Treg Enhancing Therapies to Treat Autoimmune Diseases


Abstract
:1. The Regulatory T Cell
1.1. Treg Phenotype
1.2. Treg Subsets
1.3. Treg Function
1.4. T Cell Receptor
2. Autoimmune Disease
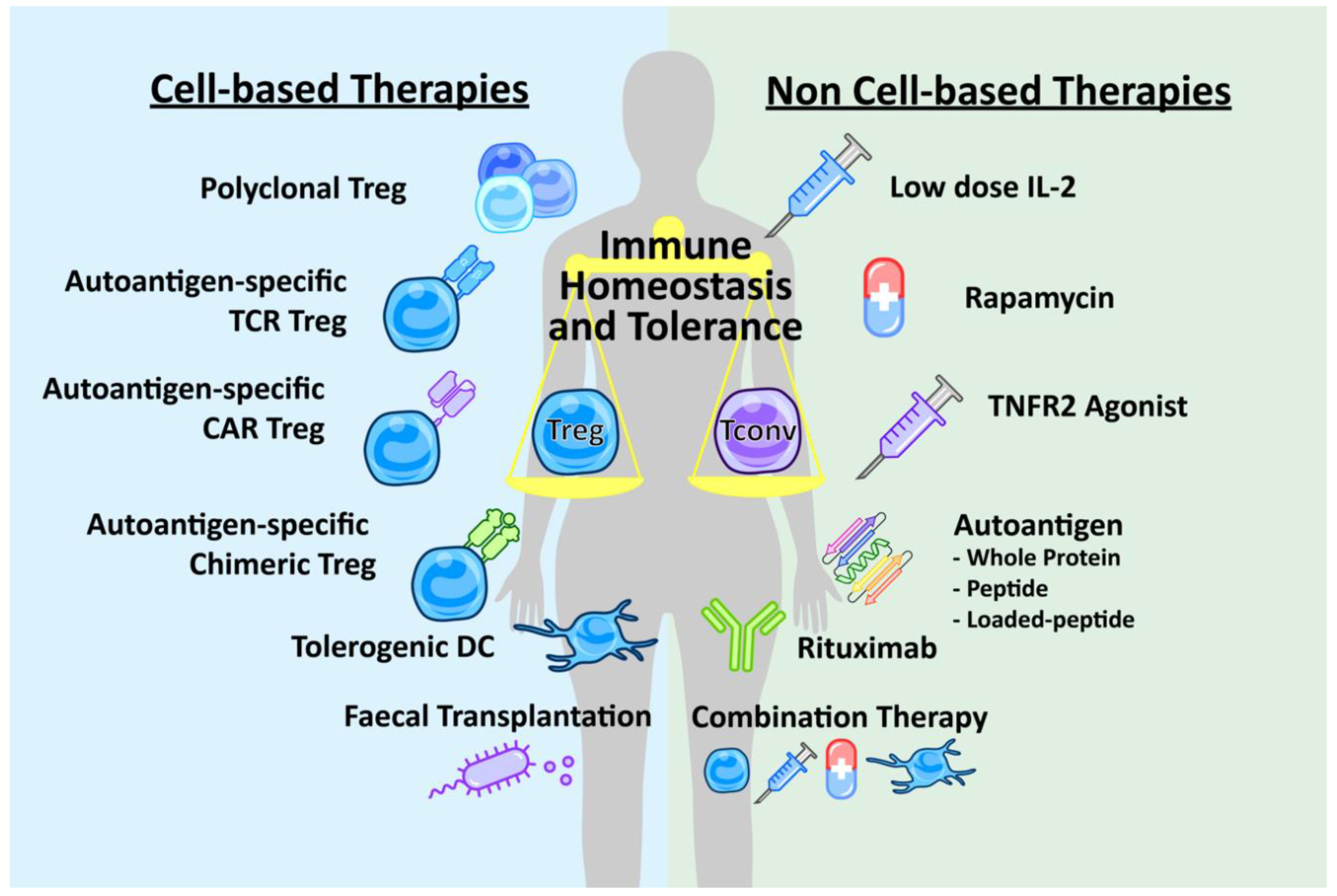
Enhancing Treg Immunosuppression to Treat Autoimmune Disease
3. Non-Cell-Based Therapies
3.1. Low-Dose IL-2
3.2. Rapamycin
3.3. TNF Receptor 2
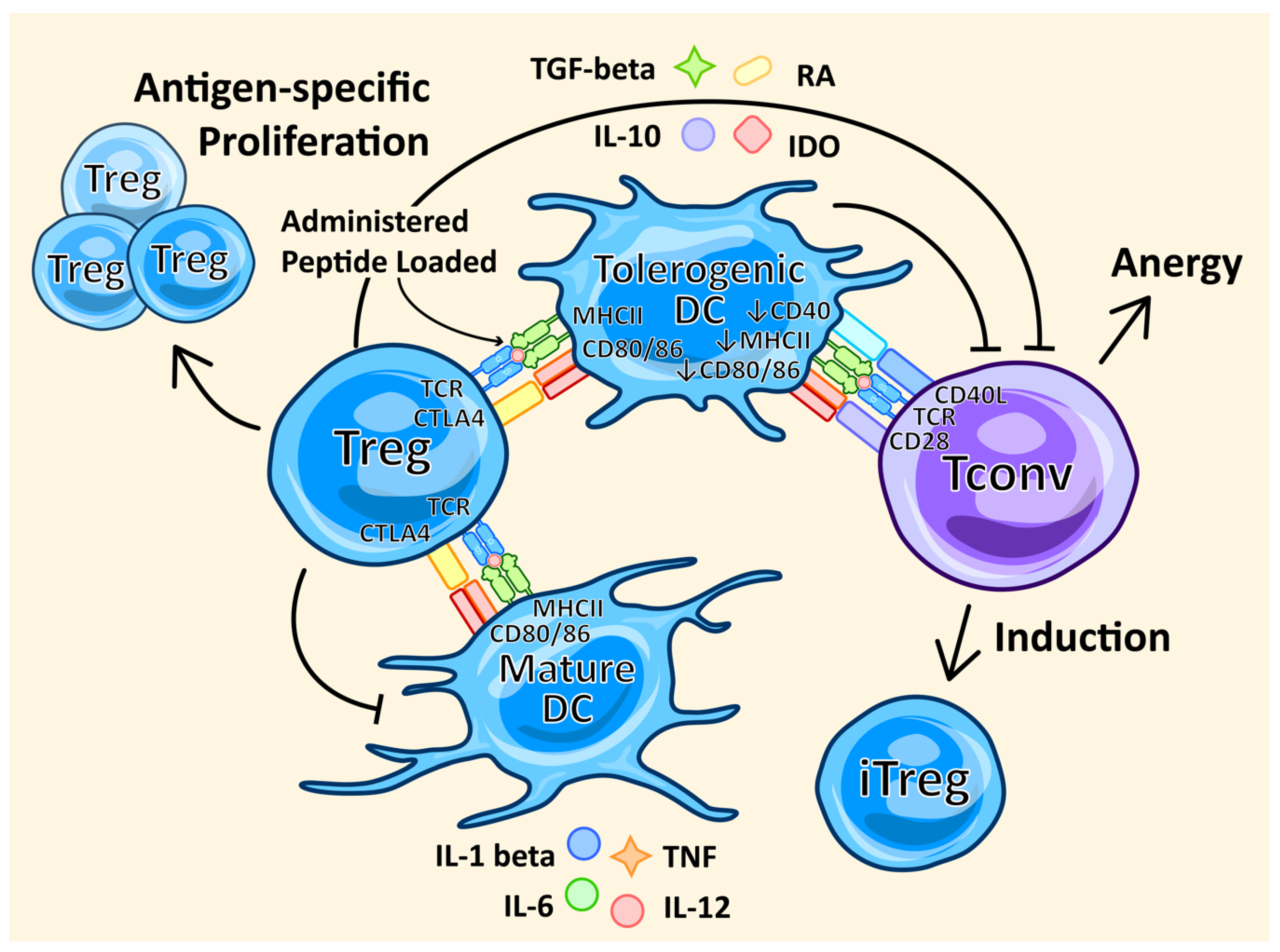
3.4. Peptide Administration
4. Cell-Based Therapies
4.1. Microbiome Therapy
4.2. Treg Therapy
4.3. Polyclonal Treg Therapy
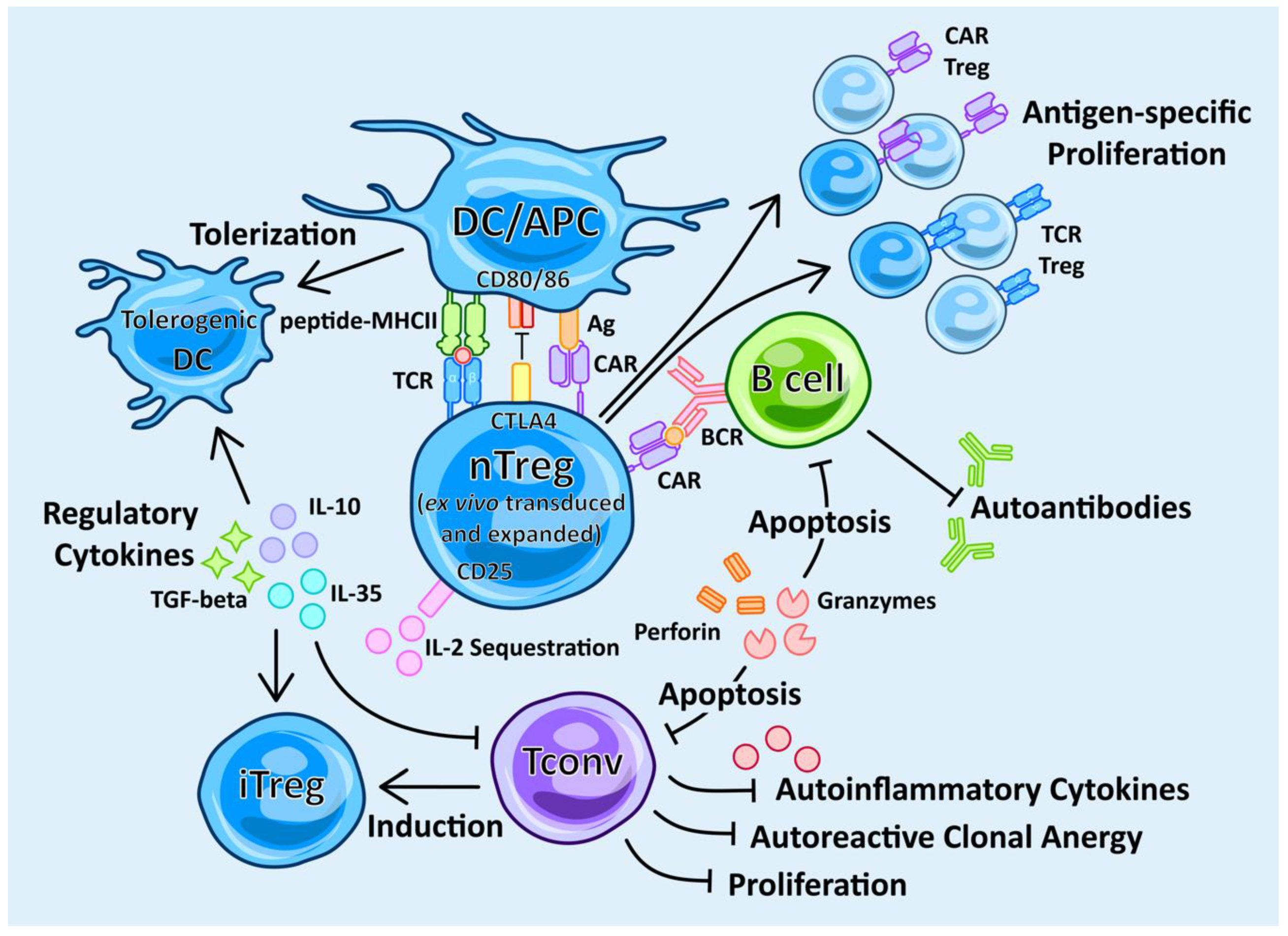
4.4. Engineered Antigen-Specific Treg (TCR-Treg) Therapy
4.5. Chimeric Treg Therapy
4.6. CAR vs. TCR Treg Therapy
4.7. DC Therapy
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Shimizu, J.; Yamazaki, S.; Sakaguchi, S. Induction of tumor immunity by removing CD25+CD4+ T cells: A common basis between tumor immunity and autoimmunity. J. Immunol. 1999, 163, 5211–5218. [Google Scholar] [PubMed]
- Sutmuller, R.P.; van Duivenvoorde, L.M.; van Elsas, A.; Schumacher, T.N.; Wildenberg, M.E.; Allison, J.P.; Toes, R.E.; Offringa, R.; Melief, C.J. Synergism of cytotoxic T lymphocyte-associated antigen 4 blockade and depletion of CD25(+) regulatory T cells in antitumor therapy reveals alternative pathways for suppression of autoreactive cytotoxic T lymphocyte responses. J. Exp. Med. 2001, 194, 823–832. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Taylor, P.A.; Noelle, R.J.; Blazar, B.R. CD4(+)CD25(+) immune regulatory cells are required for induction of tolerance to alloantigen via costimulatory blockade. J. Exp. Med. 2001, 193, 1311–1318. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Belkaid, Y.; Rouse, B.T. Natural regulatory T cells in infectious disease. Nat. Immunol. 2005, 6, 353–360. [Google Scholar] [CrossRef]
- Sakaguchi, S.; Sakaguchi, N.; Asano, M.; Itoh, M.; Toda, M. Immunologic self-tolerance maintained by activated T cells expressing IL-2 receptor alpha-chains (CD25). Breakdown of a single mechanism of self-tolerance causes various autoimmune diseases. J. Immunol. 1995, 155, 1151–1164. [Google Scholar]
- Asano, M.; Toda, M.; Sakaguchi, N.; Sakaguchi, S. Autoimmune disease as a consequence of developmental abnormality of a T cell subpopulation. J. Exp. Med. 1996, 184, 387–396. [Google Scholar] [CrossRef]
- Hori, S.; Nomura, T.; Sakaguchi, S. Control of regulatory T cell development by the transcription factor Foxp3. Science 2003, 299, 1057–1061. [Google Scholar] [CrossRef] [Green Version]
- Fontenot, J.D.; Gavin, M.A.; Rudensky, A.Y. Foxp3 programs the development and function of CD4(+)CD25(+) regulatory T cells. Nat. Immunol. 2003, 4, 330–336. [Google Scholar] [CrossRef]
- Husebye, E.S.; Anderson, M.S.; Kampe, O. Autoimmune Polyendocrine Syndromes. N. Engl. J. Med. 2018, 378, 2543–2544. [Google Scholar] [CrossRef]
- Ohkura, N.; Kitagawa, Y.; Sakaguchi, S. Development and Maintenance of Regulatory T cells. Immunity 2013, 38, 414–423. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, W.; Putnam, A.L.; Xu-Yu, Z.; Szot, G.L.; Lee, M.R.; Zhu, S.; Gottlieb, P.A.; Kapranov, P.; Gingeras, T.R.; Fazekas de St Groth, B.; et al. CD127 expression inversely correlates with FoxP3 and suppressive function of human CD4+ T reg cells. J. Exp. Med. 2006, 203, 1701–1711. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sakaguchi, S.; Miyara, M.; Costantino, C.M.; Hafler, D.A. FOXP3(+) regulatory T cells in the human immune system. Nat. Rev. Immunol. 2010, 10, 490–500. [Google Scholar] [CrossRef] [PubMed]
- Mills, K.H. Regulatory T cells: Friend or foe in immunity to infection? Nat. Rev. Immunol. 2004, 4, 841–855. [Google Scholar] [CrossRef]
- Josefowicz, S.Z.; Lu, L.F.; Rudensky, A.Y. Regulatory T Cells: Mechanisms of Differentiation and Function. Annu. Rev. Immunol. 2012, 30, 531–564. [Google Scholar] [CrossRef]
- Kanamori, M.; Nakatsukasa, H.; Okada, M.; Lu, Q.J.; Yoshimura, A. Induced Regulatory T Cells: Their Development, Stability, and Applications. Trends Immunol. 2016, 37, 803–811. [Google Scholar] [CrossRef] [PubMed]
