Non-Coding RNAs, a Novel Paradigm for the Management of Gastrointestinal Stromal Tumors



Abstract
:1. Gastrointestinal Stromal Tumors: A Brief Introduction
2. Current Treatment of Gastrointestinal Stromal Tumors
3. Clinical Needs Regarding the Management of Gastrointestinal Stromal Tumors
4. Non-Coding RNAs
5. Dysregulated miRNAs in GIST

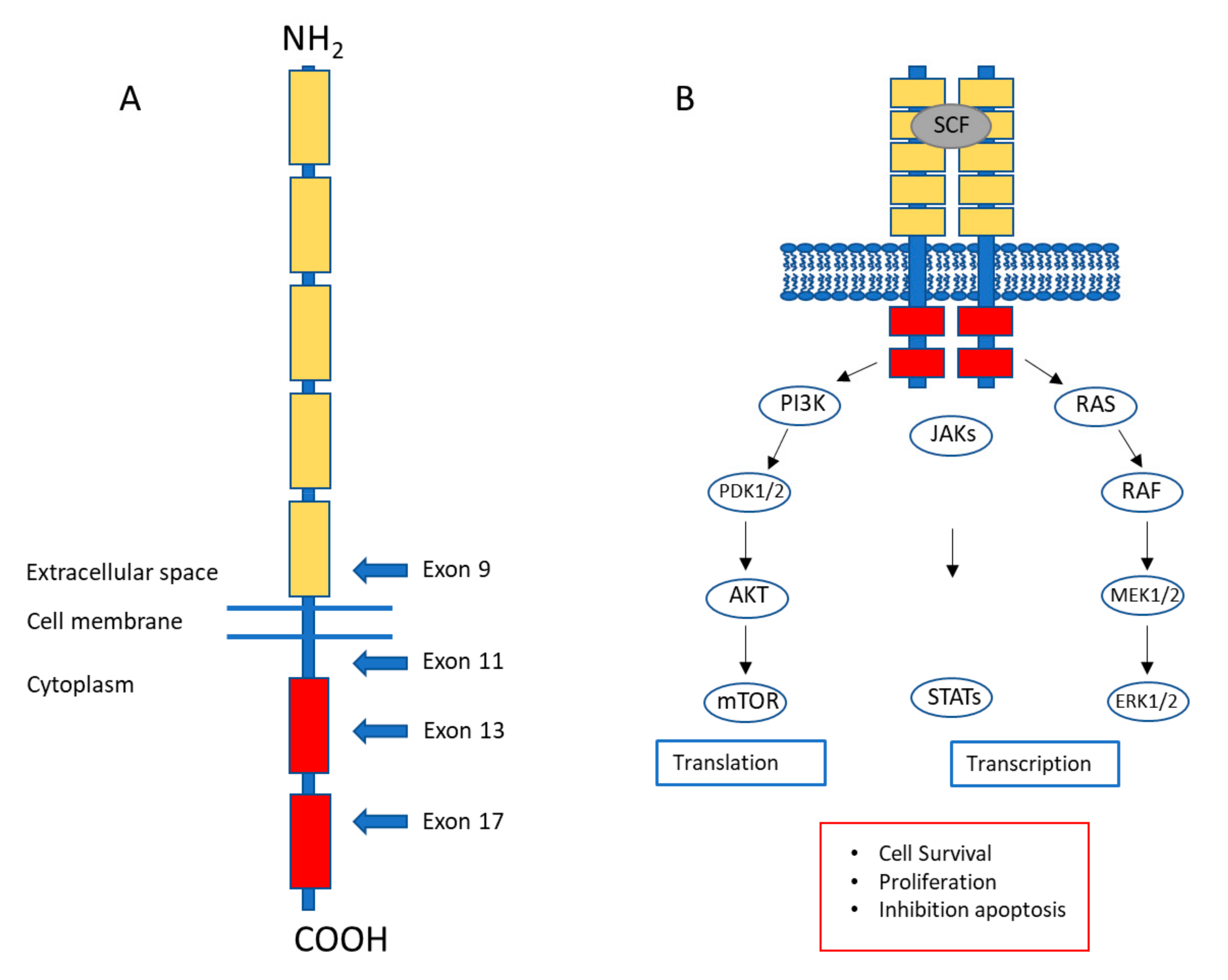{kind=link}
| miRNAs Up/Downregulated in GIST a,c | Comparison/Number of Samples | Platform | Validated miRNAs; Targets and/or Pathways; Association with Clinicopathological Parameters | Ref. |
|---|---|---|---|---|
| Upregulated: Let-7b; miR-10a; miR-22; miR-29a; miR-29b; miR-29c; miR-30a-5p; miR-30c; miR-30d; miR-30e-5p; miR-99b; miR-125a; miR-140*; miR-143; miR-145 Downregulated: miR-1; miR-92; miR-133a; miR-133b; miR-200b; miR-221; miR-222; miR-368; miR-376a | Snap-frozen tumor and tissue samples Primary GIST (n = 8) vs. SS (n = 7); LMS (n = 6); DDLPS (n = 1); RMS (n = 6); NSM (n = 5); skeletal muscle (n = 2) | Microarray Sequencing | [80] | |
| Downregulated: miR-127; miR-134; miR-136; miR-154; miR-154*; miR-299-5p; miR-299-3p; miR-323; miR-329; miR-342; miR-368; miR-369-5p; miR-369-3p; miR-376a; miR-376a*; miR-376b; miR-377; miR-379; miR-381; miR-382; miR-409-3p; miR-409-5p; miR-410; miR-411; miR-431; miR-432*; miR-433; miR-485-3p; miR-487a; miR-487b; miR-493-3p; miR-493-5p; miR-494; miR-495; miR-539; miR-625; miR-654; miR-758 | Snap-frozen tumor samples Primary GIST (n = 20) comparing 14q loss (n = 14) vs. 14q presence (n = 6) | Microarray | • Association with 14q loss | [84] |
| N.A. | Snap frozen tumor tissue Discovery: Primary GIST (n = 12) Validation: Primary GIST (n = 49) | Microarray RT-PCR | miR-132; miR-221; miR-222; miR-504
| [85] |
| Downregulated: miR-221; miR-222 | FFPE samples Primary GIST and adjacent normal tissue (n = 54 pairs) | RT-PCR | • Association with KIT positivity | [87] |
| Downregulated:miR-494 | Snap-frozen tumor samples Primary GIST (n = 31) | miR-494; KIT • Association with 14 q loss | [84,92] | |
| Upregulated: miR-29c; miR-30a; miR-330-3p; miR-497; miR-603 Downregulated: miR-21; miR-221; miR-222; miR-382; miR-938 | Snap-frozen tumor samples Primary GIST (n = 50) vs. intestinal LMS (n = 10) | Microarray RT-PCR | miR-17, miR-20a; ETV1 miR-222; KIT | [88] |
| See publication | FFPE samples Adult KIT/PDGFRA mutant GIST (n = 30) vs. adult WT GIST (n = 25) vs. pediatric WT GIST (n = 18) | RT-PCR | • Distinct miRNA signatures for GIST subtypes correlating with clinicopathological parameters. | [102] |
| Upregulated: miR-330-3p; miR-455-5p; miR-455-3p; miR-886-3p Downregulated: miR-129-1-3p; miR-129-5p; miR-214-5p; miR-424; miR-450a; miR-491-5p | Snap-frozen tumor samples Discovery: KIT/PDGFRA mutant GIST (n = 9) vs. WT GIST (n = 4) Validation: Mutant GIST (n = 13) vs. WT GIST (n = 3) | Microarray RT-PCR | miR-139-5p; miR-148a; miR-193-3p; miR-330-3p; miR-455-5p; miR-129-1-3p; miR-129-2-3p; miR-876-5p
| [103] |
| Downregulated: miR-133b | Snap-frozen tumor samples Primary GIST (n = 19) comparing high grade vs. intermediate and low grade | Microarray RT-PCR | miR-133b; inverse correlation between fascin-1 and miR-133b • Downregulated in high-grade GIST | [94] |
| Downregulated: miR-218 | Snap-frozen tumor and tissue samples, primary GIST (n = 10), normal adjacent tissue (n = 5) | RT-PCR | miR-218; KIT | [90] |
| Upregulated: miR-140-3p; miR-483-5p; miR-3151-5p Downregulated: miR-28-3p; miR-133a-3p; miR-133b; miR-195-5p; miR-378f; miR-3135b; miR-4535 | Fresh tumor samples Primary GIST (n = 9) vs. leiomyomas (n = 7) | Microarray RT-PCR | miR-140-5p, miR-140-3p | [96] |
| Downregulated: miR-221; miR-222 | FFPE samples Primary GIST (n = 24) vs. smooth muscle (n = 6) | RT-PCR | miR-221/222; KIT | [89] |
| Downregulated: miR-9-3p; miR-34a; miR-152; miR-155; miR-203; miR-335; miR-375; miR-489; miR-582; miR-615; miR-618 | GIST-T1 (n = 1), snap-frozen primary GIST samples (n = 39), FFPE primary GIST samples (n = 98) | RT-PCR | miR-34a, PDGFRA miR-335 • Association with CpG island methylation | [97] |
| Upregulated: miR-34c-5p; miR-4773 Downregulated: Let-7c; miR-218; miR-488*; miR-4683 | Snap-frozen tumor samples Primary GIST (n = 53): malignant GIS T (n = 30) vs. benign GIST (n = 9) b | RT-PCR | • Association with malignant GISTs | [95] |
| Upregulated: miR-196a Downregulated: Let-7c; miR-29b-2*; miR-29c*; miR-204; miR-204-3p; miR-218; miR-625; miR628-5p; miR-744; miR-891b | Snap-frozen tumor samples Primary GIST (n = 53): malignant GIST (n = 30) vs. borderline GIST (n = 14) b | RT-PCR | • Association with malignant GISTs | [95] |
| Upregulated: miR-455-3p; miR-483-5p; miR-509-3p; miR-675-3p Downregulated: miR-141-3p; miR-133a-3p; miR-133b; miR-182-5p; miR-192-5p; miR-200a-3p; miR-200b-3p; miR-200c-3p; miR-203a-3p; miR-215-5p; miR-375; miR-429; miR-451a; miR-486-5p; miR-490-3p | FFPE tumor and tissue samples Discovery: Pairs (n = 15) primary GIST and adjacent tissue Validation: Pairs (n = 40) primary GIST and adjacent tissue | RNA-seq RT-PCR | All listed miRNAs validated
| [98] |
| Downregulated: miR-152 | Cell lines GIST48; GIST430; GIST882; GIST-T1 | RT-PCR | miR-152; CTSL | [99] |
| Upregulated: miR-374b | FFPE samples Pairs (n = 143) of Primary GIST and adjacent tissue | RT-PCR | miR-374b; PTEN • Association of miR-374b levels with tumor diameter and pathological state | [100] |
| Downregulated: miR-494 | Snap-frozen tumor samples Primary GIST (n = 35) | Microarray | miR-494; BIRC5 | [93] |
| Upregulated: miR-29b-1-5p Downregulated: miR-134-5p; miR-323b-3p; miR-382-5p; miR-409-3p; miR-1185-1-3p; miR-3187-3p; miR-4510 | Discovery: Pairs (n = 6) primary GIST and adjacent tissue Validation: Pairs (n = 64) primary GIST and adjacent tissue | RNA-seq RT-PCR | miR-4510; APOC2 • Association of miR-4510 levels with tumor location, tumor size, mitotic index and risk classification. | [101] |
| Downregulated: miR-200b-3p; miR-375-3p | FFPE tumor and tissue samples Discovery: Pairs (n = 15) primary GIST and adjacent tissue Validation: Pairs (n = 40) primary GIST and adjacent tissue | RNA-seq RT-PCR | miR-200b-3p; EGFR miR-375-3p; KIT | [91,98] |
