Thermogenic Activation Downregulates High Mitophagy Rate in Human Masked and Mature Beige Adipocytes

 , , ,
, , ,  and
and
Abstract
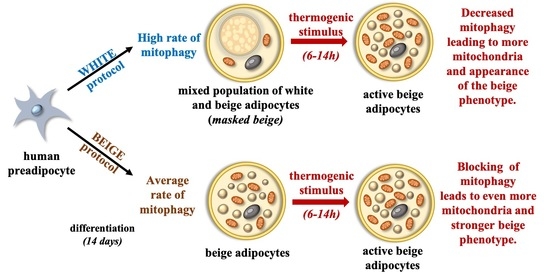
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
2.1. cAMP-Mediated Thermogenic Stimulus Results in Increased UCP1 Expression and Mitochondrial Content in Differentiated White and Beige Adipocytes
2.2. Thermogenic Activation Represses Autophagy
2.3. cAMP Stimulation Results in Reduced Mitophagy and Increased Mitochondrial Fragmentation in Masked and Mature Beige Adipocytes
2.4. Downregulation of Mitophagy upon Thermogenic Stimulus Is Mediated through PKA Activity
2.5. Thermogenic Stimulus Downregulates the Expression of Parkin and Other Mitophagy Related Genes but Silencing of Parkin Does Not Result in Higher Thermogenic Potential
3. Discussion
4. Materials and Methods
4.1. Materials
4.2. Ethics Statement
4.3. Isolation, Cell Culture, Differentiation and Treatments of hASC and Simpson–Golabi–Behmel Syndrome (SGBS) Cells
4.4. RNA Isolation, RT-PCR, qPCR
4.5. Mitochondrial DNA (mtDNA) Isolation and Quantification by qPCR
4.6. Antibodies and Immunoblotting
4.7. Immunostaining Analysis
4.8. Image Acquisition
4.9. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Harms, M.; Seale, P. Brown and beige fat: Development, function and therapeutic potential. Nat. Med. 2013, 19, 1252–1263. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Whittle, A.J.; López, M.; Vidal-Puig, A. Using brown adipose tissue to treat obesity—The central issue. Trends Mol. Med. 2011, 17, 405–411. [Google Scholar] [CrossRef] [PubMed]
- Fedorenko, A.; Lishko, P.V.; Kirichok, Y. Mechanism of fatty-acid-dependent UCP1 uncoupling in brown fat mitochondria. Cell 2012, 151, 400–413. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pellegrinelli, V.; Carobbio, S.; Vidal-Puig, A. Adipose tissue plasticity: How fat depots respond differently to pathophysiological cues. Diabetologia 2016, 59, 1075–1088. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cannon, B.; Nedergaard, J. Brown Adipose Tissue: Function and Physiological Significance. Physiol. Rev. 2004, 84, 277–359. [Google Scholar] [CrossRef]
- Petrovic, N.; Walden, T.B.; Shabalina, I.G.; Timmons, J.A.; Cannon, B.; Nedergaard, J. Chronic peroxisome proliferator-activated receptor γ (PPARγ) activation of epididymally derived white adipocyte cultures reveals a population of thermogenically competent, UCP1-containing adipocytes molecularly distinct from classic brown adipocytes. J. Biol. Chem. 2010, 285, 7153–7164. [Google Scholar] [CrossRef] [Green Version]
- Leitner, B.P.; Huang, S.; Brychta, R.J.; Duckworth, C.J.; Baskin, A.S.; McGehee, S.; Tal, I.; Dieckmann, W.; Gupta, G.; Kolodny, G.M.; et al. Mapping of human brown adipose tissue in lean and obese young men. Proc. Natl. Acad. Sci. USA 2017, 114, 8649–8654. [Google Scholar] [CrossRef] [Green Version]
- van Marken Lichtenbelt, W.D.; Schrauwen, P. Implications of nonshivering thermogenesis for energy balance regulation in humans. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2011, 301, R285–R296. [Google Scholar] [CrossRef] [Green Version]