- Komatsu, N.; Okamoto, K.; Sawa, S.; Nakashima, T.; Oh-hora, M.; Kodama, T.; Tanaka, S.; Bluestone, J.A.; Takayanagi, H. Pathogenic conversion of Foxp3+ T cells into TH17 cells in autoimmune arthritis. Nat. Med. 2014, 20, 62–68. [Google Scholar] [CrossRef] [PubMed]
- Zhou, X.; Bailey-Bucktrout, S.L.; Jeker, L.T.; Penaranda, C.; Martinez-Llordella, M.; Ashby, M.; Nakayama, M.; Rosenthal, W.; Bluestone, J.A. Instability of the transcription factor Foxp3 leads to the generation of pathogenic memory T cells in vivo. Nat. Immunol. 2009, 10, 1000–1007. [Google Scholar] [CrossRef] [PubMed]
- Thornton, A.M.; Shevach, E.M. CD4+CD25+ immunoregulatory T cells suppress polyclonal T cell activation in vitro by inhibiting interleukin 2 production. J. Exp. Med. 1998, 188, 287–296. [Google Scholar] [CrossRef] [Green Version]
- Takahashi, T.; Kuniyasu, Y.; Toda, M.; Sakaguchi, N.; Itoh, M.; Iwata, M.; Shimizu, J.; Sakaguchi, S. Immunologic self-tolerance maintained by CD25+CD4+ naturally anergic and suppressive T cells: Induction of autoimmune disease by breaking their anergic/suppressive state. Int. Immunol. 1998, 10, 1969–1980. [Google Scholar] [CrossRef] [Green Version]
- Miller, A.; Lider, O.; Weiner, H.L. Antigen-driven bystander suppression after oral administration of antigens. J. Exp. Med. 1991, 174, 791–798. [Google Scholar] [CrossRef]
- Abbas, A.K.; Trotta, E.; Simeonov, D.R.; Marson, A.; Bluestone, J.A. Revisiting IL-2: Biology and therapeutic prospects. Sci. Immunol. 2018, 3, eaat1482. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fan, M.Y.; Low, J.S.; Tanimine, N.; Finn, K.K.; Priyadharshini, B.; Germana, S.K.; Kaech, S.M.; Turka, L.A. Differential Roles of IL-2 Signaling in Developing versus Mature Tregs. Cell. Rep. 2018, 25, 1204–1213.e4. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rudolph, M.G.; Stanfield, R.L.; Wilson, I.A. How TCRs bind MHCs, peptides, and coreceptors. Annu. Rev. Immunol. 2006, 24, 419–466. [Google Scholar] [CrossRef] [PubMed]
- Elhanati, Y.; Sethna, Z.; Callan, C.G.; Mora, T.; Walczak, A.M. Predicting the spectrum of TCR repertoire sharing with a data-driven model of recombination. Immunol. Rev. 2018, 284, 167–179. [Google Scholar] [CrossRef] [Green Version]
- Ooi, J.D.; Petersen, J.; Tan, Y.H.; Huynh, M.; Willett, Z.J.; Ramarathinam, S.H.; Eggenhuizen, P.J.; Loh, K.L.; Watson, K.A.; Gan, P.Y.; et al. Dominant protection from HLA-linked autoimmunity by antigen-specific regulatory T cells. Nature 2017, 545, 243–247. [Google Scholar] [CrossRef] [Green Version]
- Holler, P.D.; Chlewicki, L.K.; Kranz, D.M. TCRs with high affinity for foreign pMHC show self-reactivity. Nat. Immunol. 2003, 4, 55–62. [Google Scholar] [CrossRef]
- Ooi, J.D.; Jiang, J.H.; Eggenhuizen, P.J.; Chua, L.L.; van Timmeren, M.; Loh, K.L.; O’Sullivan, K.M.; Gan, P.Y.; Zhong, Y.; Tsyganov, K.; et al. A plasmid-encoded peptide from Staphylococcus aureus induces anti-myeloperoxidase nephritogenic autoimmunity. Nat. Commun. 2019, 10, 1–14. [Google Scholar] [CrossRef] [Green Version]
- Klein, L.; Kyewski, B.; Allen, P.M.; Hogquist, K.A. Positive and negative selection of the T cell repertoire: What thymocytes see (and don’t see). Nat. Rev. Immunol. 2014, 14, 377–391. [Google Scholar] [CrossRef] [Green Version]
- Jordan, M.S.; Boesteanu, A.; Reed, A.J.; Petrone, A.L.; Holenbeck, A.E.; Lerman, M.A.; Naji, A.; Caton, A.J. Thymic selection of CD4(+)CD25(+) regulatory T cells induced by an agonist self-peptide. Nat. Immunol. 2001, 2, 301–306. [Google Scholar] [CrossRef]
- Malchow, S.; Leventhal, D.S.; Lee, V.; Nishi, S.; Socci, N.D.; Savage, P.A. Aire Enforces Immune Tolerance by Directing Autoreactive T Cells into the Regulatory T Cell Lineage. Immunity 2016, 44, 1102–1113. [Google Scholar] [CrossRef] [Green Version]
- Anderson, M.S.; Venanzi, E.S.; Klein, L.; Chen, Z.B.; Berzins, S.P.; Turley, S.J.; von Boehmer, H.; Bronson, R.; Dierich, A.; Benoist, C.; et al. Projection of an immunological self shadow within the thymus by the aire protein. Science 2002, 298, 1395–1401. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thornton, A.M.; Shevach, E.M. Suppressor effector function of CD4+CD25+ immunoregulatory T cells is antigen nonspecific. J. Immunol. 2000, 164, 183–190. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tai, X.G.; Van Laethem, F.; Pobezinsky, L.; Guinter, T.; Sharrow, S.O.; Adams, A.; Granger, L.; Kruhlak, M.; Lindsten, T.; Thompson, C.B.; et al. Basis of CTLA-4 function in regulatory and conventional CD4(+) T cells. Blood 2012, 119, 5155–5163. [Google Scholar] [CrossRef]