6. MiRNAs Associated with Gastrointestinal Stromal Tumor Metastasis
7. MiRNAs Related to Imatinib Resistance
8. Additional Non-Coding RNAs in Gastrointestinal Stromal Tumors
9. Biomarkers
10. Therapeutic Potential of Non-Coding RNAs
11. Future Directions
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| circRNA | circular RNA |
| DDLPS | Dedifferentiated Liposarcoma |
| EMT | Epithelial-Mesenchymal Transition |
| FFPE | Formalin-Fixed Paraffin-Embedded |
| GIST | Gastrointestinal Stromal Tumor |
| LMS | Leiomyosarcoma |
| lncRNA | Long Non-Coding RNA |
| miRNA | MicroRNA |
| NMS | Normal Smooth Muscle |
| RMS | Rhabdomyosarcoma |
| SS | Synovial Sarcoma |
References
- Ducimetiere, F.; Lurkin, A.; Ranchere-Vince, D.; Decouvelaere, A.V.; Peoc’h, M.; Istier, L.; Chalabreysse, P.; Muller, C.; Alberti, L.; Bringuier, P.-P.; et al. Incidence of sarcoma histotypes and molecular subtypes in a prospective epidemiological study with central pathology review and molecular testing. PLoS ONE 2011, 6, e20294. [Google Scholar] [CrossRef] [PubMed]
- Goettsch, W.G.; Bos, S.D.; Breekveldt-Postma, N.; Casparie, M.; Herings, R.M.; Hogendoorn, P.C. Incidence of gastrointestinal stromal tumours is underestimated: Results of a nation-wide study. Eur. J. Cancer 2005, 41, 2868–2872. [Google Scholar] [CrossRef] [Green Version]
- Joensuu, H.; Hohenberger, P.; Corless, C.L. Gastrointestinal stromal tumour. Lancet 2013, 382, 973–983. [Google Scholar] [CrossRef]
- Miettinen, M.; Lasota, J. Gastrointestinal stromal tumors—Definition, clinical, histological, immunohistochemical, and molecular genetic features and differential diagnosis. Virchows Arch. 2001, 438, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Kindblom, L.G.; Remotti, H.E.; Aldenborg, F.; Meis-Kindblom, J.M. Gastrointestinal pacemaker cell tumor (GIPACT): Gastrointestinal stromal tumors show phenotypic characteristics of the interstitial cells of Cajal. Am. J. Pathol. 1998, 152, 1259–1269. [Google Scholar] [PubMed]
- Sircar, K.; Hewlett, B.R.; Huizinga, J.D.; Chorneyko, K.; Berezin, I.; Riddell, R.H. Interstitial cells of Cajal as precursors of gastrointestinal stromal tumors. Am. J. Surg. Pathol. 1999, 23, 377–389. [Google Scholar] [CrossRef] [PubMed]
- Gomez-Pinilla, P.J.; Gibbons, S.J.; Bardsley, M.R.; Lorincz, A.; Pozo, M.J.; Pasricha, P.J.; Van de Rijn, M.; West, R.B.; Sarr, M.G.; Kendrick, M.L.; et al. Ano1 is a selective marker of interstitial cells of Cajal in the human and mouse gastrointestinal tract. Am. J. Physiol. Gastrointest. Liver Physiol. 2009, 296, G1370–G1381. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chi, P.; Chen, Y.; Zhang, L.; Guo, X.; Wongvipat, J.; Shamu, T.; Fletcher, J.A.; Dewell, S.; Maki, R.G.; Zheng, D.; et al. ETV1 is a lineage survival factor that cooperates with KIT in gastrointestinal stromal tumours. Nature 2010, 467, 849–853. [Google Scholar] [CrossRef] [Green Version]
- Heinrich, M.C.; Corless, C.L.; Duensing, A.; McGreevey, L.; Chen, C.J.; Joseph, N.; Singer, S.; Griffith, D.J.; Haley, A.; Town, A.; et al. PDGFRA activating mutations in gastrointestinal stromal tumors. Science 2003, 299, 708–710. [Google Scholar] [CrossRef]
- Hirota, S.; Isozaki, K.; Moriyama, Y.; Hashimoto, K.; Nishida, T.; Ishiguro, S.; Kawano, K.; Hanada, M.; Kurata, A.; Takeda, M.; et al. Gain-of-function mutations of c-kit in human gastrointestinal stromal tumors. Science 1998, 279, 577–580. [Google Scholar] [CrossRef]
- Lasota, J.; Miettinen, M. KIT and PDGFRA mutations in gastrointestinal stromal tumors (GISTs). Semin. Diagn. Pathol. 2006, 23, 91–102. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Corless, C.L.; Heinrich, M.C. Molecular pathobiology of gastrointestinal stromal sarcomas. Annu. Rev. Pathol. 2008, 3, 557–586. [Google Scholar] [CrossRef] [PubMed]
- Buchdunger, E.; Cioffi, C.L.; Law, N.; Stover, D.; Ohno-Jones, S.; Druker, B.J.; Lydon, N.B. Abl protein-tyrosine kinase inhibitor STI571 inhibits in vitro signal transduction mediated by c-kit and platelet-derived growth factor receptors. J. Pharmacol. Exp. Ther. 2000, 295, 139–145. [Google Scholar] [PubMed]
- Demetri, G.D.; von Mehren, M.; Blanke, C.D.; Van den Abbeele, A.D.; Eisenberg, B.; Roberts, P.J.; Heinrich, M.C.; Tuveson, D.A.; Singer, S.; Janicek, M.; et al. Efficacy and safety of imatinib mesylate in advanced gastrointestinal stromal tumors. N. Engl. J. Med. 2002, 347, 472–480. [Google Scholar] [CrossRef]
- van Oosterom, A.T.; Judson, I.; Verweij, J.; Stroobants, S.; Donato di Paola, E.; Dimitrijevic, S.; Martens, M.; Webb, A.; Sciot, R.; Van Glabbeke, M.; et al. Safety and efficacy of imatinib (STI571) in metastatic gastrointestinal stromal tumours: A phase I study. Lancet 2001, 358, 1421–1423. [Google Scholar] [CrossRef]
- Agaimy, A.; Terracciano, L.M.; Dirnhofer, S.; Tornillo, L.; Foerster, A.; Hartmann, A.; Bihl, M.P. V600E BRAF mutations are alternative early molecular events in a subset of KIT/PDGFRA wild-type gastrointestinal stromal tumours. J. Clin. Pathol. 2009, 62, 613–616. [Google Scholar] [CrossRef]
- Agaram, N.P.; Wong, G.C.; Guo, T.; Maki, R.G.; Singer, S.; Dematteo, R.P.; Besmer, P.; Antonescu, C.R. Novel V600E BRAF mutations in imatinib-naive and imatinib-resistant gastrointestinal stromal tumors. Genes Chromosomes Cancer 2008, 47, 853–859. [Google Scholar] [CrossRef] [Green Version]
- Hostein, I.; Faur, N.; Primois, C.; Boury, F.; Denard, J.; Emile, J.F.; Bringuier, P.P.; Scoazec, J.Y.; Coindre, J.M. BRAF mutation status in gastrointestinal stromal tumors. Am. J. Clin. Pathol. 2010, 133, 141–148. [Google Scholar] [CrossRef] [Green Version]
- Janeway, K.A.; Kim, S.Y.; Lodish, M.; Nose, V.; Rustin, P.; Gaal, J.; Dahia, P.L.; Liegl, B.; Ball, E.R.; Raygada, M.; et al. Defects in succinate dehydrogenase in gastrointestinal stromal tumors lacking KIT and PDGFRA mutations. Proc. Natl. Acad. Sci. USA 2011, 108, 314–318. [Google Scholar] [CrossRef] [Green Version]
- Pasini, B.; McWhinney, S.R.; Bei, T.; Matyakhina, L.; Stergiopoulos, S.; Muchow, M.; Boikos, S.A.; Ferrando, B.; Pacak, K.; Assie, G.; et al. Clinical and molecular genetics of patients with the Carney-Stratakis syndrome and germline mutations of the genes coding for the succinate dehydrogenase subunits SDHB, SDHC, and SDHD. Eur. J. Hum. Genet. 2008, 16, 79–88. [Google Scholar] [CrossRef]
