Amphiphilic Aminoglycosides as Medicinal Agents


Abstract
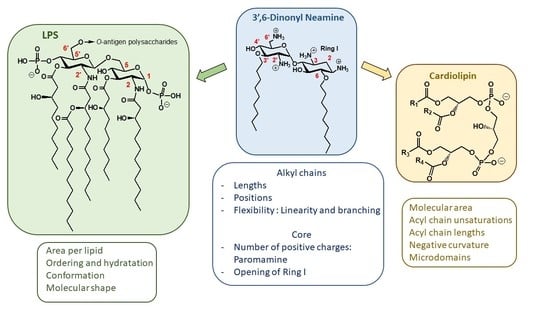
:
1. Introduction
2. Antibacterial Amphiphilic Aminoglycosides (Antibacterial AAGs)
2.1. Broad-Spectrum Antibacterial AAGs: Antibacterial NEA, PARA and 6-Amino-6-Deoxy-1-Methylglucosamine Derivatives, Structure–Activity and Structure–Cytotoxicity Relationships, Modes of Action
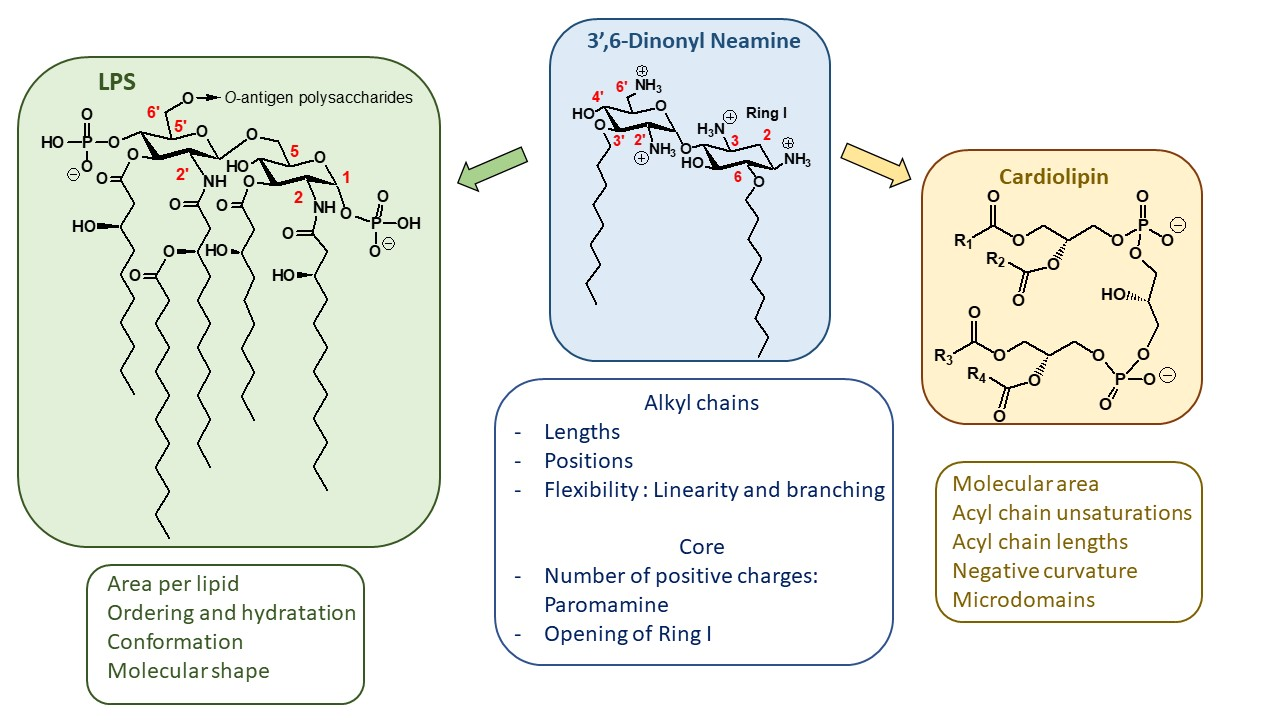
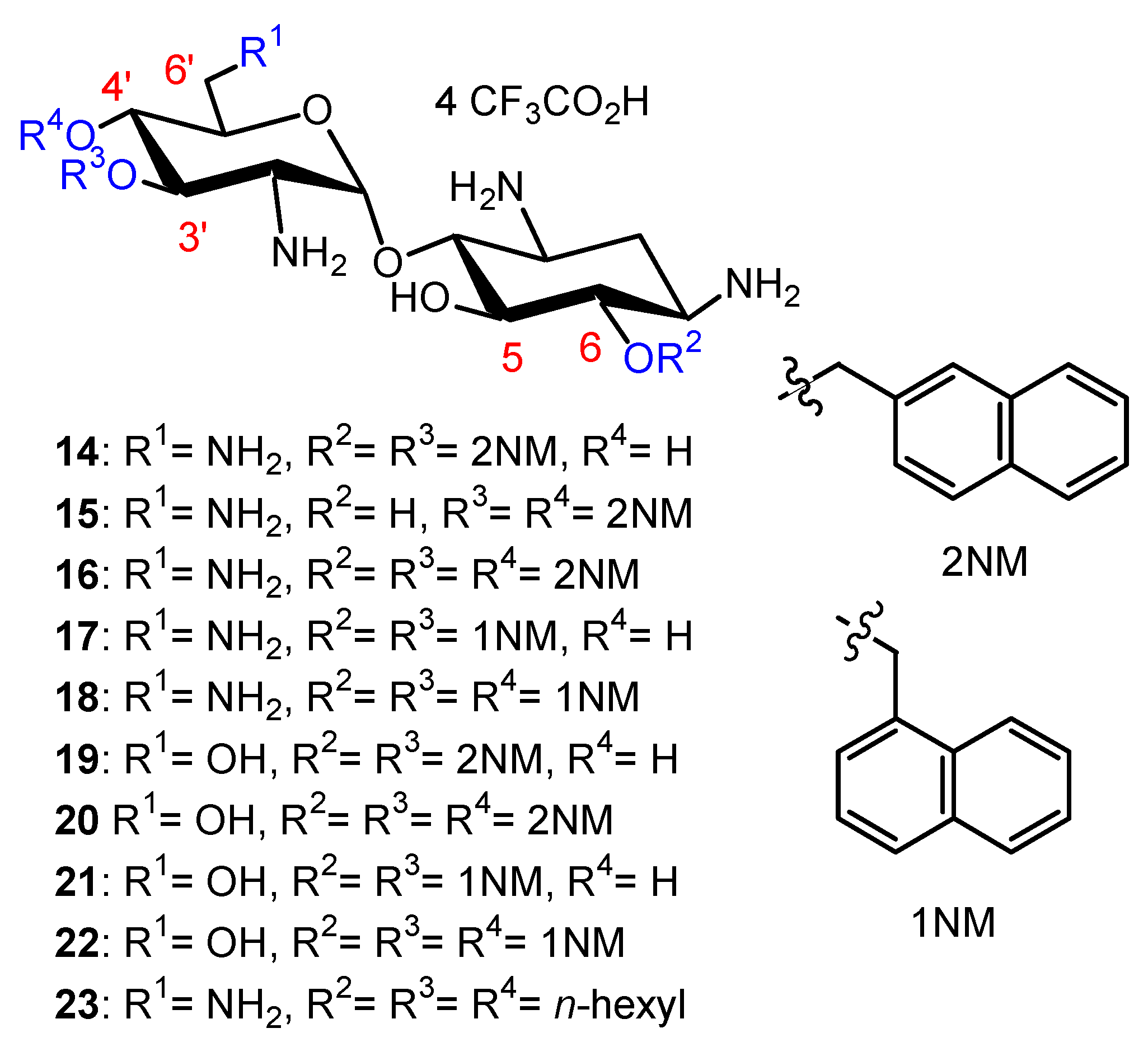
2.1.1. Synthesis of NEA and PARA Derivatives
2.1.2. Structure–Activity Relationships
First Identified Broad-Spectrum Antibacterial Amphiphilic NEA Derivatives
Comparison of the Antibacterial Activities of NEA and PARA Derivatives
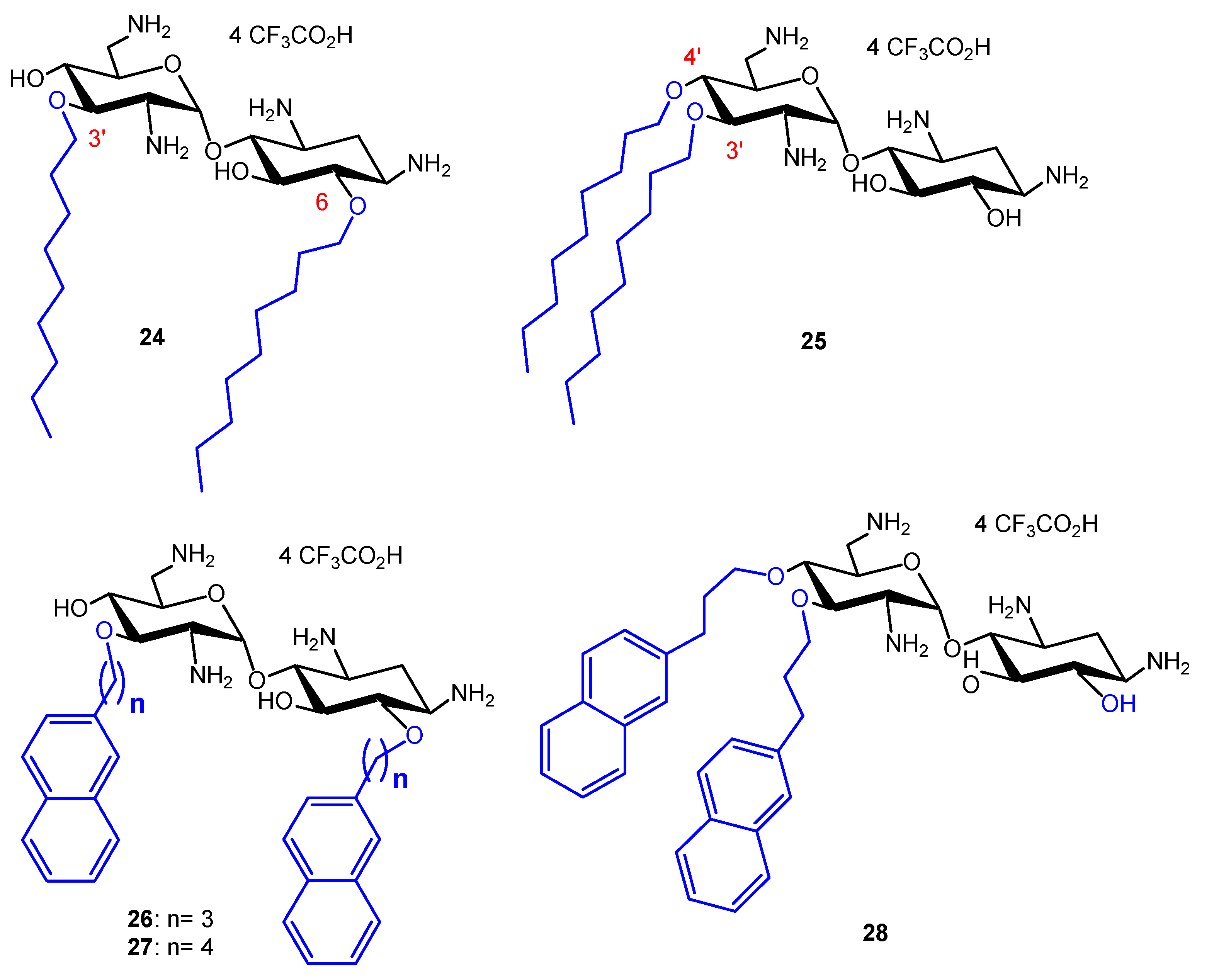
Comparison of the Antibacterial Activities of 3′,6- and 3′,4′-Dialkyl NEA Derivatives