- Van Ham, M.; Teich, R.; Philipsen, L.; Niemz, J.; Amsberg, N.; Wissing, J.; Nimtz, M.; Grobe, L.; Kliche, S.; Thiel, N.; et al. TCR signalling network organization at the immunological synapses of murine regulatory T cells. Eur. J. Immunol. 2017, 47, 2043–2058. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, C.T.; Workman, C.J.; Flies, D.; Pan, X.Y.; Marson, A.L.; Zhou, G.; Hipkiss, E.L.; Ravi, S.; Kowalski, J.; Levitsky, H.I.; et al. Role of LAG-3 in regulatory T cells. Immunity 2004, 21, 503–513. [Google Scholar] [CrossRef] [Green Version]
- Yokosuka, T.; Kobayashi, W.; Takamatsu, M.; Sakata-Sogawa, K.; Zeng, H.; Hashimoto-Tane, A.; Yagita, H.; Tokunaga, M.; Saito, T. Spatiotemporal Basis of CTLA-4 Costimulatory Molecule-Mediated Negative Regulation of T Cell Activation. Immunity 2010, 33, 326–339. [Google Scholar] [CrossRef] [Green Version]
- Marengere, L.E.M.; Waterhouse, P.; Duncan, G.S.; Mittrucker, H.W.; Feng, G.S.; Mak, T.W. Regulation of T cell receptor signaling by tyrosine phosphatase SYP association with CTLA-4. Science 1996, 272, 1170–1173. [Google Scholar] [CrossRef]
- Chuang, E.; Fisher, T.S.; Morgan, R.W.; Robbins, M.D.; Duerr, J.M.; Vander Heiden, M.G.; Gardner, J.P.; Hambor, J.E.; Neveu, M.J.; Thompson, C.B. The CD28 and CTLA-4 receptors associate with the serine/threonine phosphatase PP2A. Immunity 2000, 13, 313–322. [Google Scholar] [CrossRef] [Green Version]
- Davis, M.M.; Bjorkman, P.J. T-cell antigen receptor genes and T-cell recognition. Nature 1988, 334, 395–402. [Google Scholar] [CrossRef]
- Serra, P.; Santamaria, P. Antigen-specific therapeutic approaches for autoimmunity. Nat. Biotechnol. 2019, 37, 238–251. [Google Scholar] [CrossRef]
- Navarra, S.V.; Guzman, R.M.; Gallacher, A.E.; Hall, S.; Levy, R.A.; Jimenez, R.E.; Li, E.K.M.; Thomas, M.; Kim, H.Y.; Leon, M.G.; et al. Efficacy and safety of belimumab in patients with active systemic lupus erythematosus: A randomised, placebo-controlled, phase 3 trial. Lancet 2011, 377, 721–731. [Google Scholar] [CrossRef]
- Gottenberg, J.E.; Guillevin, L.; Lambotte, O.; Combe, B.; Allanore, Y.; Cantagrel, A.; Larroche, C.; Soubrier, M.; Bouillet, L.; Dougados, M.; et al. Tolerance and short term efficacy of rituximab in 43 patients with systemic autoimmune diseases. Ann. Rheum. Dis. 2005, 64, 913–920. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Battaglia, M.; Stabilini, A.; Roncarolo, M.G. Rapamycin selectively expands CD4+CD25+FoxP3+ regulatory T cells. Blood 2005, 105, 4743–4748. [Google Scholar] [CrossRef] [PubMed]
- Zeiser, R.; Leveson-Gower, D.B.; Zambricki, E.A.; Kambham, N.; Beilhack, A.; Loh, J.; Hou, J.Z.; Negrin, R.S. Differential impact of mammalian target of rapamycin inhibition on CD4+CD25+Foxp3+ regulatory T cells compared with conventional CD4+ T cells. Blood 2008, 111, 453–462. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Battaglia, M.; Stabilini, A.; Draghici, E.; Gregori, S.; Mocchetti, C.; Bonifacio, E.; Roncarolo, M.G. Rapamycin and interleukin-10 treatment induces T regulatory type 1 cells that mediate antigen-specific transplantation tolerance. Diabetes 2006, 55, 40–49. [Google Scholar] [CrossRef] [PubMed]
- Biswas, M.; Sarkar, D.; Kumar, S.R.; Nayak, S.; Rogers, G.L.; Markusic, D.M.; Liao, G.; Terhorst, C.; Herzog, R.W. Synergy between rapamycin and FLT3 ligand enhances plasmacytoid dendritic cell-dependent induction of CD4+CD25+FoxP3+ Treg. Blood 2015, 125, 2937–2947. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, X.; Baumel, M.; Mannel, D.N.; Howard, O.M.Z.; Oppenheim, J.J. Interaction of TNF with TNF receptor type 2 promotes expansion and function of mouse CD4(+)CD25(+) T regulatory cells. J. Immunol. 2007, 179, 154–161. [Google Scholar] [CrossRef] [Green Version]
- Pozsgay, J.; Szekanecz, Z.; Sarmay, G. Antigen-specific immunotherapies in rheumatic diseases. Nat. Rev. Rheumatol. 2017, 13, 525–537. [Google Scholar] [CrossRef]
