Respir. J. 1998, 11, 377–383. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van Der Vliet, A.; Nguyen, M.N.; Shigenaga, M.K.; Eiserich, J.P.; Marelich, G.P.; Cross, C.E. Myeloperoxidase and protein oxidation in cystic fibrosis. Am. J. Physiol. Lung Cell. Mol. Physiol. 2000, 279, L537–L546. [Google Scholar] [CrossRef]
- Wanner, A.; Salathe, M.; O’Riordan, T.G. Mucociliary clearance in the airways. Am. J. Respir. Crit. Care Med. 1996, 154, 1868–1902. [Google Scholar] [CrossRef]
- Riordan, J.R.; Rommens, J.M.; Kerem, B.; Alon, N.; Rozmahel, R.; Grzelczak, Z.; Zielenski, J.; Lok, S.; Plavsic, N.; Chou, J.L.; et al. Identification of the cystic fibrosis gene: Cloning and characterization of complementary DNA. Science 1989, 245, 1066. [Google Scholar] [CrossRef]
- Linsdell, P.; Tabcharani, J.A.; Rommens, J.M.; Hou, Y.X.; Chang, X.B.; Tsui, L.C.; Riordan, J.R.; Hanrahan, J.W. Permeability of wild-type and mutant cystic fibrosis transmembrane conductance regulator chloride channels to polyatomic anions. J. Gen. Physiol. 1997, 110, 355–364. [Google Scholar] [CrossRef] [Green Version]
- Frizzell, R.A.; Hanrahan, J.W. Physiology of epithelial chloride and fluid secretion. Cold Spring Harb. Perspect. Med. 2012, 2, a009563. [Google Scholar] [CrossRef] [Green Version]
- Conner, G.E.; Wijkstrom-Frei, C.; Randell, S.H.; Fernandez, V.E.; Salathe, M. The lactoperoxidase system links anion transport to host defense in cystic fibrosis. FEBS Lett. 2007, 581, 271–278. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Elkins, M.R.; Robinson, M.; Rose, B.R.; Harbour, C.; Moriarty, C.P.; Marks, G.B.; Belousova, E.G.; Xuan, W.; Bye, P.T.P. A Controlled Trial of Long-Term Inhaled Hypertonic Saline in Patients with Cystic Fibrosis. N. Engl. J. Med. 2006, 354, 229–240. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Konstan, M.W. Therapies aimed at airway inflammation in cystic fibrosis. Clin. Chest Med. 1998, 19, 505–513. [Google Scholar] [CrossRef]
- Chandler, J.D.; Min, E.; Huang, J.; McElroy, C.S.; Dickerhof, N.; Mocatta, T.; Fletcher, A.A.; Evans, C.M.; Liang, L.; Patel, M.; et al. Antiinflammatory and Antimicrobial Effects of Thiocyanate in a Cystic Fibrosis Mouse Model. Am. J. Respir. Cell Mol. Biol. 2015, 53, 193–205. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cegolon, L.; Salata, C.; Piccoli, E.; Juarez, V.; Palu, G.; Mastrangelo, G.; Calistri, A. In vitro antiviral activity of hypothiocyanite against A/H1N1/2009 pandemic influenza virus. Int. J. Hyg. Environ. Health 2014, 217, 17–22. [Google Scholar] [CrossRef] [PubMed]
- Gingerich, A.; Pang, L.; Hanson, J.; Dlugolenski, D.; Streich, R.; Lafontaine, E.R.; Nagy, T.; Tripp, R.A.; Rada, B. Hypothiocyanite produced by human and rat respiratory epithelial cells inactivates extracellular H1N2 influenza A virus. Inflamm. Res. 2016, 65, 71–80. [Google Scholar] [CrossRef]
- Patel, U.; Gingerich, A.; Widman, L.; Sarr, D.; Tripp, R.A.; Rada, B. Susceptibility of influenza viruses to hypothiocyanite and hypoiodite produced by lactoperoxidase in a cell-free system. PLoS ONE 2018, 13, e0199167. [Google Scholar] [CrossRef]
- Kitchens, G.G. Relationship of environmental tobacco smoke to otitis media in young children. Laryngoscope 1995, 105, 1–13. [Google Scholar] [CrossRef]