- Claussnitzer, M.; Dankel, S.N.; Kim, K.H.; Quon, G.; Meuleman, W.; Haugen, C.; Glunk, V.; Sousa, I.S.; Beaudry, J.L.; Puviindran, V.; et al. FTO obesity variant circuitry and adipocyte browning in humans. N. Engl. J. Med. 2015, 373, 895–907. [Google Scholar] [CrossRef] [Green Version]
- Sidossis, L.; Kajimura, S. Brown and beige fat in humans: Thermogenic adipocytes that control energy and glucose homeostasis. J. Clin. Investig. 2015, 125, 478–486. [Google Scholar] [CrossRef]
- Kajimura, S.; Spiegelman, B.M.; Seale, P. Brown and beige fat: Physiological roles beyond heat generation. Cell Metab. 2015, 22, 546–559. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liesa, M.; Shirihai, O.S. Mitochondrial dynamics in the regulation of nutrient utilization and energy expenditure. Cell Metab. 2013, 17, 491–506. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wikstrom, J.D.; Mahdaviani, K.; Liesa, M.; Sereda, S.B.; Si, Y.; Las, G.; Twig, G.; Petrovic, N.; Zingaretti, C.; Graham, A.; et al. Hormone-induced mitochondrial fission is utilized by brown adipocytes as an amplification pathway for energy expenditure. EMBO J. 2014, 33, 418–436. [Google Scholar] [CrossRef] [PubMed]
- Pisani, D.F.; Barquissau, V.; Chambard, J.C.; Beuzelin, D.; Ghandour, R.A.; Giroud, M.; Mairal, A.; Pagnotta, S.; Cinti, S.; Langin, D.; et al. Mitochondrial fission is associated with UCP1 activity in human brite/beige adipocytes. Mol. Metab. 2018, 7, 35–44. [Google Scholar] [CrossRef] [PubMed]
- Palikaras, K.; Tavernarakis, N. Mitochondrial homeostasis: The interplay between mitophagy and mitochondrial biogenesis. Exp. Gerontol. 2014, 56, 182–188. [Google Scholar] [CrossRef]
- Kelly, D.P.; Scarpulla, R.C. Transcriptional regulatory circuits controlling mitochondrial biogenesis and function. Genes Dev. 2004, 18, 357–368. [Google Scholar] [CrossRef] [Green Version]
- Kim, I.; Rodriguez-Enriquez, S.; Lemasters, J.J. Selective degradation of mitochondria by mitophagy. Arch. Biochem. Biophys. 2007, 462, 245–253. [Google Scholar] [CrossRef] [Green Version]
- Füllgrabe, J.; Klionsky, D.J.; Joseph, B. The return of the nucleus: Transcriptional and epigenetic control of autophagy. Nat. Rev. Mol. Cell Biol. 2014, 15, 65–74. [Google Scholar] [CrossRef]
- Lamb, C.A.; Yoshimori, T.; Tooze, S.A. The autophagosome: Origins unknown, biogenesis complex. Nat. Rev. Mol. Cell Biol. 2013, 14, 759–774. [Google Scholar] [CrossRef]
- Palikaras, K.; Lionaki, E.; Tavernarakis, N. Mechanisms of mitophagy in cellular homeostasis, physiology and pathology. Nat. Cell Biol. 2018, 20, 1013–1022. [Google Scholar] [CrossRef]
- Narendra, D.; Tanaka, A.; Suen, D.F.; Youle, R.J. Parkin is recruited selectively to impaired mitochondria and promotes their autophagy. J. Cell Biol. 2008, 183, 795–803. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Matsuda, N.; Sato, S.; Shiba, K.; Okatsu, K.; Saisho, K.; Gautier, C.A.; Sou, Y.S.; Saiki, S.; Kawajiri, S.; Sato, F.; et al. PINK1 stabilized by mitochondrial depolarization recruits Parkin to damaged mitochondria and activates latent Parkin for mitophagy. J. Cell Biol. 2010, 189, 211–221. [Google Scholar] [CrossRef] [PubMed]