- Zhang, N.; Nandakumar, K.S. Recent advances in the development of vaccines for chronic inflammatory autoimmune diseases. Vaccine 2018, 36, 3208–3220. [Google Scholar] [CrossRef]
- Clemente-Casares, X.; Blanco, J.; Ambalavanan, P.; Yamanouchi, J.; Singha, S.; Fandos, C.; Tsai, S.; Wang, J.; Garabatos, N.; Izquierdo, C.; et al. Expanding antigen-specific regulatory networks to treat autoimmunity. Nature 2016, 530, 434–440. [Google Scholar] [CrossRef]
- Serra, P.; Santamaria, P. Nanoparticle-based approaches to immune tolerance for the treatment of autoimmune diseases. Eur. J. Immunol. 2018, 48, 751–756. [Google Scholar] [CrossRef] [PubMed]
- Desreumaux, P.; Foussat, A.; Allez, M.; Beaugerie, L.; Hebuterne, X.; Bouhnik, Y.; Nachury, M.; Brun, V.; Bastian, H.; Belmonte, N.; et al. Safety and Efficacy of Antigen-Specific Regulatory T-Cell Therapy for Patients With Refractory Crohn’s Disease. Gastroenterology 2012, 143, 1207–1217.e2. [Google Scholar] [CrossRef] [PubMed]
- Bluestone, J.A.; Buckner, J.H.; Fitch, M.; Gitelman, S.E.; Gupta, S.; Hellerstein, M.K.; Herold, K.C.; Lares, A.; Lee, M.R.; Li, K.V.; et al. Type 1 diabetes immunotherapy using polyclonal regulatory T cells. Sci. Transl. Med. 2015, 7, 315ra189. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Harris, D.T.; Kranz, D.M. Adoptive T Cell Therapies: A Comparison of T Cell Receptors and Chimeric Antigen Receptors. Trends Pharmacol. Sci. 2016, 37, 220–230. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dawson, N.A.J.; Levings, M.K. Antigen-specific regulatory T cells: Are police CARs the answer? Transl. Res. 2017, 187, 53–58. [Google Scholar] [CrossRef]
- Giannoukakis, N.; Phillips, B.; Finegold, D.; Harnaha, J.; Trucco, M. Phase I (safety) study of autologous tolerogenic dendritic cells in type 1 diabetic patients. Diabetes Care 2011, 34, 2026–2032. [Google Scholar] [CrossRef] [Green Version]
- Yamazaki, S.; Inaba, K.; Tarbell, K.V.; Steinman, R.M. Dendritic cells expand antigen-specific Foxp3(+)CD25(+)CD4(+) regulatory T cells including suppressors of alloreactivity. Immunol. Rev. 2006, 212, 314–329. [Google Scholar] [CrossRef]
- Sadlack, B.; Lohler, J.; Schorle, H.; Klebb, G.; Haber, H.; Sickel, E.; Noelle, R.J.; Horak, I. Generalized Autoimmune-Disease in Interleukin-2-Deficient Mice Is Triggered by an Uncontrolled Activation and Proliferation of Cd4(+) T-Cells. Eur. J. Immunol. 1995, 25, 3053–3059. [Google Scholar] [CrossRef]
- Furtado, G.C.; de Lafaille, M.A.C.; Kutchukhidze, N.; Lafaille, J.J. Interleukin 2 signaling is required for CD4(+) regulatory T cell function. J. Exp. Med. 2002, 196, 851–857. [Google Scholar] [CrossRef] [Green Version]
- Tang, Q.Z. Therapeutic Window of Interleukin-2 for Autoimmune Diseases. Diabetes 2015, 64, 1912–1913. [Google Scholar] [CrossRef] [Green Version]
- Saadoun, D.; Rosenzwajg, M.; Joly, F.; Six, A.; Carrat, F.; Thibault, V.; Sene, D.; Cacoub, P.; Klatzmann, D. Regulatory T-Cell Responses to Low-Dose Interleukin-2 in HCV-Induced Vasculitis. N. Engl. J. Med. 2011, 365, 2067–2077. [Google Scholar] [CrossRef] [PubMed]
- Koreth, J.; Matsuoka, K.; Kim, H.T.; McDonough, S.M.; Bindra, B.; Alyea, E.P.; Armand, P.; Cutler, C.; Ho, V.T.; Treister, N.S.; et al. Interleukin-2 and Regulatory T Cells in Graft-versus-Host Disease. N. Engl. J. Med. 2011, 365, 2055–2066. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- He, J.; Zhang, X.; Wei, Y.B.; Sun, X.L.; Chen, Y.P.; Deng, J.; Jin, Y.B.; Gan, Y.Z.; Hu, X.; Jia, R.L.; et al. Low-dose interleukin-2 treatment selectively modulates CD4(+) T cell subsets in patients with systemic lupus erythematosus. Nat. Med. 2016, 22, 991–993. [Google Scholar] [CrossRef] [PubMed]
- Todd, J.A.; Evangelou, M.; Cutler, A.J.; Pekalski, M.L.; Walker, N.M.; Stevens, H.E.; Porter, L.; Smyth, D.J.; Rainbow, D.B.; Ferreira, R.C.; et al. Regulatory T Cell Responses in Participants with Type 1 Diabetes after a Single Dose of Interleukin-2: A Non-Randomised, Open Label, Adaptive Dose-Finding Trial. PLoS Med. 2016, 13, e1002139. [Google Scholar] [CrossRef]