- Jones, L.L.; Hassanien, A.; Cook, D.G.; Britton, J.; Leonardi-Bee, J. Parental Smoking and the Risk of Middle Ear Disease in Children: A Systematic Review and Meta-analysis. Arch. Pediatrics Adolesc. Med. 2012, 166, 18–27. [Google Scholar] [CrossRef]
- Yilmaz, G.; Caylan, N.D.; Karacan, C.D. Effects of Active and Passive Smoking on Ear Infections. Curr. Infect. Dis. Rep. 2012, 14, 166–174. [Google Scholar] [CrossRef]
- Feldman, C.; Anderson, R. New insights into pneumococcal disease. Respirology 2009, 14, 167–179. [Google Scholar] [CrossRef] [PubMed]
- Bates, M.N.; Khalakdina, A.; Pai, M.; Chang, L.; Lessa, F.; Smith, K.R. Risk of tuberculosis from exposure to tobacco smoke: A systematic review and meta-analysis. Arch. Intern. Med. 2007, 167, 335–342. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lin, H.H.; Ezzati, M.; Murray, M. Tobacco smoke, indoor air pollution and tuberculosis: A systematic review and meta-analysis. PLoS Med. 2007, 4, e20. [Google Scholar] [CrossRef] [PubMed]
- Slama, K.; Chiang, C.Y.; Enarson, D.A.; Hassmiller, K.; Fanning, A.; Gupta, P.; Ray, C. Tobacco and tuberculosis: A qualitative systematic review and meta-analysis. Int. J. Tuberc. Lung Dis. 2007, 11, 1049–1061. [Google Scholar] [PubMed]
- Gajalakshmi, V.; Peto, R.; Kanaka, T.S.; Jha, P. Smoking and mortality from tuberculosis and other diseases in India: Retrospective study of 43000 adult male deaths and 35000 controls. Lancet 2003, 362, 507–515. [Google Scholar] [CrossRef]
- Wang, J.; Shen, H. Review of cigarette smoking and tuberculosis in China: Intervention is needed for smoking cessation among tuberculosis patients. BMC Public Health 2009, 9, 292. [Google Scholar] [CrossRef] [Green Version]
- Brunet, L.; Pai, M.; Davids, V.; Ling, D.; Paradis, G.; Lenders, L.; Meldau, R.; van Zyl Smit, R.; Calligaro, G.; Allwood, B.; et al. High prevalence of smoking among patients with suspected tuberculosis in South Africa. Eur. Respir. J. 2011, 38, 139–146. [Google Scholar] [CrossRef] [Green Version]
- Bonacci, R.A.; Cruz-Hervert, L.P.; García-García, L.; Reynales-Shigematsu, L.M.; Ferreyra-Reyes, L.; Bobadilla-del-Valle, M.; Canizales-Quintero, S.; Ferreira-Guerrero, E.; Báez-Saldaña, R.; Téllez-Vázquez, N.; et al. Impact of cigarette smoking on rates and clinical prognosis of pulmonary tuberculosis in Southern Mexico. J. Infect. 2013, 66, 303–312. [Google Scholar] [CrossRef] [Green Version]
- Sanz Herrero, F.; Blanquer Olivas, J. Microbiology and risk factors for community-acquired pneumonia. Semin. Respir. Crit. Care Med. 2012, 33, 220–231. [Google Scholar] [CrossRef]
- Nuorti, J.P.; Butler, J.C.; Farley, M.M.; Harrison, L.H.; McGeer, A.; Kolczak, M.S.; Breiman, R.F. Cigarette Smoking and Invasive Pneumococcal Disease. N. Engl. J. Med. 2000, 342, 681–689. [Google Scholar] [CrossRef]
- Mathe, G.; Gouveia, J.; Hercend, T.; Gros, F.; Dorval, T.; Hazon, J.; Misset, J.L.; Schwarzenberg, L.; Ribaud, P.; Lemaigre, G.; et al. Correlation between precancerous bronchial metaplasia and cigarette consumption, and preliminary results of retinoid treatment. Cancer Detect. Prev. 1982, 5, 461–466. [Google Scholar] [PubMed]
- Agius, A.M.; Wake, M.; Pahor, A.L.; Smallman, L.A. Smoking and middle ear ciliary beat frequency in otitis media with effusion. Acta Oto-Laryngol. 1995, 115, 44–49. [Google Scholar] [CrossRef] [PubMed]