- Lazarou, M.; Sliter, D.A.; Kane, L.A.; Sarraf, S.A.; Wang, C.; Burman, J.L.; Sideris, D.P.; Fogel, A.I.; Youle, R.J. The ubiquitin kinase PINK1 recruits autophagy receptors to induce mitophagy. Nature 2015, 524, 309–314. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hamacher-Brady, A.; Brady, N.R. Mitophagy programs: Mechanisms and physiological implications of mitochondrial targeting by autophagy. Cell. Mol. Life Sci. 2016, 73, 775–795. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schweers, R.L.; Zhang, J.; Randall, M.S.; Loyd, M.R.; Li, W.; Dorsey, F.C.; Kundu, M.; Opferman, J.T.; Cleveland, J.L.; Miller, J.L.; et al. NIX is required for programmed mitochondrial clearance during reticulocyte maturation. Proc. Natl. Acad. Sci. USA 2007, 104, 19500–19505. [Google Scholar] [CrossRef] [Green Version]
- Sandoval, H.; Thiagarajan, P.; Dasgupta, S.K.; Schumacher, A.; Prchal, J.T.; Chen, M.; Wang, J. Essential role for Nix in autophagic maturation of erythroid cells. Nature 2008, 454, 232–235. [Google Scholar] [CrossRef]
- Altshuler-Keylin, S.; Shinoda, K.; Hasegawa, Y.; Ikeda, K.; Hong, H.; Kang, Q.; Yang, Y.; Perera, R.M.; Debnath, J.; Kajimura, S. Beige Adipocyte Maintenance Is Regulated by Autophagy-Induced Mitochondrial Clearance. Cell Metab. 2016, 24, 402–419. [Google Scholar] [CrossRef] [Green Version]
- Lu, X.; Altshuler-Keylin, S.; Wang, Q.; Chen, Y.; Sponton, C.H.; Ikeda, K.; Maretich, P.; Yoneshiro, T.; Kajimura, S. Mitophagy controls beige adipocyte maintenance through a Parkin-dependent and UCP1-independent mechanism. Sci. Signal. 2018, 11, eaap8526. [Google Scholar] [CrossRef] [Green Version]
- Kristof, E.; Doan-Xuan, Q.M.; Bai, P.; Bacso, Z.; Fesüs, L. Laser-scanning cytometry can quantify human adipocyte browning and proves effectiveness of irisin. Sci. Rep. 2015, 5, 12540. [Google Scholar] [CrossRef]
- Klusóczki, Á.; Veréb, Z.; Vámos, A.; Fischer-Posovszky, P.; Wabitsch, M.; Bacso, Z.; Fésüs, L.; Kristóf, E. Differentiating SGBS adipocytes respond to PPARγ stimulation, irisin and BMP7 by functional browning and beige characteristics. Sci. Rep. 2019, 9, 5823. [Google Scholar] [CrossRef]
- Fischer-Posovszky, P.; Newell, F.S.; Wabitsch, M.; Tornqvist, H.E. Human SGBS cells—A unique tool for studies of human fat cell biology. Obes. Facts 2008, 1, 184–189. [Google Scholar] [CrossRef] [PubMed]
- Elabd, C.; Chiellini, C.; Carmona, M.; Galitzky, J.; Cochet, O.; Petersen, R.; Pénicaud, L.; Kristiansen, K.; Bouloumié, A.; Casteilla, L.; et al. Human multipotent adipose-derived stem cells differentiate into functional brown adipocytes. Stem Cells 2009, 27, 2753–2760. [Google Scholar] [CrossRef] [PubMed]
- Wabitsch, M.; Brenner, R.E.; Melzner, I.; Braun, M.; Möller, P.; Heinze, E.; Debatin, K.M.; Hauner, H. Characterization of a human preadipocyte cell strain with high capacity for adipose differentiation. Int. J. Obes. 2001, 25, 8–15. [Google Scholar] [CrossRef] [Green Version]
- Doan-Xuan, Q.M.; Sarvari, A.K.; Fischer-Posovszky, P.; Wabitsch, M.; Balajthy, Z.; Fesus, L.; Bacso, Z. High content analysis of differentiation and cell death in human adipocytes. Cytom. Part A 2013, 83, 933–943. [Google Scholar]
- Yoshii, S.R.; Mizushima, N. Monitoring and measuring autophagy. Int. J. Mol. Sci. 2017, 18, 1865. [Google Scholar] [CrossRef]
- Mizushima, N.; Yoshimori, T.; Levine, B. Methods in Mammalian Autophagy Research. Cell 2010, 140, 313–326. [Google Scholar] [CrossRef] [Green Version]