- Casali, P.G.; Abecassis, N.; Aro, H.T.; Bauer, S.; Biagini, R.; Bielack, S.; Bonvalot, S.; Boukovinas, I.; Bovee, J.; Brodowicz, T.; et al. Gastrointestinal stromal tumours: ESMO-EURACAN Clinical Practice Guidelines for diagnosis, treatment and follow-up. Ann. Oncol. 2018, 29 (Suppl. 4), iv68–iv78. [Google Scholar] [CrossRef] [PubMed]
- Joensuu, H.; Eriksson, M.; Sundby Hall, K.; Reichardt, A.; Hartmann, J.T.; Pink, D.; Ramadori, G.; Hohenberger, P.; Al-Batran, S.E.; Schlemmer, M.; et al. Adjuvant Imatinib for High-Risk GI Stromal Tumor: Analysis of a Randomized Trial. J. Clin. Oncol. 2016, 34, 244–250. [Google Scholar] [CrossRef] [PubMed]
- Szucs, Z.; Thway, K.; Fisher, C.; Bulusu, R.; Constantinidou, A.; Benson, C.; van der Graaf, W.T.; Jones, R.L. Molecular subtypes of gastrointestinal stromal tumors and their prognostic and therapeutic implications. Future Oncol. 2017, 13, 93–107. [Google Scholar] [CrossRef] [Green Version]
- Corless, C.L.; Ballman, K.V.; Antonescu, C.R.; Kolesnikova, V.; Maki, R.G.; Pisters, P.W.; Blackstein, M.E.; Blanke, C.D.; Demetri, G.D.; Heinrich, M.C.; et al. Pathologic and molecular features correlate with long-term outcome after adjuvant therapy of resected primary GI stromal tumor: The ACOSOG Z9001 trial. J. Clin. Oncol. 2014, 32, 1563–1570. [Google Scholar] [CrossRef]
- Joensuu, H.; Wardelmann, E.; Sihto, H.; Eriksson, M.; Sundby Hall, K.; Reichardt, A.; Hartmann, J.T.; Pink, D.; Cameron, S.; Hohenberger, P.; et al. Effect of KIT and PDGFRA Mutations on Survival in Patients with Gastrointestinal Stromal Tumors Treated with Adjuvant Imatinib: An Exploratory Analysis of a Randomized Clinical Trial. JAMA Oncol. 2017, 3, 602–609. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gastrointestinal Stromal Tumor Meta-Analysis Group (MetaGIST). Comparison of two doses of imatinib for the treatment of unresectable or metastatic gastrointestinal stromal tumors: A meta-analysis of 1,640 patients. J. Clin. Oncol. 2010, 28, 1247–1253. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Corless, C.L.; Schroeder, A.; Griffith, D.; Town, A.; McGreevey, L.; Harrell, P.; Shiraga, S.; Bainbridge, T.; Morich, J.; Heinrich, M.C. PDGFRA mutations in gastrointestinal stromal tumors: Frequency, spectrum and in vitro sensitivity to imatinib. J. Clin. Oncol. 2005, 23, 5357–5364. [Google Scholar] [CrossRef] [PubMed]
- Debiec-Rychter, M.; Sciot, R.; Le Cesne, A.; Schlemmer, M.; Hohenberger, P.; van Oosterom, A.T.; Blay, J.Y.; Leyvraz, S.; Stul, M.; Casali, P.G.; et al. KIT mutations and dose selection for imatinib in patients with advanced gastrointestinal stromal tumours. Eur. J. Cancer 2006, 42, 1093–1103. [Google Scholar] [CrossRef]
- Nishida, T. Therapeutic strategies for wild-type gastrointestinal stromal tumor: Is it different from KIT or PDGFRA-mutated GISTs? Transl. Gastroenterol. Hepatol. 2017, 2, 92. [Google Scholar] [CrossRef] [Green Version]
- Amirnasr, A.; Gits, C.M.M.; van Kuijk, P.F.; Smid, M.; Vriends, A.L.M.; Rutkowski, P.; Sciot, R.; Schoffski, P.; Debiec-Rychter, M.; Sleijfer, S.; et al. Molecular Comparison of Imatinib-Naive and Resistant Gastrointestinal Stromal Tumors: Differentially Expressed microRNAs and mRNAs. Cancers (Basel) 2019, 11. [Google Scholar] [CrossRef] [Green Version]
- Antonescu, C.R.; Besmer, P.; Guo, T.; Arkun, K.; Hom, G.; Koryotowski, B.; Leversha, M.A.; Jeffrey, P.D.; Desantis, D.; Singer, S.; et al. Acquired resistance to imatinib in gastrointestinal stromal tumor occurs through secondary gene mutation. Clin. Cancer Res. 2005, 11, 4182–4190. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gajiwala, K.S.; Wu, J.C.; Christensen, J.; Deshmukh, G.D.; Diehl, W.; DiNitto, J.P.; English, J.M.; Greig, M.J.; He, Y.A.; Jacques, S.L.; et al. KIT kinase mutants show unique mechanisms of drug resistance to imatinib and sunitinib in gastrointestinal stromal tumor patients. Proc. Natl. Acad. Sci. USA 2009, 106, 1542–1547. [Google Scholar] [CrossRef] [Green Version]
- Debiec-Rychter, M.; Cools, J.; Dumez, H.; Sciot, R.; Stul, M.; Mentens, N.; Vranckx, H.; Wasag, B.; Prenen, H.; Roesel, J.; et al. Mechanisms of resistance to imatinib mesylate in gastrointestinal stromal tumors and activity of the PKC412 inhibitor against imatinib-resistant mutants. Gastroenterology 2005, 128, 270–279. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liegl, B.; Kepten, I.; Le, C.; Zhu, M.; Demetri, G.D.; Heinrich, M.C.; Fletcher, C.D.; Corless, C.L.; Fletcher, J.A. Heterogeneity of kinase inhibitor resistance mechanisms in GIST. J. Pathol. 2008, 216, 64–74. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mahadevan, D.; Cooke, L.; Riley, C.; Swart, R.; Simons, B.; Della Croce, K.; Wisner, L.; Iorio, M.; Shakalya, K.; Garewal, H.; et al. A novel tyrosine kinase switch is a mechanism of imatinib resistance in gastrointestinal stromal tumors. Oncogene 2007, 26, 3909–3919. [Google Scholar] [CrossRef] [Green Version]
- Sakurama, K.; Noma, K.; Takaoka, M.; Tomono, Y.; Watanabe, N.; Hatakeyama, S.; Ohmori, O.; Hirota, S.; Motoki, T.; Shirakawa, Y.; et al. Inhibition of focal adhesion kinase as a potential therapeutic strategy for imatinib-resistant gastrointestinal stromal tumor. Mol. Cancer Ther. 2009, 8, 127–134. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Takahashi, T.; Serada, S.; Ako, M.; Fujimoto, M.; Miyazaki, Y.; Nakatsuka, R.; Ikezoe, T.; Yokoyama, A.; Taguchi, T.; Shimada, K.; et al. New findings of kinase switching in gastrointestinal stromal tumor under imatinib using phosphoproteomic analysis. Int. J. Cancer 2013, 133, 2737–2743. [Google Scholar] [CrossRef]
- Demetri, G.D.; van Oosterom, A.T.; Garrett, C.R.; Blackstein, M.E.; Shah, M.H.; Verweij, J.; McArthur, G.; Judson, I.R.; Heinrich, M.C.; Morgan, J.A.; et al. Efficacy and safety of sunitinib in patients with advanced gastrointestinal stromal tumour after failure of imatinib: A randomised controlled trial. Lancet 2006, 368, 1329–1338. [Google Scholar] [CrossRef]