- Mullen, J.B.; Wright, J.L.; Wiggs, B.R.; Paré, P.D.; Hogg, J.C. Structure of central airways in current smokers and ex-smokers with and without mucus hypersecretion: Relationship to lung function. Thorax 1987, 42, 843–848. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cecchi, I.; Arias de la Rosa, I.; Menegatti, E.; Roccatello, D.; Collantes-Estevez, E.; Lopez-Pedrera, C.; Barbarroja, N. Neutrophils: Novel key players in Rheumatoid Arthritis. Current and future therapeutic targets. Autoimmun. Rev. 2018, 17, 1138–1149. [Google Scholar] [CrossRef]
- Wipke, B.T.; Allen, P.M. Essential role of neutrophils in the initiation and progression of a murine model of rheumatoid arthritis. J. Immunol. 2001, 167, 1601–1608. [Google Scholar] [CrossRef] [Green Version]
- Tanaka, D.; Kagari, T.; Doi, H.; Shimozato, T. Essential role of neutrophils in anti-type II collagen antibody and lipopolysaccharide-induced arthritis. Immunology 2006, 119, 195–202. [Google Scholar] [CrossRef]
- Eyles, J.L.; Hickey, M.J.; Norman, M.U.; Croker, B.A.; Roberts, A.W.; Drake, S.F.; James, W.G.; Metcalf, D.; Campbell, I.K.; Wicks, I.P. A key role for G-CSF-induced neutrophil production and trafficking during inflammatory arthritis. Blood 2008, 112, 5193–5201. [Google Scholar] [CrossRef] [Green Version]
- Odobasic, D.; Yang, Y.; Muljadi, R.C.; O’Sullivan, K.M.; Kao, W.; Smith, M.; Morand, E.F.; Holdsworth, S.R. Endogenous myeloperoxidase is a mediator of joint inflammation and damage in experimental arthritis. Arthritis Rheumatol 2014, 66, 907–917. [Google Scholar] [CrossRef]
- Nzeusseu Toukap, A.; Delporte, C.; Noyon, C.; Franck, T.; Rousseau, A.; Serteyn, D.; Raes, M.; Vanhaeverbeek, M.; Moguilevsky, N.; Neve, J.; et al. Myeloperoxidase and its products in synovial fluid of patients with treated or untreated rheumatoid arthritis. Free Radic. Res. 2014, 48, 461–465. [Google Scholar] [CrossRef] [Green Version]
- Sur Chowdhury, C.; Giaglis, S.; Walker, U.A.; Buser, A.; Hahn, S.; Hasler, P. Enhanced neutrophil extracellular trap generation in rheumatoid arthritis: Analysis of underlying signal transduction pathways and potential diagnostic utility. Arthritis Res. 2014, 16, R122. [Google Scholar] [CrossRef] [Green Version]
- Itoh, Y. Metalloproteinases in Rheumatoid Arthritis: Potential Therapeutic Targets to Improve Current Therapies. Prog. Mol. Biol. Transl. Sci. 2017, 148, 327–338. [Google Scholar] [CrossRef] [PubMed]
- Kandasamy, A.D.; Chow, A.K.; Ali, M.A.; Schulz, R. Matrix metalloproteinase-2 and myocardial oxidative stress injury: Beyond the matrix. Cardiovasc. Res. 2010, 85, 413–423. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fu, X.; Kassim, S.Y.; Parks, W.C.; Heinecke, J.W. Hypochlorous acid oxygenates the cysteine switch domain of pro-matrilysin (MMP-7). A mechanism for matrix metalloproteinase activation and atherosclerotic plaque rupture by myeloperoxidase. J. Biol. Chem. 2001, 276, 41279–41287. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nagy, P.; Jameson, G.N.; Winterbourn, C.C. Kinetics and mechanisms of the reaction of hypothiocyanous acid with 5-thio-2-nitrobenzoic acid and reduced glutathione. Chem. Res. Toxicol. 2009, 22, 1833–1840. [Google Scholar] [CrossRef] [PubMed]
- Costenbader, K.H.; Feskanich, D.; Mandl, L.A.; Karlson, E.W. Smoking intensity, duration, and cessation, and the risk of rheumatoid arthritis in women. Am. J. Med. 2006, 119, 503.e1–503.e9. [Google Scholar] [CrossRef]