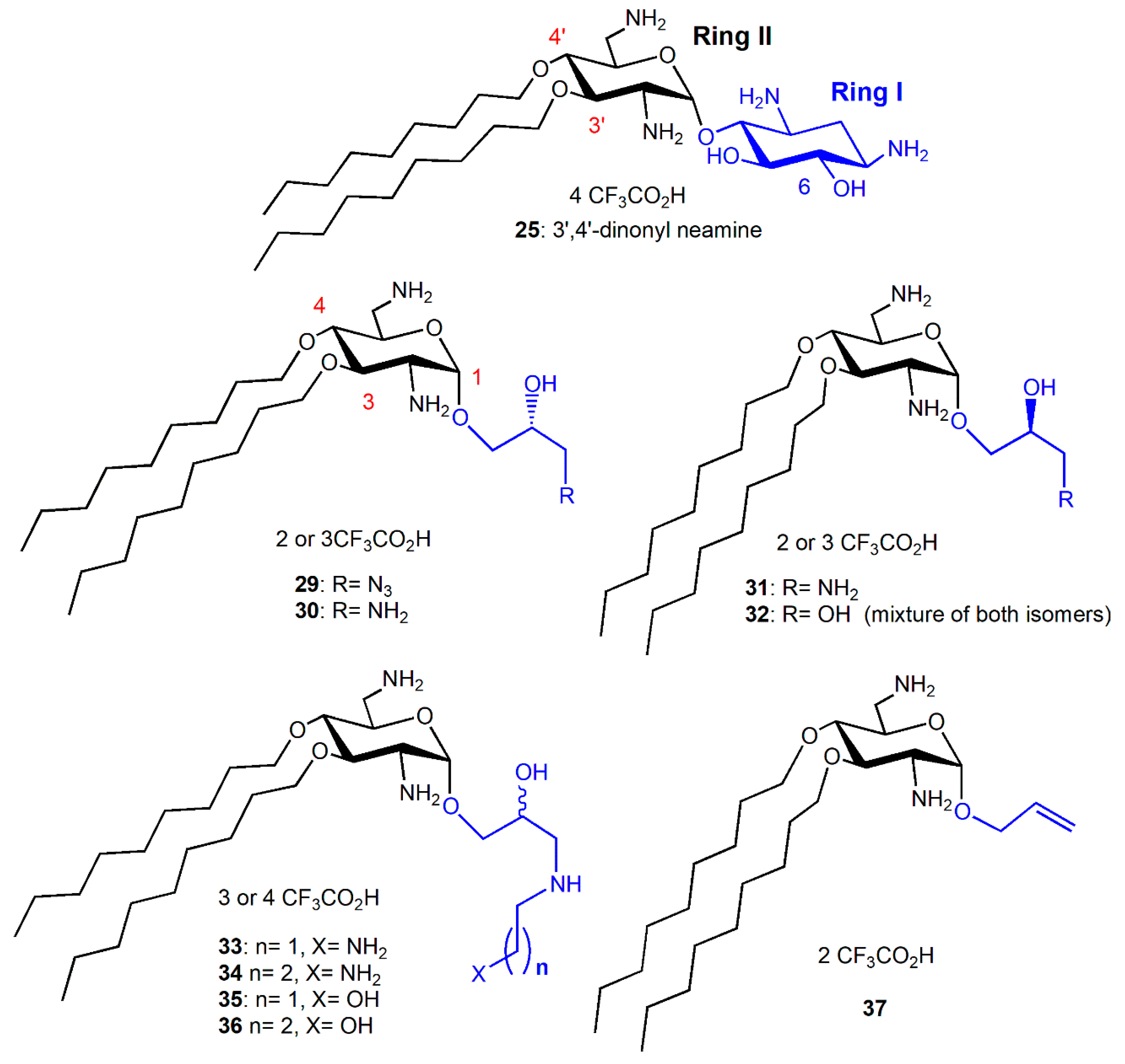
6-Amino-6-Deoxy-1-Methylglucosamine (1-Methyl Neosamine) Derivatives, Analogues of 3′,4′-Dialkyl NEA Derivatives
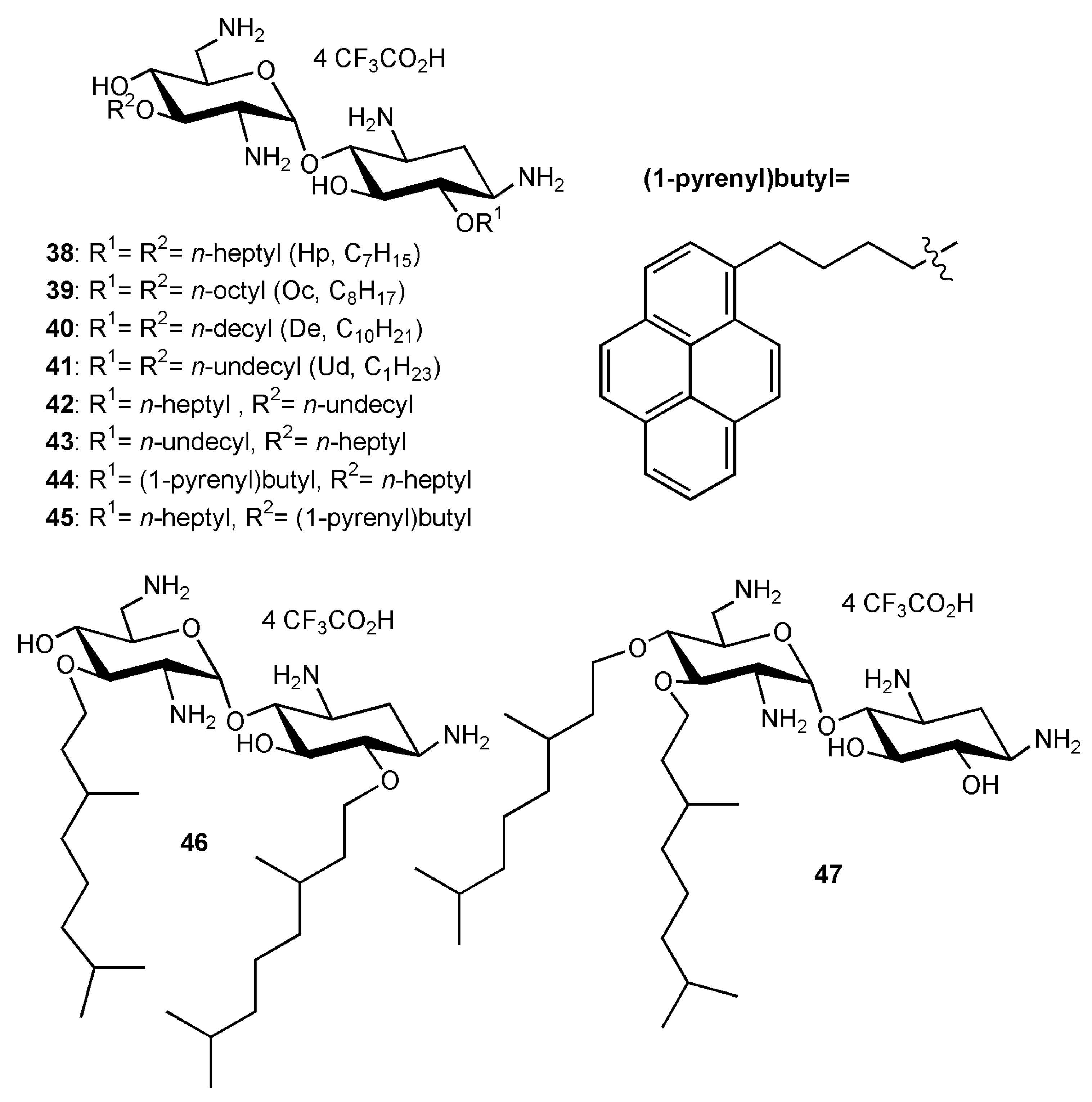
Fine-Tuning of the Structure–Activity Relationships (SAR)
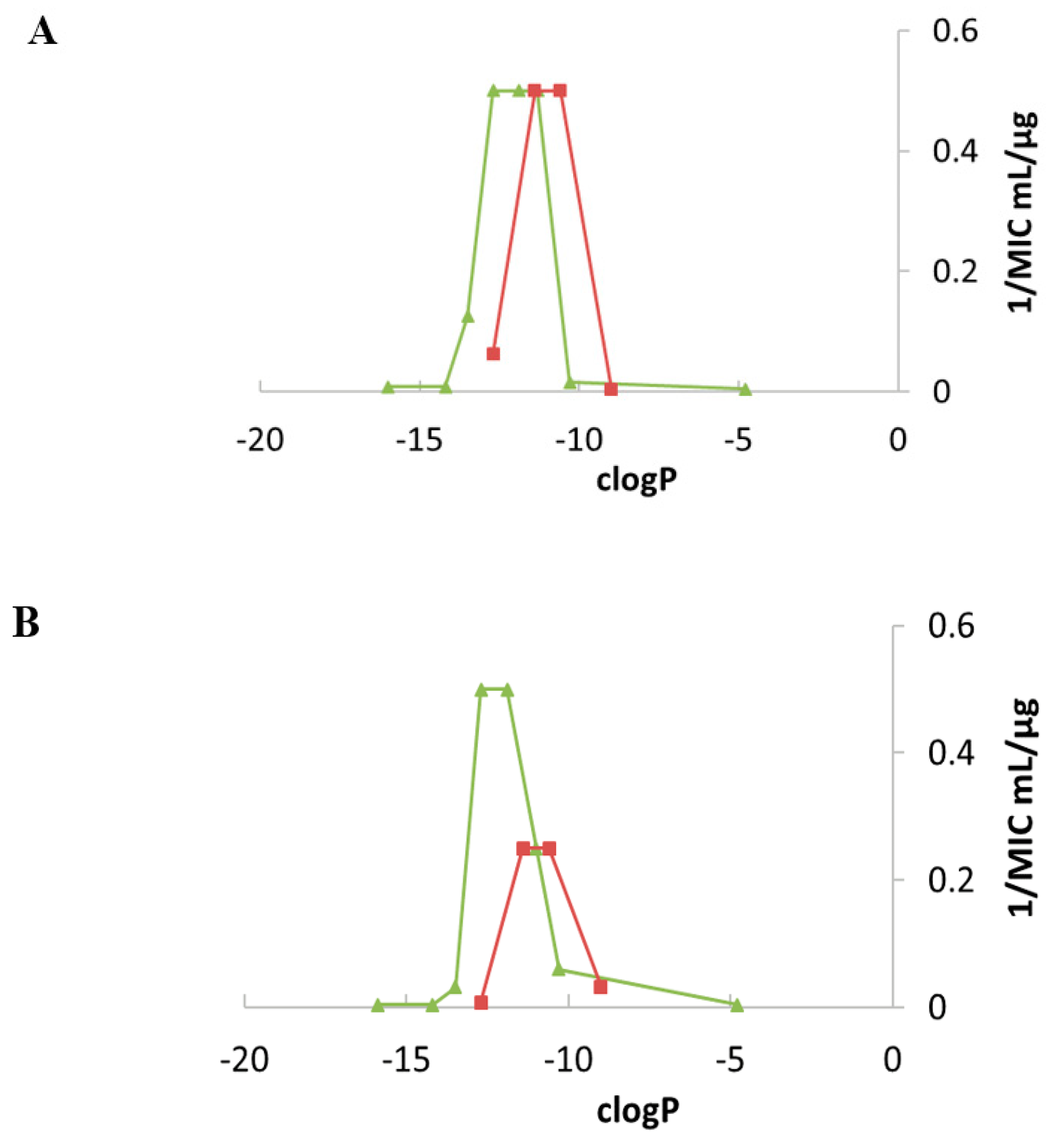
Emergence of Resistance to Amphiphilic NEA Derivatives: MIC Changes against P. aeruginosa upon a Long Exposures to AAGs
Solubility of AAGs at High Concentration in Aqueous Solutions and Dosage of 24 for Studies In Vivo (Unpublished Results)
2.1.3. Targets and Modes of Action against Gram-Negative Bacteria
Anionic LPS
Cardiolipin (CL)
2.1.4. Targets and Modes of Action against Gram-Positive Bacteria
2.2. Recent Reports on AAGs in the Field of Antibacterial Agents
2.3. AAG Positioning as Potential Antibacterial Drug Candidates, Toxicity