- George, S.; Blay, J.Y.; Casali, P.G.; Le Cesne, A.; Stephenson, P.; Deprimo, S.E.; Harmon, C.S.; Law, C.N.; Morgan, J.A.; Ray-Coquard, I.; et al. Clinical evaluation of continuous daily dosing of sunitinib malate in patients with advanced gastrointestinal stromal tumour after imatinib failure. Eur. J. Cancer 2009, 45, 1959–1968. [Google Scholar] [CrossRef]
- Demetri, G.D.; Reichardt, P.; Kang, Y.K.; Blay, J.Y.; Rutkowski, P.; Gelderblom, H.; Hohenberger, P.; Leahy, M.; von Mehren, M.; Joensuu, H.; et al. Efficacy and safety of regorafenib for advanced gastrointestinal stromal tumours after failure of imatinib and sunitinib (GRID): An international, multicentre, randomised, placebo-controlled, phase 3 trial. Lancet 2013, 381, 295–302. [Google Scholar] [CrossRef] [Green Version]
- Blay, J.Y.; Serrano, C.; Heinrich, M.C.; Zalcberg, J.; Bauer, S.; Gelderblom, H.; Schoffski, P.; Jones, R.L.; Attia, S.; D’Amato, G.; et al. Ripretinib in patients with advanced gastrointestinal stromal tumours (INVICTUS): A double-blind, randomised, placebo-controlled, phase 3 trial. Lancet Oncol. 2020, 21, 923–934. [Google Scholar] [CrossRef]
- Heinrich, M.C.; Jones, R.L.; von Mehren, M.; Schoffski, P.; Serrano, C.; Kang, Y.K.; Cassier, P.A.; Mir, O.; Eskens, F.; Tap, W.D.; et al. Avapritinib in advanced PDGFRA D842V-mutant gastrointestinal stromal tumour (NAVIGATOR): A multicentre, open-label, phase 1 trial. Lancet Oncol. 2020, 21, 935–946. [Google Scholar] [CrossRef]
- Lander, E.S.; Linton, L.M.; Birren, B.; Nusbaum, C.; Zody, M.C.; Baldwin, J.; Devon, K.; Dewar, K.; Doyle, M.; FitzHugh, W.; et al. Initial sequencing and analysis of the human genome. Nature 2001, 409, 860–921. [Google Scholar]
- Venter, J.C.; Adams, M.D.; Myers, E.W.; Li, P.W.; Mural, R.J.; Sutton, G.G.; Smith, H.O.; Yandell, M.; Evans, C.A.; Holt, R.A.; et al. The sequence of the human genome. Science 2001, 291, 1304–1351. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Salzberg, S.L. Open questions: How many genes do we have? BMC Biol. 2018, 16, 94. [Google Scholar] [CrossRef] [PubMed]
- Pray, L. Eukaryotic Genome Complexity. Nat. Educ. 2008, 1, 96. [Google Scholar]
- Carninci, P.; Kasukawa, T.; Katayama, S.; Gough, J.; Frith, M.C.; Maeda, N.; Oyama, R.; Ravasi, T.; Lenhard, B.; Wells, C.; et al. The transcriptional landscape of the mammalian genome. Science 2005, 309, 1559–1563. [Google Scholar] [PubMed] [Green Version]
- Djebali, S.; Davis, C.A.; Merkel, A.; Dobin, A.; Lassmann, T.; Mortazavi, A.; Tanzer, A.; Lagarde, J.; Lin, W.; Schlesinger, F.; et al. Landscape of transcription in human cells. Nature 2012, 489, 101–108. [Google Scholar] [CrossRef]
- Frith, M.C.; Pheasant, M.; Mattick, J.S. The amazing complexity of the human transcriptome. Eur. J. Hum. Genet. 2005, 13, 894–897. [Google Scholar] [CrossRef]
- Mattick, J.S. RNA regulation: A new genetics? Nat. Rev. Genet. 2004, 5, 316–323. [Google Scholar] [CrossRef]
- Lee, R.C.; Feinbaum, R.L.; Ambros, V. Elegans heterochronic gene lin-4 encodes small RNAs with antisense complementarity to lin-14. Cell 1993, 75, 843–854. [Google Scholar] [CrossRef]
- Lagos-Quintana, M.; Rauhut, R.; Meyer, J.; Borkhardt, A.; Tuschl, T. New microRNAs from mouse and human. RNA 2003, 9, 175–179. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pasquinelli, A.E.; Reinhart, B.J.; Slack, F.; Martindale, M.Q.; Kuroda, M.I.; Maller, B.; Hayward, D.C.; Ball, E.E.; Degnan, B.; Muller, P.; et al. Conservation of the sequence and temporal expression of let-7 heterochronic regulatory RNA. Nature 2000, 408, 86–89. [Google Scholar] [CrossRef] [PubMed]
- Bartel, D.P. Metazoan MicroRNAs. Cell 2018, 173, 20–51. [Google Scholar] [CrossRef]
- Dragomir, M.P.; Knutsen, E.; Calin, G.A. SnapShot: Unconventional miRNA Functions. Cell 2018, 174, 1038.e1. [Google Scholar] [CrossRef]
- Orom, U.A.; Nielsen, F.C.; Lund, A.H. MicroRNA-10a binds the 5’UTR of ribosomal protein mRNAs and enhances their translation. Mol. Cell 2008, 30, 460–471. [Google Scholar] [CrossRef]
- Friedman, R.C.; Farh, K.K.; Burge, C.B.; Bartel, D.P. Most mammalian mRNAs are conserved targets of microRNAs. Genome. Res. 2009, 19, 92–105. [Google Scholar] [CrossRef] [Green Version]
- Lewis, B.P.; Burge, C.B.; Bartel, D.P. Conserved seed pairing, often flanked by adenosines, indicates that thousands of human genes are microRNA targets. Cell 2005, 120, 15–20. [Google Scholar] [CrossRef] [Green Version]
- Adams, B.D.; Kasinski, A.L.; Slack, F.J. Aberrant regulation and function of microRNAs in cancer. Curr. Biol. 2014, 24, R762–R766. [Google Scholar] [CrossRef] [Green Version]
- Calin, G.A.; Croce, C.M. MicroRNA signatures in human cancers. Nat. Rev. Cancer 2006, 6, 857–866. [Google Scholar] [CrossRef]
- Di Leva, G.; Garofalo, M.; Croce, C.M. MicroRNAs in cancer. Annu. Rev. Pathol. 2014, 9, 287–314. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Croce, C.M. Causes and consequences of microRNA dysregulation in cancer. Nat. Rev. Genet. 2009, 10, 704–714. [Google Scholar] [CrossRef]
- Zaravinos, A. The Regulatory Role of MicroRNAs in EMT and Cancer. J. Oncol. 2015, 2015, 865816. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fanini, F.; Fabbri, M. MicroRNAs and cancer resistance: A new molecular plot. Clin. Pharmacol. Ther. 2016, 99, 485–493. [Google Scholar] [CrossRef] [PubMed]
- Kosaka, N.; Iguchi, H.; Ochiya, T. Circulating microRNA in body fluid: A new potential biomarker for cancer diagnosis and prognosis. Cancer Sci. 2010, 101, 2087–2092. [Google Scholar] [CrossRef]
- Mitchell, P.S.; Parkin, R.K.; Kroh, E.M.; Fritz, B.R.; Wyman, S.K.; Pogosova-Agadjanyan, E.L.; Peterson, A.; Noteboom, J.; O’Briant, K.C.; Allen, A.; et al. Circulating microRNAs as stable blood-based markers for cancer detection. Proc. Natl. Acad. Sci. USA 2008, 105, 10513–10518. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rupaimoole, R.; Slack, F.J. MicroRNA therapeutics: Towards a new era for the management of cancer and other diseases. Nat. Rev. Drug Discov. 2017, 16, 203–222. [Google Scholar] [CrossRef]
- Slack, F.J.; Chinnaiyan, A.M. The Role of Non-coding RNAs in Oncology. Cell 2019, 179, 1033–1055. [Google Scholar] [CrossRef]