- Pankiv, S.; Clausen, T.H.; Lamark, T.; Brech, A.; Bruun, J.A.; Outzen, H.; Øvervatn, A.; Bjørkøy, G.; Johansen, T. p62/SQSTM1 binds directly to Atg8/LC3 to facilitate degradation of ubiquitinated protein aggregates by autophagy. J. Biol. Chem. 2007, 282, 24131–24145. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dolman, N.J.; Chambers, K.M.; Mandavilli, B.; Batchelor, R.H.; Janes, M.S. Tools and techniques to measure mitophagy using fluorescence microscopy. Autophagy 2013, 9, 1653–1662. [Google Scholar] [CrossRef] [Green Version]
- Vereb, G.; Matkó, J.; Vámosi, G.; Ibrahim, S.M.; Magyar, E.; Varga, S.; Szöllősi, J.; Jenei, A.; Gaspar, R.; Waldmann, T.A.; et al. Cholesterol-dependent clustering of IL-2Rα and its colocalization with HLA and CD48 on T lymphoma cells suggest their functional association with lipid rafts. Proc. Natl. Acad. Sci. USA 2000, 97, 6013–6018. [Google Scholar] [CrossRef] [Green Version]
- Mocsár, G.; Volkó, J.; Rönnlund, D.; Widengren, J.; Nagy, P.; Szöllősi, J.; Tóth, K.; Goldman, C.K.; Damjanovich, S.; Waldmann, T.A.; et al. MHC I Expression Regulates Co-clustering and Mobility of Interleukin-2 and -15 Receptors in T Cells. Biophys. J. 2016, 111, 100–112. [Google Scholar] [CrossRef]
- Rodger, C.E.; McWilliams, T.G.; Ganley, I.G. Mammalian mitophagy—From in vitro molecules to in vivo models. FEBS J. 2018, 285, 1185–1202. [Google Scholar] [CrossRef] [Green Version]
- Tanaka, K. The PINK1–Parkin axis: An Overview. Neurosci. Res. 2020, in press. [Google Scholar] [CrossRef]
- Altshuler-Keylin, S.; Kajimura, S. Mitochondrial homeostasis in adipose tissue remodeling. Sci. Signal. 2017, 10, eaai9248. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cairó, M.; Villarroya, J.; Cereijo, R.; Campderrós, L.; Giralt, M.; Villarroya, F. Thermogenic activation represses autophagy in brown adipose tissue. Int. J. Obes. 2016, 40, 1591–1599. [Google Scholar] [CrossRef] [PubMed]
- Cairó, M.; Campderrós, L.; Gavaldà-Navarro, A.; Cereijo, R.; Delgado-Anglés, A.; Quesada-López, T.; Giralt, M.; Villarroya, J.; Villarroya, F. Parkin controls brown adipose tissue plasticity in response to adaptive thermogenesis. EMBO Rep. 2019, 20, e46832. [Google Scholar] [CrossRef]
- Kajimura, S.; Saito, M. A New Era in Brown Adipose Tissue Biology: Molecular Control of Brown Fat Development and Energy Homeostasis. Annu. Rev. Physiol. 2014, 76, 225–249. [Google Scholar] [CrossRef] [Green Version]
- Cherra, S.J.; Kulich, S.M.; Uechi, G.; Balasubramani, M.; Mountzouris, J.; Day, B.W.; Chu, C.T. Regulation of the autophagy protein LC3 by phosphorylation. J. Cell Biol. 2010, 190, 533–539. [Google Scholar] [CrossRef] [PubMed]
- He, C.; Klionsky, D.J. Regulation Mechanisms and Signaling Pathways of Autophagy. Annu. Rev. Genet. 2009, 43, 67–93. [Google Scholar] [CrossRef] [Green Version]
- Liu, D.; Bordicchia, M.; Zhang, C.; Fang, H.; Wei, W.; Li, J.L.; Guilherme, A.; Guntur, K.; Czech, M.P.; Collins, S. Activation of mTORC1 is essential for β-adrenergic stimulation of adipose browning. J. Clin. Investig. 2016, 126, 1704–1716. [Google Scholar] [CrossRef] [Green Version]