- Rosenzwajg, M.; Churlaud, G.; Mallone, R.; Six, A.; Derian, N.; Chaara, W.; Lorenzon, R.; Long, S.A.; Buckner, J.H.; Afonso, G.; et al. Low-dose interleukin-2 fosters a dose-dependent regulatory T cell tuned milieu in T1D patients. J. Autoimmun. 2015, 58, 48–58. [Google Scholar] [CrossRef] [Green Version]
- Hartemann, A.; Bensimon, G.; Payan, C.A.; Jacqueminet, S.; Bourron, O.; Nicolas, N.; Fonfrede, M.; Rosenzwajg, M.; Bernard, C.; Klatzmann, D. Low-dose interleukin 2 in patients with type 1 diabetes: A phase 1/2 randomised, double-blind, placebo-controlled trial. Lancet Diabetes Endocrinol. 2013, 1, 295–305. [Google Scholar] [CrossRef]
- Trotta, E.; Bessette, P.H.; Silveria, S.L.; Ely, L.K.; Jude, K.M.; Le, D.T.; Holst, C.R.; Coyle, A.; Potempa, M.; Lanier, L.L.; et al. A human anti-IL-2 antibody that potentiates regulatory T cells by a structure-based mechanism. Nat. Med. 2018, 24, 1005–1014. [Google Scholar] [CrossRef] [Green Version]
- Sockolosky, J.T.; Trotta, E.; Parisi, G.; Picton, L.; Su, L.L.; Le, A.C.; Chhabra, A.; Silveria, S.L.; George, B.M.; King, I.C.; et al. Selective targeting of engineered T cells using orthogonal IL-2 cytokine-receptor complexes. Science 2018, 359, 1037–1042. [Google Scholar] [CrossRef] [Green Version]
- McDermott, D.F. The application of high-dose interleukin-2 for metastatic renal cell carcinoma. Med. Oncol. 2009, 26, 13–17. [Google Scholar] [CrossRef]
- Thomson, A.W.; Turnquist, H.R.; Raimondi, G. Immunoregulatory functions of mTOR inhibition. Nat. Rev. Immunol. 2009, 9, 324–337. [Google Scholar] [CrossRef] [Green Version]
- Hackstein, H.; Taner, T.; Zahorchak, A.F.; Morelli, A.E.; Logar, A.J.; Gessner, A.; Thomson, A.W. Rapamycin inhibits IL-4--induced dendritic cell maturation in vitro and dendritic cell mobilization and function in vivo. Blood 2003, 101, 4457–4463. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Subleski, J.J.; Hamano, R.; Howard, O.M.Z.; Wiltrout, R.H.; Oppenheim, J.J. Co-expression of TNFR2 and CD25 identifies more of the functional CD4(+)FOXP3(+) regulatory T cells in human peripheral blood. Eur. J. Immunol. 2010, 40, 1099–1106. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, X.; Wu, X.Q.; Zhou, Q.; Howard, O.M.Z.; Netea, M.G.; Oppenheim, J.J. TNFR2 Is Critical for the Stabilization of the CD4(+)Foxp3(+) Regulatory T Cell Phenotype in the Inflammatory Environment. J. Immunol. 2013, 190, 1076–1084. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Christen, U.; Wolfe, T.; Mohrle, U.; Hughes, A.C.; Rodrigo, E.; Green, E.A.; Flavell, R.A.; von Herrath, M.G. A dual role for TNF-alpha in type 1 diabetes: Islet-specific expression abrogates the ongoing autoimmune process when induced late but not early during pathogenesis. J. Immunol. 2001, 166, 7023–7032. [Google Scholar] [CrossRef]
- Ban, L.; Zhang, J.; Wang, L.; Kuhtreiber, W.; Burger, D.; Faustman, D.L. Selective death of autoreactive T cells in human diabetes by TNF or TNF receptor 2 agonism. Proc. Natl. Acad. Sci. USA 2008, 105, 13644–13649. [Google Scholar] [CrossRef] [Green Version]
- Gan, P.Y.; Tan, D.; Ooi, J.D.; Kitching, A.R.; Holdsworth, S.R. Nasal Administration of the Immunodominant Mpo Epitope (Mpo409-428) Prevents the Induction of Anti-Mpo Autoimmunity and Attenuates Anca Associated Anti-Mpo Glomerulonephritis. Nephrology 2014, 19, 41. [Google Scholar]
- Thomson, A.W.; Knolle, P.A. Antigen-presenting cell function in the tolerogenic liver environment. Nat. Rev. Immunol. 2010, 10, 753–766. [Google Scholar] [CrossRef]
- Luth, S.; Huber, S.; Schramm, C.; Buch, T.; Zander, S.; Stadelmann, C.; Bruck, W.; Wraith, D.C.; Herkel, J.; Lohse, A.W. Ectopic expression of neural autoantigen in mouse liver suppresses experimental autoimmune neuroinflammation by inducing antigen-specific Tregs. J. Clin. Investig. 2008, 118, 3403–3410. [Google Scholar] [CrossRef] [Green Version]
- Akbarpour, M.; Goudy, K.S.; Cantore, A.; Russo, F.; Sanvito, F.; Naldini, L.; Annoni, A.; Roncarolo, M.G. Insulin B chain 9-23 gene transfer to hepatocytes protects from type 1 diabetes by inducing Ag-specific FoxP3+ Tregs. Sci. Transl. Med. 2015, 7, 289ra81. [Google Scholar] [CrossRef]
- Slingerland, A.E.; Schwabkey, Z.; Wiesnoski, D.H.; Jenq, R.R. Clinical Evidence for the Microbiome in Inflammatory Diseases. Front. Immunol. 2017, 8, 400. [Google Scholar] [CrossRef] [Green Version]