- Iyer, M.K.; Niknafs, Y.S.; Malik, R.; Singhal, U.; Sahu, A.; Hosono, Y.; Barrette, T.R.; Prensner, J.R.; Evans, J.R.; Zhao, S.; et al. The landscape of long noncoding RNAs in the human transcriptome. Nat. Genet. 2015, 47, 199–208. [Google Scholar] [CrossRef]
- Kopp, F.; Mendell, J.T. Functional Classification and Experimental Dissection of Long Noncoding RNAs. Cell 2018, 172, 393–407. [Google Scholar] [CrossRef] [Green Version]
- Marchese, F.P.; Raimondi, I.; Huarte, M. The multidimensional mechanisms of long noncoding RNA function. Genome Biol. 2017, 18, 206. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ransohoff, J.D.; Wei, Y.; Khavari, P.A. The functions and unique features of long intergenic non-coding RNA. Nat. Rev. Mol. Cell Biol. 2018, 19, 143–157. [Google Scholar] [CrossRef] [PubMed]
- Wang, K.C.; Chang, H.Y. Molecular mechanisms of long noncoding RNAs. Mol. Cell 2011, 43, 904–914. [Google Scholar] [CrossRef] [Green Version]
- Peng, W.X.; Koirala, P.; Mo, Y.Y. LncRNA-mediated regulation of cell signaling in cancer. Oncogene 2017, 36, 5661–5667. [Google Scholar] [CrossRef]
- Schmitt, A.M.; Chang, H.Y. Long Noncoding RNAs in Cancer Pathways. Cancer Cell 2016, 29, 452–463. [Google Scholar] [CrossRef] [Green Version]
- Salzman, J.; Gawad, C.; Wang, P.L.; Lacayo, N.; Brown, P.O. Circular RNAs are the predominant transcript isoform from hundreds of human genes in diverse cell types. PLoS ONE 2012, 7, e30733. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Salzman, J. Circular RNA Expression: Its Potential Regulation and Function. Trends Genet. 2016, 32, 309–316. [Google Scholar] [CrossRef] [Green Version]
- Li, X.; Yang, L.; Chen, L.L. The Biogenesis, Functions, and Challenges of Circular RNAs. Mol. Cell 2018, 71, 428–442. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vo, J.N.; Cieslik, M.; Zhang, Y.; Shukla, S.; Xiao, L.; Zhang, Y.; Wu, Y.M.; Dhanasekaran, S.M.; Engelke, C.G.; Cao, X.; et al. The Landscape of Circular RNA in Cancer. Cell 2019, 176, 869–881. [Google Scholar] [CrossRef] [Green Version]
- Subramanian, S.; Lui, W.O.; Lee, C.H.; Espinosa, I.; Nielsen, T.O.; Heinrich, M.C.; Corless, C.L.; Fire, A.Z.; van de Rijn, M. MicroRNA expression signature of human sarcomas. Oncogene 2008, 27, 2015–2026. [Google Scholar] [CrossRef] [Green Version]
- Gunawan, B.; von Heydebreck, A.; Sander, B.; Schulten, H.J.; Haller, F.; Langer, C.; Armbrust, T.; Bollmann, M.; Gasparov, S.; Kovac, D.; et al. An oncogenetic tree model in gastrointestinal stromal tumours (GISTs) identifies different pathways of cytogenetic evolution with prognostic implications. J. Pathol. 2007, 211, 463–470. [Google Scholar] [CrossRef] [PubMed]
- Assamaki, R.; Sarlomo-Rikala, M.; Lopez-Guerrero, J.A.; Lasota, J.; Andersson, L.C.; Llombart-Bosch, A.; Miettinen, M.; Knuutila, S. Array comparative genomic hybridization analysis of chromosomal imbalances and their target genes in gastrointestinal stromal tumors. Genes Chromosomes Cancer 2007, 46, 564–576. [Google Scholar] [CrossRef] [PubMed]
- Wozniak, A.; Sciot, R.; Guillou, L.; Pauwels, P.; Wasag, B.; Stul, M.; Vermeesch, J.R.; Vandenberghe, P.; Limon, J.; Debiec-Rychter, M. Array CGH analysis in primary gastrointestinal stromal tumors: Cytogenetic profile correlates with anatomic site and tumor aggressiveness, irrespective of mutational status. Genes Chromosomes Cancer 2007, 46, 261–276. [Google Scholar] [CrossRef] [PubMed]
- Choi, H.J.; Lee, H.; Kim, H.; Kwon, J.E.; Kang, H.J.; You, K.T.; Rhee, H.; Noh, S.H.; Paik, Y.K.; Hyung, W.J.; et al. MicroRNA expression profile of gastrointestinal stromal tumors is distinguished by 14q loss and anatomic site. Int. J. Cancer 2010, 126, 1640–1650. [Google Scholar] [CrossRef]
- Haller, F.; von Heydebreck, A.; Zhang, J.D.; Gunawan, B.; Langer, C.; Ramadori, G.; Wiemann, S.; Sahin, O. Localization- and mutation-dependent microRNA (miRNA) expression signatures in gastrointestinal stromal tumours (GISTs), with a cluster of co-expressed miRNAs located at 14q32.31. J. Pathol. 2010, 220, 71–86. [Google Scholar] [CrossRef]
- Felli, N.; Fontana, L.; Pelosi, E.; Botta, R.; Bonci, D.; Facchiano, F.; Liuzzi, F.; Lulli, V.; Morsilli, O.; Santoro, S.; et al. MicroRNAs 221 and 222 inhibit normal erythropoiesis and erythroleukemic cell growth via kit receptor down-modulation. Proc. Natl. Acad. Sci. USA 2005, 102, 18081–18086. [Google Scholar] [CrossRef] [Green Version]
- Koelz, M.; Lense, J.; Wrba, F.; Scheffler, M.; Dienes, H.P.; Odenthal, M. Down-regulation of miR-221 and miR-222 correlates with pronounced Kit expression in gastrointestinal stromal tumors. Int. J. Oncol. 2011, 38, 503–511. [Google Scholar] [CrossRef] [Green Version]
- Gits, C.M.; van Kuijk, P.F.; Jonkers, M.B.; Boersma, A.W.; van Ijcken, W.F.; Wozniak, A.; Sciot, R.; Rutkowski, P.; Schoffski, P.; Taguchi, T.; et al. MiR-17-92 and miR-221/222 cluster members target KIT and ETV1 in human gastrointestinal stromal tumours. Br. J. Cancer 2013, 109, 1625–1635. [Google Scholar] [CrossRef]
- Ihle, M.A.; Trautmann, M.; Kuenstlinger, H.; Huss, S.; Heydt, C.; Fassunke, J.; Wardelmann, E.; Bauer, S.; Schildhaus, H.U.; Buettner, R.; et al. miRNA-221 and miRNA-222 induce apoptosis via the KIT/AKT signalling pathway in gastrointestinal stromal tumours. Mol. Oncol. 2015, 9, 1421–1433. [Google Scholar] [CrossRef]
- Fan, R.; Zhong, J.; Zheng, S.; Wang, Z.; Xu, Y.; Li, S.; Zhou, J.; Yuan, F. MicroRNA-218 inhibits gastrointestinal stromal tumor cell and invasion by targeting KIT. Tumour Biol. 2014, 35, 4209–4217. [Google Scholar] [CrossRef]
- Gyvyte, U.; Lukosevicius, R.; Inciuraite, R.; Streleckiene, G.; Gudoityte, G.; Bekampyte, J.; Valentini, S.; Salteniene, V.; Ruzgys, P.; Satkauskas, S.; et al. The Role of miR-375-3p and miR-200b-3p in Gastrointestinal Stromal Tumors. Int. J. Mol. Sci. 2020, 21, 5151. [Google Scholar] [CrossRef] [PubMed]