- Whitehouse, M.W.; Jones, M. Pro-inflammatory activity in rats of thiocyanate, a metabolite of the hydrocyanic acid inhaled from tobacco smoke. Inflamm. Res. 2009, 58, 693–704. [Google Scholar] [CrossRef]
- de Brito Rocha, S.; Baldo, D.C.; Andrade, L.E.C. Clinical and pathophysiologic relevance of autoantibodies in rheumatoid arthritis. Adv. Rheumatol. 2019, 59, 2. [Google Scholar] [CrossRef]
- Shi, J.; van Veelen, P.A.; Mahler, M.; Janssen, G.M.; Drijfhout, J.W.; Huizinga, T.W.; Toes, R.E.; Trouw, L.A. Carbamylation and antibodies against carbamylated proteins in autoimmunity and other pathologies. Autoimmun. Rev. 2014, 13, 225–230. [Google Scholar] [CrossRef]
- Delporte, C.; Zouaoui Boudjeltia, K.; Furtmuller, P.G.; Maki, R.A.; Dieu, M.; Noyon, C.; Soudi, M.; Dufour, D.; Coremans, C.; Nuyens, V.; et al. Myeloperoxidase-catalyzed oxidation of cyanide to cyanate: A potential carbamylation route involved in the formation of atherosclerotic plaques? J. Biol. Chem. 2018, 293, 6374–6386. [Google Scholar] [CrossRef] [Green Version]
- Verheul, M.K.; Fearon, U.; Trouw, L.A.; Veale, D.J. Biomarkers for rheumatoid and psoriatic arthritis. Clin. Immunol. 2015, 161, 2–10. [Google Scholar] [CrossRef]
- Shi, J.; Knevel, R.; Suwannalai, P.; van der Linden, M.P.; Janssen, G.M.; van Veelen, P.A.; Levarht, N.E.; van der Helm-van Mil, A.H.; Cerami, A.; Huizinga, T.W.; et al. Autoantibodies recognizing carbamylated proteins are present in sera of patients with rheumatoid arthritis and predict joint damage. Proc. Natl. Acad. Sci. USA 2011, 108, 17372–17377. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nishida, A.; Inoue, R.; Inatomi, O.; Bamba, S.; Naito, Y.; Andoh, A. Gut microbiota in the pathogenesis of inflammatory bowel disease. Clin. J. Gastroenterol. 2018, 11, 1–10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marchal-Bressenot, A.; Salleron, J.; Boulagnon-Rombi, C.; Bastien, C.; Cahn, V.; Cadiot, G.; Diebold, M.D.; Danese, S.; Reinisch, W.; Schreiber, S.; et al. Development and validation of the Nancy histological index for UC. Gut 2017, 66, 43–49. [Google Scholar] [CrossRef] [PubMed]
- Buell, M.G.; Berin, M.C. Neutrophil-independence of the initiation of colonic injury. Comparison of results from three models of experimental colitis in the rat. Dig. Dis. Sci. 1994, 39, 2575–2588. [Google Scholar] [CrossRef] [PubMed]
- Natsui, M.; Kawasaki, K.; Takizawa, H.; Hayashi, S.I.; Matsuda, Y.; Sugimura, K.; Seki, K.; Narisawa, R.; Sendo, F.; Asakura, H. Selective depletion of neutrophils by a monoclonal antibody, RP-3, suppresses dextran sulphate sodium-induced colitis in rats. J. Gastroenterol. Hepatol. 1997, 12, 801–808. [Google Scholar] [CrossRef]
- Chami, B.; San Gabriel, P.T.; Kum-Jew, S.; Wang, X.; Dickerhof, N.; Dennis, J.M.; Witting, P.K. The nitroxide 4-methoxy-tempo inhibits the pathogenesis of dextran sodium sulfate-stimulated experimental colitis. Redox. Biol. 2020, 28, 101333. [Google Scholar] [CrossRef]
- Saiki, T.; Mitsuyama, K.; Toyonaga, A.; Ishida, H.; Tanikawa, K. Detection of pro- and anti-inflammatory cytokines in stools of patients with inflammatory bowel disease. Scand. J. Gastroenterol. 1998, 33, 616–622. [Google Scholar] [CrossRef]
- Hansberry, D.R.; Shah, K.; Agarwal, P.; Agarwal, N. Fecal Myeloperoxidase as a Biomarker for Inflammatory Bowel Disease. Cureus 2017, 9, e1004. [Google Scholar] [CrossRef] [Green Version]