- Arpaia, N.; Campbell, C.; Fan, X.Y.; Dikiy, S.; van der Veeken, J.; deRoos, P.; Liu, H.; Cross, J.R.; Pfeffer, K.; Coffer, P.J.; et al. Metabolites produced by commensal bacteria promote peripheral regulatory T-cell generation. Nature 2013, 504, 451–455. [Google Scholar] [CrossRef] [PubMed]
- Verma, R.; Lee, C.; Jeun, E.J.; Yi, J.; Kim, K.S.; Ghosh, A.; Byun, S.; Lee, C.G.; Kang, H.J.; Kim, G.C.; et al. Cell surface polysaccharides of Bifidobacterium bifidum induce the generation of Foxp3(+) regulatory T cells. Sci. Immunol. 2018, 3, eaat6975. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dominguez-Villar, M.; Hafler, D.A. Regulatory T cells in autoimmune disease. Nat. Immunol. 2018, 19, 665–673. [Google Scholar] [CrossRef] [PubMed]
- Ferreira, L.M.R.; Muller, Y.D.; Bluestone, J.A.; Tang, Q.Z. Next-generation regulatory T cell therapy. Nat. Rev. Drug Discov. 2019, 18, 749–769. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hall, B.M.; Pearce, N.W.; Gurley, K.E.; Dorsch, S.E. Specific Unresponsiveness in Rats with Prolonged Cardiac Allograft Survival after Treatment with Cyclosporine. 3. Further Characterization of the Cd4+ Suppressor-Cell and Its Mechanisms of Action. J. Exp. Med. 1990, 171, 141–157. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Groux, H.; OGarra, A.; Bigler, M.; Rouleau, M.; Antonenko, S.; deVries, J.E.; Roncarolo, M.G. A CD4(+) T-cell subset inhibits antigen-specific T-cell responses and prevents colitis. Nature 1997, 389, 737–742. [Google Scholar] [CrossRef]
- Marek-Trzonkowska, N.; Mysliwiec, M.; Dobyszuk, A.; Grabowska, M.; Derkowska, I.; Juscinska, J.; Owczuk, R.; Szadkowska, A.; Witkowski, P.; Mlynarski, W.; et al. Therapy of type 1 diabetes with CD4(+)CD25(high) CD127-regulatory T cells prolongs survival of pancreatic islets - Results of one year follow-up. Clin. Immunol. 2014, 153, 23–30. [Google Scholar] [CrossRef]
- Dall’Era, M.; Pauli, M.L.; Remedios, K.; Taravati, K.; Sandova, P.M.; Putnam, A.L.; Lares, A.; Haemel, A.; Tang, Q.Z.; Hellerstein, M.; et al. Adoptive Treg Cell Therapy in a Patient With Systemic Lupus Erythematosus. Arthritis Rheumatol. 2019, 71, 431–440. [Google Scholar] [CrossRef]
- Scotta, C.; Fanelli, G.; Hoong, S.J.; Romano, M.; Lamperti, E.N.; Sukthankar, M.; Guggino, G.; Fazekasova, H.; Ratnasothy, K.; Becker, P.D.; et al. Impact of immunosuppressive drugs on the therapeutic efficacy of ex vivo expanded human regulatory T cells. Haematologica 2016, 101, 91–100. [Google Scholar] [CrossRef]
- Tang, Q.Z.; Henriksen, K.J.; Bi, M.Y.; Finger, E.B.; Szot, G.; Ye, J.Q.; Masteller, E.L.; McDevitt, H.; Bonyhadi, M.; Bluestone, J.A. In vitro-expanded antigen-specific regulatory T cells suppress autoimmune diabetes. J. Exp. Med. 2004, 199, 1455–1465. [Google Scholar] [CrossRef] [Green Version]
- Green, E.A.; Choi, Y.W.; Flavell, R.A. Pancreatic lymph node-derived CD4(+)CD25(+) Treg cells: Highly potent regulators of diabetes that require TRANCE-RANK signals. Immunity 2002, 16, 183–191. [Google Scholar] [CrossRef] [Green Version]
- Wang, G.C.; Dash, P.; McCullers, J.A.; Doherty, P.C.; Thomas, P.G. T cell receptor alphabeta diversity inversely correlates with pathogen-specific antibody levels in human cytomegalovirus infection. Sci. Transl. Med. 2012, 4, 128ra42. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Simone, M.; Rossetti, G.; Pagani, M. Single Cell T Cell Receptor Sequencing: Techniques and Future Challenges. Front. Immunol. 2018, 9, 1638. [Google Scholar] [CrossRef] [PubMed]
- Cebula, A.; Kuczma, M.; Szurek, E.; Pietrzak, M.; Savage, N.; Elhefnawy, W.R.; Rempala, G.; Kraj, P.; Ignatowicz, L. Dormant pathogenic CD4(+) T cells are prevalent in the peripheral repertoire of healthy mice. Nat. Commun. 2019, 10, 4882. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yeh, W.I.; Seay, H.R.; Newby, B.; Posgai, A.L.; Moniz, F.B.; Michels, A.; Mathews, C.E.; Bluestone, J.A.; Brusko, T.M. Avidity and Bystander Suppressive Capacity of Human Regulatory T Cell Expressing De Novo Autoreactive T-Cell Receptors in Type 1 Diabetes. Front. Immunol. 2017, 8, 1313. [Google Scholar] [CrossRef]