3. Other Biological Activities of AAGs
3.1. Recent Advances in the Field of Antifungal AAGs
3.2. AAG Vehicles for Nucleic Acids, Effects on DNA or RNA
3.2.1. Gene and siRNA Delivery
3.2.2. Peptide (Polyamide) Nucleic Acid (PNA)-AG Conjugates to Target RNA
3.2.3. Some Particular Effects of AAGs on DNA and RNA
3.3. AAGs as Intracellular Delivery Vectors of Drugs
3.4. A New Target of AAGs
4. Discussion and Conclusions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| AAG | Amphiphilic aminoglycoside |
| AG | Aminoglycoside |
| Bu | n-butyl |
| CIP | Ciprofloxacin |
| CL | Cardiolipin |
| clogP | Calculated log(octanol/water partition coefficient) |
| COL | Colistin |
| DiMOc | 3,7-(dimethyl)octyl |
| DBP | Dibasic naphthyl peptide |
| De | Decyl |
| EPI | Efflux pump inhibitor |
| Hx | n-hexyl |
| Hp | n-heptyl |
| IM | Inner membrane |
| KANA | Kanamycin A 3 |
| KANB | Kanamycin B 4 |
| LPS | Lipopolysaccharides |
| MDR | Multidrug-resistant |
| MIC | Minimum inhibitory concentration |
| MOX | Moxifloxacin |
| MRSA | Methicillin resistant S. aureus |
| 2NB | 2-naphthylbutyl |
| NEA | Neamine 6 |
| NEB | Nebramine 8 |
| NEO | Neomycin B 1 |
| 2NH | 2-naphthylhexyl |
| 2NM | 2-naphthylmethyl |
| NMP | 1-(1′-naphthylmethyl)piperazine |
| Nn | n-nonyl |
| 2NP | 2-naphthylpropyl |
| Oc | n-octyl |
| Ocd | n-octadecyl (C18) |
| OM | Outer membrane |
| PAR | Paroxetine |
| PARA | Paromamine 7 |
| PARO | Paromomycin 2 |
| PDA | Polydopamine |
| PE | Phosphatidylethanolamine |
| PG | Phosphatidylglycerol |
| PMB | para-methoxybenzyl |
| POPE | 1-palmitoyl-2-oleoyl-sn-glycero-3-phosphatidylethanolamine |
| POPG | 1-palmitoyl-2-oleoyl-sn-glycero-3-phospho-(1′-rac-glycerol) |