- Kim, W.K.; Park, M.; Kim, Y.K.; Tae, Y.K.; Yang, H.K.; Lee, J.M.; Kim, H. MicroRNA-494 downregulates KIT and inhibits gastrointestinal stromal tumor cell proliferation. Clin. Cancer Res. 2011, 17, 7584–7594. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yun, S.; Kim, W.K.; Kwon, Y.; Jang, M.; Bauer, S.; Kim, H. Survivin is a novel transcription regulator of KIT and is downregulated by miRNA-494 in gastrointestinal stromal tumors. Int. J. Cancer 2018, 142, 2080–2093. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yamamoto, H.; Kohashi, K.; Fujita, A.; Oda, Y. Fascin-1 overexpression and miR-133b downregulation in the progression of gastrointestinal stromal tumor. Mod. Pathol. 2013, 26, 563–571. [Google Scholar] [CrossRef]
- Tong, H.X.; Zhou, Y.H.; Hou, Y.Y.; Zhang, Y.; Huang, Y.; Xie, B.; Wang, J.Y.; Jiang, Q.; He, J.Y.; Shao, Y.B.; et al. Expression profile of microRNAs in gastrointestinal stromal tumors revealed by high throughput quantitative RT-PCR microarray. World J. Gastroenterol. 2015, 21, 5843–5855. [Google Scholar] [CrossRef]
- Fujita, K.; Kobara, H.; Mori, H.; Fujihara, S.; Chiyo, T.; Matsunaga, T.; Nishiyama, N.; Ayaki, M.; Yachida, T.; Morishita, A.; et al. Differences in miRNA expression profiles between GIST and leiomyoma in human samples acquired by submucosal tunneling biopsy. Endosc. Int. Open 2015, 3, E665–E671. [Google Scholar] [CrossRef] [Green Version]
- Isosaka, M.; Niinuma, T.; Nojima, M.; Kai, M.; Yamamoto, E.; Maruyama, R.; Nobuoka, T.; Nishida, T.; Kanda, T.; Taguchi, T.; et al. A Screen for Epigenetically Silenced microRNA Genes in Gastrointestinal Stromal Tumors. PLoS ONE 2015, 10, e0133754. [Google Scholar] [CrossRef]
- Gyvyte, U.; Juzenas, S.; Salteniene, V.; Kupcinskas, J.; Poskiene, L.; Kucinskas, L.; Jarmalaite, S.; Stuopelyte, K.; Steponaitiene, R.; Hemmrich-Stanisak, G.; et al. MiRNA profiling of gastrointestinal stromal tumors by next-generation sequencing. Oncotarget 2017, 8, 37225–37238. [Google Scholar] [CrossRef]
- Lu, H.J.; Yan, J.; Jin, P.Y.; Zheng, G.H.; Qin, S.M.; Wu, D.M.; Lu, J.; Zheng, Y.L. MicroRNA-152 inhibits tumor cell growth while inducing apoptosis via the transcriptional repression of cathepsin L in gastrointestinal stromal tumor. Cancer Biomark. 2018, 21, 711–722. [Google Scholar] [CrossRef]
- Long, Z.W.; Wu, J.H.; Cai, H.; Wang, Y.N.; Zhou, Y. MiR-374b Promotes Proliferation and Inhibits Apoptosis of Human GIST Cells by Inhibiting PTEN through Activation of the PI3K/Akt Pathway. Mol. Cells 2018, 41, 532–544. [Google Scholar]
- Chen, Y.; Qin, C.; Cui, X.; Geng, W.; Xian, G.; Wang, Z. miR-4510 acts as a tumor suppressor in gastrointestinal stromal tumor by targeting APOC2. J. Cell Physiol. 2020, 235, 5711–5721. [Google Scholar] [CrossRef] [PubMed]
- Kelly, L.; Bryan, K.; Kim, S.Y.; Janeway, K.A.; Killian, J.K.; Schildhaus, H.U.; Miettinen, M.; Helman, L.; Meltzer, P.S.; van de Rijn, M.; et al. Post-transcriptional dysregulation by miRNAs is implicated in the pathogenesis of gastrointestinal stromal tumor [GIST]. PLoS ONE 2013, 8, e64102. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pantaleo, M.A.; Ravegnini, G.; Astolfi, A.; Simeon, V.; Nannini, M.; Saponara, M.; Urbini, M.; Gatto, L.; Indio, V.; Sammarini, G.; et al. Integrating miRNA and gene expression profiling analysis revealed regulatory networks in gastrointestinal stromal tumors. Epigenomics 2016, 8, 1347–1366. [Google Scholar] [CrossRef] [PubMed]
- Bachet, J.B.; Tabone-Eglinger, S.; Dessaux, S.; Besse, A.; Brahimi-Adouane, S.; Emile, J.F.; Blay, J.Y.; Alberti, L. Gene expression patterns of hemizygous and heterozygous KIT mutations suggest distinct oncogenic pathways: A study in NIH3T3 cell lines and GIST samples. PLoS ONE 2013, 8, e61103. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hafner, M.; Landthaler, M.; Burger, L.; Khorshid, M.; Hausser, J.; Berninger, P.; Rothballer, A.; Ascano, M., Jr.; Jungkamp, A.C.; Munschauer, M.; et al. Transcriptome-wide identification of RNA-binding protein and microRNA target sites by PAR-CLIP. Cell 2010, 141, 129–141. [Google Scholar] [CrossRef] [Green Version]
- Hou, Y.Y.; Lu, S.H.; Zhou, Y.; Qi, W.D.; Shi, Y.; Tan, Y.S.; Zhu, X.Z. Stage and histological grade of gastrointestinal stromal tumors based on a new approach are strongly associated with clinical behaviors. Mod. Pathol. 2009, 22, 556–569. [Google Scholar] [CrossRef] [Green Version]
- Shi, Y.; Hou, Y.Y.; Lu, S.H.; Zhou, Y.; Xu, J.F.; Ji, Y.; Hou, J.; Xu, C.; Liu, Y.L.; Tan, Y.S.; et al. Clinical and pathological studies of borderline gastrointestinal stromal tumors. Chin. Med. J. (England) 2010, 123, 2514–2520. [Google Scholar]
- Niinuma, T.; Suzuki, H.; Nojima, M.; Nosho, K.; Yamamoto, H.; Takamaru, H.; Yamamoto, E.; Maruyama, R.; Nobuoka, T.; Miyazaki, Y.; et al. Upregulation of miR-196a and HOTAIR drive malignant character in gastrointestinal stromal tumors. Cancer Res. 2012, 72, 1126–1136. [Google Scholar] [CrossRef] [Green Version]
- Niinuma, T.; Kai, M.; Kitajima, H.; Yamamoto, E.; Harada, T.; Maruyama, R.; Nobuoka, T.; Nishida, T.; Kanda, T.; Hasegawa, T.; et al. Downregulation of miR-186 is associated with metastatic recurrence of gastrointestinal stromal tumors. Oncol. Lett. 2017, 14, 5703–5710. [Google Scholar] [CrossRef]
- Akcakaya, P.; Caramuta, S.; Ahlen, J.; Ghaderi, M.; Berglund, E.; Ostman, A.; Branstrom, R.; Larsson, C.; Lui, W.O. microRNA expression signatures of gastrointestinal stromal tumours: Associations with imatinib resistance and patient outcome. Br. J. Cancer 2014, 111, 2091–2102. [Google Scholar] [CrossRef] [Green Version]
- Liu, S.; Cui, J.; Liao, G.; Zhang, Y.; Ye, K.; Lu, T.; Qi, J.; Wan, G. MiR-137 regulates epithelial-mesenchymal transition in gastrointestinal stromal tumor. Tumour Biol. 2014, 35, 9131–9138. [Google Scholar] [CrossRef] [PubMed]
- Ding, J.; Xia, Y.; Yu, Z.; Wen, J.; Zhang, Z.; Zhang, Z.; Liu, Z.; Jiang, Z.; Liu, H.; Liao, G. Identification of upstream miRNAs of SNAI2 and their influence on the metastasis of gastrointestinal stromal tumors. Cancer Cell Int. 2019, 19, 289. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Joensuu, H.; DeMatteo, R.P. The management of gastrointestinal stromal tumors: A model for targeted and multidisciplinary therapy of malignancy. Annu. Rev. Med. 2012, 63, 247–258. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gao, X.; Shen, K.; Wang, C.; Ling, J.; Wang, H.; Fang, Y.; Shi, Y.; Hou, Y.; Qin, J.; Sun, Y.; et al. MiR-320a downregulation is associated with imatinib resistance in gastrointestinal stromal tumors. Acta. Biochim. Biophys. Sin. (Shanghai) 2014, 46, 72–75. [Google Scholar] [CrossRef] [Green Version]