- Hull, C.M.; Nickolay, L.E.; Estorninho, M.; Richardson, M.W.; Riley, J.L.; Peakman, M.; Maher, J.; Tree, T.I.M. Generation of human islet-specific regulatory T cells by TCR gene transfer. J. Autoimmun. 2017, 79, 63–73. [Google Scholar] [CrossRef] [Green Version]
- Kim, Y.C.; Zhang, A.H.; Su, Y.; Rieder, S.A.; Rossi, R.J.; Ettinger, R.A.; Pratt, K.P.; Shevach, E.M.; Scott, D.W. Engineered antigen-specific human regulatory T cells: Immunosuppression of FVIII-specific T- and B-cell responses. Blood 2015, 125, 1107–1115. [Google Scholar] [CrossRef] [Green Version]
- Provasi, E.; Genovese, P.; Lombardo, A.; Magnani, Z.; Liu, P.Q.; Reik, A.; Chu, V.; Paschon, D.E.; Zhang, L.; Kuball, J.; et al. Editing T cell specificity towards leukemia by zinc finger nucleases and lentiviral gene transfer. Nat. Med. 2012, 18, 807–815. [Google Scholar] [CrossRef]
- Mastaglio, S.; Genovese, P.; Magnani, Z.; Ruggiero, E.; Landoni, E.; Camisa, B.; Schiroli, G.; Provasi, E.; Lombardo, A.; Reik, A.; et al. NY-ESO-1 TCR single edited stem and central memory T cells to treat multiple myeloma without graft-versus-host disease. Blood 2017, 130, 606–618. [Google Scholar] [CrossRef]
- Ochi, T.; Fujiwara, H.; Okamoto, S.; An, J.; Nagai, K.; Shirakata, T.; Mineno, J.; Kuzushima, K.; Shiku, H.; Yasukawa, M. Novel adoptive T-cell immunotherapy using a WT1-specific TCR vector encoding silencers for endogenous TCRs shows marked antileukemia reactivity and safety. Blood 2011, 118, 1495–1503. [Google Scholar] [CrossRef]
- Legut, M.; Dolton, G.; Mian, A.A.; Ottmann, O.G.; Sewell, A.K. CRISPR-mediated TCR replacement generates superior anticancer transgenic T cells. Blood 2018, 131, 311–322. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cohen, C.J.; Li, Y.F.; El-Gamil, M.; Robbins, P.F.; Rosenberg, S.A.; Morgan, R.A. Enhanced antitumor activity of T cells engineered to express T-cell receptors with a second disulfide bond. Cancer Res. 2007, 67, 3898–3903. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mekala, D.J.; Geiger, T.L. Immunotherapy of autoimmune encephalomyelitis with redirected CD4(+)CD25(+) T lymphocytes. Blood 2005, 105, 2090–2092. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moisini, L.; Nguyen, P.; Fugger, L.; Geiger, T.L. Redirecting therapeutic T cells against myelin-specific T lymphocytes using a humanized myelin basic protein-HLA-DR2-zeta chimeric receptor. J. Immunol. 2008, 180, 3601–3611. [Google Scholar] [CrossRef] [PubMed]
- Qian, Z.H.; Latham, K.A.; Whittington, K.B.; Miller, D.C.; Brand, D.D.; Rosloniec, E.F. Engineered Regulatory T Cells Coexpressing MHC Class II: Peptide Complexes Are Efficient Inhibitors of Autoimmune T Cell Function and Prevent the Development of Autoimmune Arthritis. J. Immunol. 2013, 190, 5382–5391. [Google Scholar] [CrossRef] [Green Version]
- Brusko, T.M.; Koya, R.C.; Zhu, S.; Lee, M.R.; Putnam, A.L.; McClymont, S.A.; Nishimura, M.I.; Han, S.H.; Chang, L.J.; Atkinson, M.A.; et al. Human Antigen-Specific Regulatory T Cells Generated by T Cell Receptor Gene Transfer. PLoS ONE 2010, 5, e11726. [Google Scholar] [CrossRef]
- Fransson, M.; Piras, E.; Burman, J.; Nilsson, B.; Essand, M.; Lu, B.F.; Harris, R.A.; Magnusson, P.U.; Brittebo, E.; Loskog, A.S.I. CAR/FoxP3-engineered T regulatory cells target the CNS and suppress EAE upon intranasal delivery. J Neuroinflamm 2012, 9, 112. [Google Scholar] [CrossRef] [Green Version]
- Maldini, C.R.; Ellis, G.I.; Riley, J.L. CAR T cells for infection, autoimmunity and allotransplantation. Nat. Rev. Immunol. 2018, 18, 605–616. [Google Scholar] [CrossRef]
- Ellebrecht, C.T.; Bhoj, V.G.; Nace, A.; Choi, E.J.; Mao, X.M.; Cho, M.J.; Di Zenzo, G.; Lanzavecchia, A.; Seykora, J.T.; Cotsarelis, G.; et al. Reengineering chimeric antigen receptor T cells for targeted therapy of autoimmune disease. Science 2016, 353, 179–184. [Google Scholar] [CrossRef] [Green Version]
- Bluhm, J.; Kieback, E.; Marino, S.F.; Oden, F.; Westermann, J.; Chmielewski, M.; Abken, H.; Uckert, W.; Hopken, U.E.; Rehm, A. CAR T Cells with Enhanced Sensitivity to B Cell Maturation Antigen for the Targeting of B Cell Non-Hodgkin’s Lymphoma and Multiple Myeloma. Mol. Ther. 2018, 26, 1906–1920. [Google Scholar] [CrossRef] [Green Version]