- Chang, C.R.; Blackstone, C. Cyclic AMP-dependent protein kinase phosphorylation of Drp1 regulates its GTPase activity and mitochondrial morphology. J. Biol. Chem. 2007, 282, 21583–21587. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Han, X.J.; Lu, Y.F.; Li, S.A.; Kaitsuka, T.; Sato, Y.; Tomizawa, K.; Nairn, A.C.; Takei, K.; Matsui, H.; Matsushita, M. CaM kinase I alpha-induced phosphorylation of Drp1 regulates mitochondrial morphology. J. Cell Biol. 2008, 182, 573–585. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Taylor, D.; Gottlieb, R.A. Parkin-mediated mitophagy is downregulated in browning of white adipose tissue. Obesity 2017, 25, 704–712. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Corsa, C.A.S.; Pearson, G.L.; Renberg, A.; Askar, M.M.; Vozheiko, T.; MacDougald, O.A.; Soleimanpour, S.A. The E3 ubiquitin ligase parkin is dispensable for metabolic homeostasis in murine pancreatic β cells and adipocytes. J. Biol. Chem. 2019, 294, 7296–7307. [Google Scholar] [CrossRef] [PubMed]
- Müller, T.D.; Lee, S.J.; Jastroch, M.; Kabra, D.; Stemmer, K.; Aichler, M.; Abplanalp, B.; Ananthakrishnan, G.; Bhardwaj, N.; Collins, S.; et al. P62 Links β-adrenergic input to mitochondrial function and thermogenesis. J. Clin. Investig. 2013, 123, 469–478. [Google Scholar] [CrossRef]
- Ju, L.; Chen, S.; Alimujiang, M.; Bai, N.; Yan, H.; Fang, Q.; Han, J.; Ma, X.; Yang, Y.; Jia, W. A novel role for Bcl2l13 in promoting beige adipocyte biogenesis. Biochem. Biophys. Res. Commun. 2018, 506, 485–491. [Google Scholar] [CrossRef]
- Tol, M.J.; Ottenhoff, R.; van Eijk, M.; Zelcer, N.; Aten, J.; Houten, S.M.; Geerts, D.; Van Roomen, C.; Bierlaagh, M.C.; Scheij, S.; et al. A PPARγ-Bnip3 axis couples adipose mitochondrial fusion-fission balance to systemic insulin sensitivity. Diabetes 2016, 65, 2591–2605. [Google Scholar] [CrossRef] [Green Version]
- Singh, R.; Xiang, Y.; Wang, Y.; Baikati, K.; Cuervo, A.M.; Luu, Y.K.; Tang, Y.; Pessin, J.E.; Schwartz, G.J.; Czaja, M.J. Autophagy regulates adipose mass and differentiation in mice. J. Clin. Investig. 2009, 119, 3329–3339. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.; Goldman, S.; Baerga, R.; Zhao, Y.; Komatsu, M.; Jin, S. Adipose-specific deletion of autophagy-related gene 7 (atg7) in mice reveals a role in adipogenesis. Proc. Natl. Acad. Sci. USA 2009, 106, 19860–19865. [Google Scholar] [CrossRef] [Green Version]
- Martinez-Lopez, N.; Garcia-Macia, M.; Sahu, S.; Athonvarangkul, D.; Liebling, E.; Merlo, P.; Cecconi, F.; Schwartz, G.J.; Singh, R. Autophagy in the CNS and Periphery Coordinate Lipophagy and Lipolysis in the Brown Adipose Tissue and Liver. Cell Metab. 2016, 23, 113–127. [Google Scholar] [CrossRef] [Green Version]
- Öst, A.; Svensson, K.; Ruishalme, I.; Brännmark, C.; Franck, N.; Krook, H.; Sandström, P.; Kjolhede, P.; Strålfors, P. Attenuated mTOR signaling and enhanced autophagy in adipocytes from obese patients with type 2 diabetes. Mol. Med. 2010, 16, 235–246. [Google Scholar] [CrossRef]
- Kovsan, J.; Blüher, M.; Tarnovscki, T.; Klöting, N.; Kirshtein, B.; Madar, L.; Shai, I.; Golan, R.; Harman-Boehm, I.; Schön, M.R.; et al. Altered autophagy in human adipose tissues in obesity. J. Clin. Endocrinol. Metab. 2011, 96, E268–E277. [Google Scholar] [CrossRef] [PubMed]