- Fan, R.; Zhong, J.; Zheng, S.; Wang, Z.; Xu, Y.; Li, S.; Zhou, J.; Yuan, F. microRNA-218 increase the sensitivity of gastrointestinal stromal tumor to imatinib through PI3K/AKT pathway. Clin. Exp. Med. 2015, 15, 137–144. [Google Scholar] [CrossRef]
- Huang, W.K.; Akcakaya, P.; Gangaev, A.; Lee, L.; Zeljic, K.; Hajeri, P.; Berglund, E.; Ghaderi, M.; Ahlen, J.; Branstrom, R.; et al. miR-125a-5p regulation increases phosphorylation of FAK that contributes to imatinib resistance in gastrointestinal stromal tumors. Exp. Cell Res. 2018, 371, 287–296. [Google Scholar] [CrossRef]
- Zhang, Z.; Jiang, N.Y.; Guan, R.Y.; Zhu, Y.K.; Jiang, F.Q.; Piao, D. Identification of critical microRNAs in gastrointestinal stromal tumor patients treated with Imatinib. Neoplasma 2018, 65, 683–692. [Google Scholar] [CrossRef] [Green Version]
- Shi, Y.; Gao, X.; Hu, Q.; Li, X.; Xu, J.; Lu, S.; Liu, Y.; Xu, C.; Jiang, D.; Lin, J.; et al. PIK3C2A is a gene-specific target of microRNA-518a-5p in imatinib mesylate-resistant gastrointestinal stromal tumor. Lab. Investig. 2016, 96, 652–660. [Google Scholar] [CrossRef] [Green Version]
- Kou, Y.; Yang, R.; Wang, Q. Serum miR-518e-5p is a potential biomarker for secondary imatinib-resistant gastrointestinal stromal tumor. J. Biosci. 2018, 43, 1015–1023. [Google Scholar] [CrossRef] [PubMed]
- Calin, G.A.; Croce, C.M. MicroRNA-cancer connection: The beginning of a new tale. Cancer Res. 2006, 66, 7390–7394. [Google Scholar] [CrossRef] [Green Version]
- Xu, J.; Li, C.X.; Li, Y.S.; Lv, J.Y.; Ma, Y.; Shao, T.T.; Xu, L.D.; Wang, Y.Y.; Du, L.; Zhang, Y.P.; et al. MiRNA-miRNA synergistic network: Construction via co-regulating functional modules and disease miRNA topological features. Nucleic Acids Res. 2011, 39, 825–836. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bure, I.; Geer, S.; Knopf, J.; Roas, M.; Henze, S.; Strobel, P.; Agaimy, A.; Wiemann, S.; Hoheisel, J.D.; Hartmann, A.; et al. Long noncoding RNA HOTAIR is upregulated in an aggressive subgroup of gastrointestinal stromal tumors (GIST) and mediates the establishment of gene-specific DNA methylation patterns. Genes Chromosomes Cancer 2018, 57, 584–597. [Google Scholar] [CrossRef]
- Lee, N.K.; Lee, J.H.; Kim, W.K.; Yun, S.; Youn, Y.H.; Park, C.H.; Choi, Y.Y.; Kim, H.; Lee, S.K. Promoter methylation of PCDH10 by HOTAIR regulates the progression of gastrointestinal stromal tumors. Oncotarget 2016, 7, 75307–75318. [Google Scholar] [CrossRef] [PubMed]
- van Mierlo, G.; Veenstra, G.J.C.; Vermeulen, M.; Marks, H. The Complexity of PRC2 Subcomplexes. Trends Cell Biol. 2019, 29, 660–671. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hu, J.C.; Wang, Q.; Jiang, L.X.; Cai, L.; Zhai, H.Y.; Yao, Z.W.; Zhang, M.L.; Feng, Y. Effect of long non-coding RNA AOC4P on gastrointestinal stromal tumor cells. Onco Targets Ther. 2018, 11, 6259–6269. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cao, K.; Li, M.; Miao, J.; Lu, X.; Kang, X.; Zhu, H.; Du, S.; Li, X.; Zhang, Q.; Guan, W.; et al. CCDC26 knockdown enhances resistance of gastrointestinal stromal tumor cells to imatinib by interacting with c-KIT. Am. J. Transl. Res. 2018, 10, 274–282. [Google Scholar]
- Yan, J.; Chen, D.; Chen, X.; Sun, X.; Dong, Q.; Hu, C.; Zhou, F.; Chen, W. Downregulation of lncRNA CCDC26 contributes to imatinib resistance in human gastrointestinal stromal tumors through IGF-1R upregulation. Braz. J. Med. Biol. Res. 2019, 52, e8399. [Google Scholar] [CrossRef]
- Badalamenti, G.; Barraco, N.; Incorvaia, L.; Galvano, A.; Fanale, D.; Cabibi, D.; Calo, V.; Curro, G.; Bazan, V.; Russo, A. Are Long Noncoding RNAs New Potential Biomarkers in Gastrointestinal Stromal Tumors (GISTs)? The Role of H19 and MALAT1. J. Oncol. 2019, 2019, 5458717. [Google Scholar] [CrossRef] [Green Version]
- Gyvyte, U.; Kupcinskas, J.; Juzenas, S.; Inciuraite, R.; Poskiene, L.; Salteniene, V.; Link, A.; Fassan, M.; Franke, A.; Kupcinskas, L.; et al. Identification of long intergenic non-coding RNAs (lincRNAs) deregulated in gastrointestinal stromal tumors (GISTs). PLoS ONE 2018, 13, e0209342. [Google Scholar] [CrossRef]
- Yan, J.; Chen, D.; Chen, X.; Sun, X.; Dong, Q.; Du, Z.; Wang, T. Identification of imatinib-resistant long non-coding RNAs in gastrointestinal stromal tumors. Oncol. Lett. 2019, 17, 2283–2295. [Google Scholar] [CrossRef]
- Bai, F.; Zhang, N.; Fang, W.; He, X.; Zheng, Y.; Gu, D. PCAT6 mediates cellular biological functions in gastrointestinal stromal tumor via upregulation of PRDX5 and activation of Wnt pathway. Mol. Carcinog. 2020, 59, 661–669. [Google Scholar] [CrossRef]
- Patop, I.L.; Kadener, S. circRNAs in Cancer. Curr. Opin. Genet. Dev. 2018, 48, 121–127. [Google Scholar] [CrossRef] [PubMed]
- Jia, N.; Tong, H.; Zhang, Y.; Katayama, H.; Wang, Y.; Lu, W.; Zhang, S.; Wang, J. CeRNA Expression Profiling Identifies KIT-Related circRNA-miRNA-mRNA Networks in Gastrointestinal Stromal Tumour. Front. Genet. 2019, 10, 825. [Google Scholar] [CrossRef] [PubMed]
- Kosela-Paterczyk, H.; Paziewska, A.; Kulecka, M.; Balabas, A.; Kluska, A.; Dabrowska, M.; Piatkowska, M.; Zeber-Lubecka, N.; Ambrozkiewicz, F.; Karczmarski, J.; et al. Signatures of circulating microRNA in four sarcoma subtypes. J. Cancer 2020, 11, 874–882. [Google Scholar] [CrossRef] [Green Version]
- Lui, P.Y.; Jin, D.Y.; Stevenson, N.J. MicroRNA: Master controllers of intracellular signaling pathways. Cell. Mol. Life Sci. 2015, 72, 3531–3542. [Google Scholar] [CrossRef] [PubMed]
- Mayeux, R. Biomarkers: Potential uses and limitations. NeuroRx 2004, 1, 182–188. [Google Scholar] [CrossRef]
- Nair, V.S.; Maeda, L.S.; Ioannidis, J.P. Clinical outcome prediction by microRNAs in human cancer: A systematic review. J. Natl. Cancer Inst. 2012, 104, 528–540. [Google Scholar] [CrossRef] [Green Version]
- Simon, R.M.; Paik, S.; Hayes, D.F. Use of archived specimens in evaluation of prognostic and predictive biomarkers. J. Natl. Cancer Inst. 2009, 101, 1446–1452. [Google Scholar] [CrossRef] [Green Version]