- Huang, J.; Brameshuber, M.; Zeng, X.; Xie, J.M.; Li, Q.J.; Chien, Y.H.; Valitutti, S.; Davis, M.M. A Single Peptide-Major Histocompatibility Complex Ligand Triggers Digital Cytokine Secretion in CD4(+) T Cells. Immunity 2013, 39, 846–857. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Domogalla, M.P.; Rostan, P.V.; Raker, V.K.; Steinbrink, K. Tolerance through Education: How Tolerogenic Dendritic Cells Shape Immunity. Front. Immunol. 2017, 8, 1764. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Banchereau, J.; Briere, F.; Caux, C.; Davoust, J.; Lebecque, S.; Liu, Y.J.; Pulendran, B.; Palucka, K. Immunobiology of dendritic cells. Annu. Rev. Immunol. 2000, 18, 767–811. [Google Scholar] [CrossRef] [PubMed]
- Hill, J.A.; Hall, J.A.; Sun, C.M.; Cai, Q.; Ghyselinck, N.; Chambon, P.; Belkaid, Y.; Mathis, D.; Benoist, C. Retinoic acid enhances Foxp3 induction indirectly by relieving inhibition from CD4+CD44hi Cells. Immunity 2008, 29, 758–770. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Munn, D.H.; Sharma, M.D.; Lee, J.R.; Jhaver, K.G.; Johnson, T.S.; Keskin, D.B.; Marshall, B.; Chandler, P.; Antonia, S.J.; Burgess, R.; et al. Potential regulatory function of human dendritic cells expressing indoleamine 2,3-dioxygenase. Science 2002, 297, 1867–1870. [Google Scholar] [CrossRef]
- Phillips, B.E.; Garciafigueroa, Y.; Trucco, M.; Giannoukakis, N. Clinical Tolerogenic Dendritic Cells: Exploring Therapeutic Impact on Human Autoimmune Disease. Front. Immunol. 2017, 8, 1279. [Google Scholar] [CrossRef] [Green Version]




{kind=link}
{kind=link}
{kind=link}
| Study ID | Phase | Disease | Therapy | Status/Outcome |
|---|---|---|---|---|
| ISRCTN06128462 | I | T1D | Autologous polyclonal expanded nTregs | Completed. Safe and well tolerated. Most patients responded to the therapy. Patients required lower amounts of exogenous insulin. Autoantibody status unchanged. |
| NCT02691247 | II | T1D | Autologous polyclonal expanded nTregs | Completed. Well tolerated. No improvement in C peptide levels after one year. |
| NCT02772679 | I | T1D | Autologous polyclonal expanded nTregs & IL-2 | Active, not recruiting |
| NCT03011021 | I/II | T1D | Umbilical cord blood polyclonal expanded nTregs | Recruiting |
| NCT01210664 | I | T1D | Autologous polyclonal expanded nTregs | Completed. Safe and well tolerated. C peptide levels remained after 2 years. No change in autoantibodies |
| NCT03444064 | I | T1D | Autologous polyclonal expanded nTregs | Recruiting |
| NCT02932826 | I/II | T1D | Umbilical cord blood polyclonal expanded nTregs | Recruiting |
| NCT02704338 | I/II | Autoimmune hepatitis | Autologous polyclonal expanded nTregs | Unknown |
| NCT02327221 | II | Crohn’s Disease | In vitro differentiated and expanded autologous TR1 cells specific for OVA | Adverse events in a few patients. Remission was associated with lower dose of TR1 cells |
| NCT03185000 | I/II | Crohn’s Disease | Autologous polyclonal expanded naive nTregs | Not yet recruiting |
| NCT02428309 | I | Cutaneous lupus | Autologous polyclonal expanded nTregs | Terminated due to participant recruitment feasibility |
| NCT03239470 | I | Pemphigus | Autologous polyclonal expanded nTregs | Active, not recruiting |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Eggenhuizen, P.J.; Ng, B.H.; Ooi, J.D. Treg Enhancing Therapies to Treat Autoimmune Diseases. Int. J. Mol. Sci. 2020, 21, 7015. https://doi.org/10.3390/ijms21197015
Eggenhuizen PJ, Ng BH, Ooi JD. Treg Enhancing Therapies to Treat Autoimmune Diseases. International Journal of Molecular Sciences. 2020; 21(19):7015. https://doi.org/10.3390/ijms21197015
Chicago/Turabian StyleEggenhuizen, Peter J., Boaz H. Ng, and Joshua D. Ooi. 2020. "Treg Enhancing Therapies to Treat Autoimmune Diseases" International Journal of Molecular Sciences 21, no. 19: 7015. https://doi.org/10.3390/ijms21197015
APA StyleEggenhuizen, P. J., Ng, B. H., & Ooi, J. D. (2020). Treg Enhancing Therapies to Treat Autoimmune Diseases. International Journal of Molecular Sciences, 21(19), 7015. https://doi.org/10.3390/ijms21197015