- Jansen, H.J.; van Essen, P.; Koenen, T.; Joosten, L.A.B.; Netea, M.G.; Tack, C.J.; Stienstra, R. Autophagy activity is up-regulated in adipose tissue of obese individuals and modulates proinflammatory cytokine expression. Endocrinology 2012, 153, 5866–5874. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Haim, Y.; Blüuher, M.; Slutsky, N.; Goldstein, N.; Klöting, N.; Harman-Boehm, I.; Kirshtein, B.; Ginsberg, D.; Gericke, M.; Guiu Jurado, E.; et al. Elevated autophagy gene expression in adipose tissue of obese humans: A potential non-cell-cycle-dependent function of E2F1. Autophagy 2015, 11, 2074–2088. [Google Scholar] [CrossRef] [PubMed]
- Kosacka, J.; Kern, M.; Klöting, N.; Paeschke, S.; Rudich, A.; Haim, Y.; Gericke, M.; Serke, H.; Stumvoll, M.; Bechmann, I.; et al. Autophagy in adipose tissue of patients with obesity and type 2 diabetes. Mol. Cell. Endocrinol. 2015, 409, 21–32. [Google Scholar] [CrossRef] [PubMed]
- Cypess, A.M.; White, A.P.; Vernochet, C.; Schulz, T.J.; Xue, R.; Sass, C.A.; Huang, T.L.; Roberts-Toler, C.; Weiner, L.S.; Sze, C.; et al. Anatomical localization, gene expression profiling and functional characterization of adult human neck brown fat. Nat. Med. 2013, 19, 635–639. [Google Scholar] [CrossRef]
- Szatmári-Tóth, M.; Kristóf, E.; Veréb, Z.; Akhtar, S.; Facskó, A.; Fésüs, L.; Kauppinen, A.; Kaarniranta, K.; Petrovski, G. Clearance of autophagy-associated dying retinal pigment epithelial cells—A possible source for inflammation in age-related macular degeneration. Cell Death Dis. 2016, 7, e2367. [Google Scholar] [CrossRef]
- Nizsalóczki, E.; Nagy, P.; Mocsár, G.; Szabó, Á.; Csomós, I.; Waldmann, T.A.; Vámosi, G.; Mátyus, L.; Bodnár, A. Minimum degree of overlap between IL-9R and IL-2R on human T lymphoma cells: A quantitative CLSM and FRET analysis. Cytom. Part A 2018, 93, 1106–1117. [Google Scholar] [CrossRef] [Green Version]






© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Szatmári-Tóth, M.; Shaw, A.; Csomós, I.; Mocsár, G.; Fischer-Posovszky, P.; Wabitsch, M.; Balajthy, Z.; Lányi, C.; Győry, F.; Kristóf, E.; et al. Thermogenic Activation Downregulates High Mitophagy Rate in Human Masked and Mature Beige Adipocytes. Int. J. Mol. Sci. 2020, 21, 6640. https://doi.org/10.3390/ijms21186640
Szatmári-Tóth M, Shaw A, Csomós I, Mocsár G, Fischer-Posovszky P, Wabitsch M, Balajthy Z, Lányi C, Győry F, Kristóf E, et al. Thermogenic Activation Downregulates High Mitophagy Rate in Human Masked and Mature Beige Adipocytes. International Journal of Molecular Sciences. 2020; 21(18):6640. https://doi.org/10.3390/ijms21186640
Chicago/Turabian StyleSzatmári-Tóth, Mária, Abhirup Shaw, István Csomós, Gábor Mocsár, Pamela Fischer-Posovszky, Martin Wabitsch, Zoltán Balajthy, Cecília Lányi, Ferenc Győry, Endre Kristóf, and et al. 2020. "Thermogenic Activation Downregulates High Mitophagy Rate in Human Masked and Mature Beige Adipocytes" International Journal of Molecular Sciences 21, no. 18: 6640. https://doi.org/10.3390/ijms21186640
APA StyleSzatmári-Tóth, M., Shaw, A., Csomós, I., Mocsár, G., Fischer-Posovszky, P., Wabitsch, M., Balajthy, Z., Lányi, C., Győry, F., Kristóf, E., & Fésüs, L. (2020). Thermogenic Activation Downregulates High Mitophagy Rate in Human Masked and Mature Beige Adipocytes. International Journal of Molecular Sciences, 21(18), 6640. https://doi.org/10.3390/ijms21186640