- Hanash, S.M.; Baik, C.S.; Kallioniemi, O. Emerging molecular biomarkers--blood-based strategies to detect and monitor cancer. Nat. Rev. Clin. Oncol. 2011, 8, 142–150. [Google Scholar] [CrossRef]
- Pecot, C.V.; Calin, G.A.; Coleman, R.L.; Lopez-Berestein, G.; Sood, A.K. RNA interference in the clinic: Challenges and future directions. Nat. Rev. Cancer 2011, 11, 59–67. [Google Scholar] [CrossRef] [Green Version]
- Wu, S.Y.; Lopez-Berestein, G.; Calin, G.A.; Sood, A.K. RNAi therapies: Drugging the undruggable. Sci. Transl. Med. 2014, 6, 240. [Google Scholar] [CrossRef] [Green Version]
- Levin, A.A. Treating Disease at the RNA Level with Oligonucleotides. N. Engl. J. Med. 2019, 380, 57–70. [Google Scholar] [CrossRef] [PubMed]
- Stenvang, J.; Petri, A.; Lindow, M.; Obad, S.; Kauppinen, S. Inhibition of microRNA function by antimiR oligonucleotides. Silence 2012, 3, 1. [Google Scholar] [CrossRef] [Green Version]
- van Rooij, E.; Purcell, A.L.; Levin, A.A. Developing microRNA therapeutics. Circ. Res. 2012, 110, 496–507. [Google Scholar] [CrossRef] [PubMed]
- Adams, B.D.; Parsons, C.; Walker, L.; Zhang, W.C.; Slack, F.J. Targeting noncoding RNAs in disease. J. Clin. Investig. 2017, 127, 761–771. [Google Scholar] [CrossRef]
- Esposito, R.; Bosch, N.; Lanzos, A.; Polidori, T.; Pulido-Quetglas, C.; Johnson, R. Hacking the Cancer Genome: Profiling Therapeutically Actionable Long Non-coding RNAs Using CRISPR-Cas9 Screening. Cancer Cell 2019, 35, 545–557. [Google Scholar] [CrossRef] [Green Version]

| miRNAs a,b | Up/Downregulation | Functional Role | Ref. |
|---|---|---|---|
| miR-146b; miR-150; miR-132; miR-342; miR-16; miR-500; miR-212; miR-335; miR-21; miR-199a | Downregulation in high-risk GIST | [84] | |
| miR-196a | Upregulation in high-risk GIST | [108] | |
| miR-137 | Downregulation in GIST vs. normal adjacent tissue | Regulation of EMT through targeting TWIST1 | [111] |
| miR-30c-1-3p; miR-200b-3p; miR-363-3p | Downregulation in SNAI2 high GISTs | Regulation of invasion and migration through targeting SNAI2 | [112] |
| miR-186 | Downregulation in primary GISTs that exhibit metastatic recurrence | miR-186 is linked to migration and genes implicated in metastasis | [109] |
| miR-301a-3p miR-150-3p; miR-1207-5p; miR-1915 | Upregulation in metastatic GIST Downregulation in metastatic GIST | [110] |
| miRNAs Up/Downregulated in Imatinib-Resistant GIST a | Comparison/Number of Samples | Platform | Validated miRNAs; Targets and/or Pathways | Ref. |
|---|---|---|---|---|
| Upregulated: miR-15a; miR-16; miR-151-5p; miR-195 Downregulated: miR-140-5p; miR-140-3p; miR-320a; miR-483-5p; miR-574-3p; miR-1280 | Tumor samples Discovery: primary GIST (imatinib-naïve) (n = 3) vs. Imatinib-resistant GIST(n = 4) Validation: primary GIST (imatinib-naïve) (n = 16) vs. Imatinib-resistant GIST(n = 12) | Microarray RT-PCR | miR-320a | [114] |
| Downregulated: miR-218 | Cell lines GIST882 vs. GIST430 | RT-PCR | miR-218; PI3K/AKT signaling | [115] |
| Upregulated: miR-107; miR-125a-5p; miR-134; miR-301a-3p; miR-365 | Tumor samples GIST responsive on imatinib (n = 9–16) vs. GIST progressive on imatinib (n = 4–14) | Microarray RT-PCR | miR-125a-5p; PTPN18 (modulation pFAK levels) | [110,116] |
| Upregulated: miR-491-3p; miR-1260b; miR-2964a-5p; miR-3907 Downregulated: miR-221-3p; miR-518a-5p; miR-595; miR-3145-3p; miR-3655; miR-4466 | Tumor samples Paired (n = 20) primary GIST (imatinib-naïve) vs. Imatinib-resistant GIST | Microarray RT-PCR | miR-518a-5p; PIK3C2A | [118] |
| Upregulated: miR-518e-5p; miR-548e | Serum samples Imatinib-sensitive GIST(n = 37) vs. Imatinib-resistant GIST(n = 39) | Microarray RT-PCR | miR-518e-5p | [119] |
| Upregulated: miR-28-5p; miR-125a-5p | Tumor samples GIST responsive on imatinib vs. GIST progressive on imatinib | In silico analyses of microarray data b | miR-28-5p; miR-125a-5p | [117] |
| Upregulated: miR-92a; miR-118-5p; miR-335; miR-526a/miR-520c-5p/miR-518d-5p; miR-708* Downregulated: miR-24; miR-186; miR-455-3p; miR-675; miR-1296 | Tumor samples GIST imatinib-naïve (n = 33) vs. GIST imatinib-resistant (n = 20) | Microarray RT-PCR | [30] |
| Lnc RNA | Up/Down Regulation | Functional Role | Ref. |
|---|---|---|---|
| HOTAIR | Upregulation in high-risk GIST cf. low and intermediate GIST |
| [108,122,123] |
| AOC4P | Upregulation in high-risk GIST cf. low and intermediate GIST |
| [125] |
| CCDC26 | Low expression linked to imatinib resistance |
| [126,127] |
| FENDRR, H19 | Upregulation in GIST cf. adjacent normal tissue |
| [129] |
| H19 | High expression in advanced GIST with TTP < 6 months | [128] | |
| MALAT1 | High expression in advanced GIST with TTP < 6 months | • Correlation with c-KIT mutational status | [128] |
| TERT-2, OMD-1, ATP7A-2, RERE-4, TCP1-5, FAM108B1-3, C15orf54-4, ATP7A-1 TCF4-6, SNRPN-2 | Upregulation in imatinib-resistant GIST Downregulation in imatinib-resistant GIST | • HIF1 pathway regulation | [130] |
| PCAT6 | Upregulation in GIST cf. adjacent normal tissue |
| [131] |
| circ_0069765, circ_0084097, circ_0079471 | Upregulation in GIST cf. adjacent normal tissue | • Role in predicted network of circRNAs, host genes (KIT, PLAT, ETV1, resp.) and miR-142-5p, miR-144-3p and 485-3p. | [133] |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Amirnasr, A.; Sleijfer, S.; Wiemer, E.A.C. Non-Coding RNAs, a Novel Paradigm for the Management of Gastrointestinal Stromal Tumors. Int. J. Mol. Sci. 2020, 21, 6975. https://doi.org/10.3390/ijms21186975
Amirnasr A, Sleijfer S, Wiemer EAC. Non-Coding RNAs, a Novel Paradigm for the Management of Gastrointestinal Stromal Tumors. International Journal of Molecular Sciences. 2020; 21(18):6975. https://doi.org/10.3390/ijms21186975
Chicago/Turabian StyleAmirnasr, Azadeh, Stefan Sleijfer, and Erik A. C. Wiemer. 2020. "Non-Coding RNAs, a Novel Paradigm for the Management of Gastrointestinal Stromal Tumors" International Journal of Molecular Sciences 21, no. 18: 6975. https://doi.org/10.3390/ijms21186975
APA StyleAmirnasr, A., Sleijfer, S., & Wiemer, E. A. C. (2020). Non-Coding RNAs, a Novel Paradigm for the Management of Gastrointestinal Stromal Tumors. International Journal of Molecular Sciences, 21(18), 6975. https://doi.org/10.3390/ijms21186975