- Mancini, S.; Mariani, F.; Sena, P.; Benincasa, M.; Roncucci, L. Myeloperoxidase expression in human colonic mucosa is related to systemic oxidative balance in healthy subjects. Redox Rep. 2017, 22, 399–407. [Google Scholar] [CrossRef] [Green Version]
- Chami, B.; Martin, N.J.J.; Dennis, J.M.; Witting, P.K. Myeloperoxidase in the inflamed colon: A novel target for treating inflammatory bowel disease. Arch. Biochem. Biophys. 2018, 645, 61–71. [Google Scholar] [CrossRef]
- Knutson, C.G.; Mangerich, A.; Zeng, Y.; Raczynski, A.R.; Liberman, R.G.; Kang, P.; Ye, W.; Prestwich, E.G.; Lu, K.; Wishnok, J.S.; et al. Chemical and cytokine features of innate immunity characterize serum and tissue profiles in inflammatory bowel disease. Proc. Natl. Acad. Sci. USA 2013, 110, E2332–E2341. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lakatos, P.L.; Lakatos, L. Risk for colorectal cancer in ulcerative colitis: Changes, causes and management strategies. World J. Gastroenterol. 2008, 14, 3937–3947. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morgan, P.E.; Pattison, D.I.; Talib, J.; Summers, F.A.; Harmer, J.A.; Celermajer, D.S.; Hawkins, C.L.; Davies, M.J. High plasma thiocyanate levels in smokers are a key determinant of thiol oxidation induced by myeloperoxidase. Free Radic. Biol. Med. 2011, 51, 1815–1822. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Burton, T.; Rayner, B.S.; San Gabriel, P.T.; Shi, H.; El Kazzi, M.; Wang, X.; Dennis, J.M.; Ahmad, G.; Schroder, A.L.; et al. The role of sodium thiocyanate supplementation during dextran sodium sulphate-stimulated experimental colitis. Arch. Biochem. Biophys. 2020, 692, 108490. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
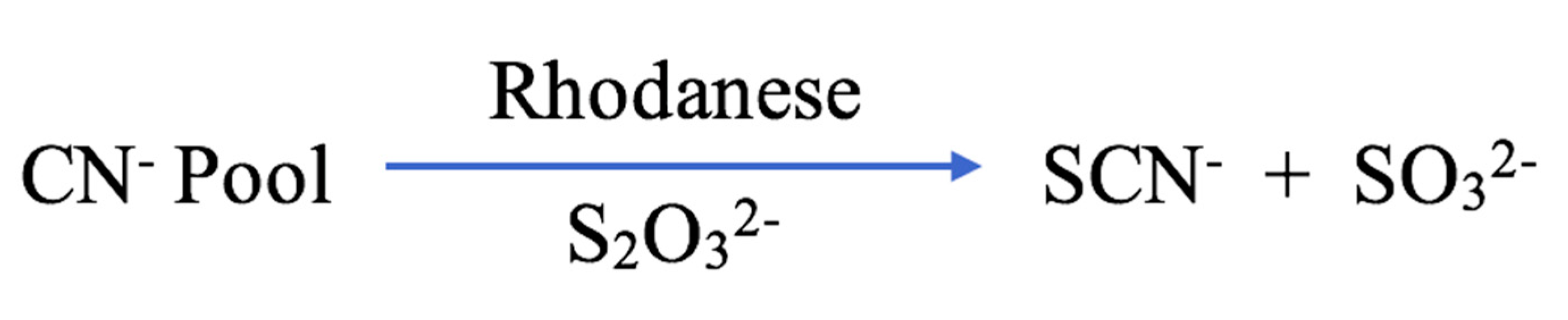{kind=link}
{kind=link}
{kind=link}
| Non-Smokers (mM) | Smokers (mM) | Vegan/Vegetarian (mM) | References | |
|---|---|---|---|---|
| Tears | 0.15 | - | - | [32] |
| Whole saliva | 0.5–2 | 2–3.6 | - | [4,5,6] |
| Nasal airway fluids | 1–1.2 | - | - | [33] |
| Lung airway fluid | 0.03–0.65 | - | - | [18,20] |
| Breastmilk | 0.0001–0.004 | - | - | [34,35] |
| Gastric fluids | 0.25–0.3 | - | - | [2] |
| Plasma | 0.03–0.05 | 0.1–0.2 | - | [4,6] |
| Urine | 0.009–0.024 | 0.33–0.275 | 0.002–0.05/0.001–0.034 | [7,10] |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
San Gabriel, P.T.; Liu, Y.; Schroder, A.L.; Zoellner, H.; Chami, B. The Role of Thiocyanate in Modulating Myeloperoxidase Activity during Disease. Int. J. Mol. Sci. 2020, 21, 6450. https://doi.org/10.3390/ijms21176450
San Gabriel PT, Liu Y, Schroder AL, Zoellner H, Chami B. The Role of Thiocyanate in Modulating Myeloperoxidase Activity during Disease. International Journal of Molecular Sciences. 2020; 21(17):6450. https://doi.org/10.3390/ijms21176450
Chicago/Turabian StyleSan Gabriel, Patrick T., Yuyang Liu, Angie L. Schroder, Hans Zoellner, and Belal Chami. 2020. "The Role of Thiocyanate in Modulating Myeloperoxidase Activity during Disease" International Journal of Molecular Sciences 21, no. 17: 6450. https://doi.org/10.3390/ijms21176450
APA StyleSan Gabriel, P. T., Liu, Y., Schroder, A. L., Zoellner, H., & Chami, B. (2020). The Role of Thiocyanate in Modulating Myeloperoxidase Activity during Disease. International Journal of Molecular Sciences, 21(17), 6450. https://doi.org/10.3390/ijms21176450