| PNA | Peptide (polyamide) nucleic acid |
| SAR | Structure-activity relationships |
| TOB | Tobramycin 5 |
| Tr | Trityl |
| Ud | n-undecyl (C11) |
| XDR | Extensively drug-resistant |
References
- Chandrika, N.T.; Garneau-Tsodikova, S. Comprehensive review of chemical strategies for the preparation of new aminoglycosides and their biological activities. Chem. Soc. Rev. 2018, 47, 1189–1249. [Google Scholar]
- Garneau-Tsodikova, S.; Labby, K.J. Mechanisms of resistance to aminoglycoside antibiotics: Overview and perspectives. MedChemComm 2016, 7, 11–27. [Google Scholar]
- Zhang, J.; Chiang, F.I.; Takemoto, J.Y.; Bensaci, M.; Litke, A.; Czyryca, P.G.; Chang, C.W.T. Surprising alteration of antibacterial activity of 5′’-modified neomycin against resistant bacteria. J. Med. Chem. 2008, 51, 7563–7573. [Google Scholar] [PubMed] [Green Version]
- Bera, S.; Zhanel, G.G.; Schweizer, F. Design, synthesis and antibacterial activities of neomycin-lipid conjugates: Polycationic lipids with potent gram-positive activity. J. Med. Chem. 2008, 51, 6160–6164. [Google Scholar]
- Baussanne, I.; Bussière, A.; Halder, S.; Ganem-Elbaz, C.; Ouberai, M.; Riou, M.; Paris, J.M.; Ennifar, E.; Mingeot-Leclercq, M.P.; Décout, J.L.; et al. Synthesis and antimicrobial evaluation of amphiphilic neamine derivatives. J. Med. Chem. 2010, 53, 119–127. [Google Scholar]
- Herzog, I.M.; Green, K.D.; Berkov-Zrihen, Y.; Feldman, M.; Vidavski, R.R.; Eldar-Boock, A.; Satchi-Fainaro, R.; Eldar, A.; Garneau-Tsodikova, S.; Fridman, M.; et al. 6″-Thioether tobramycin analogues: Towards selective targeting of bacterial membranes. Angew. Chem. Int. Ed. Engl. 2012, 51, 1–6. [Google Scholar]
- Gorityala, B.K.; Guchhait, G.; Schweizer, F. Carbohydrates in drug design and discovery. In Chapter Amphiphilic Aminoglycoside Antimicrobials in Antibacterial Discovery; Jimenez-Barbero, J., Canada, F.J., Martin-Santamaria, S., Eds.; Royal Society of Chemistry: London, UK, 2015; pp. 255–285. [Google Scholar]
- Mingeot-Leclercq, M.P.; Décout, J.L. Bacterial lipid membranes as promising targets to fight antimicrobial resistance, molecular foundations and illustration through the renewal of aminoglycoside antibiotics and emergence of amphiphilic aminoglycosides. MedChemComm 2016, 7, 586–611. [Google Scholar]
- François, B.; Szychowski, J.; Adhikari, S.S.; Pachamuthu, K.; Swayze, E.E.; Griffey, R.H.; Migawa, M.T.; Westhof, E.; Hanessian, S. Antibacterial aminoglycosides with a modified mode of binding to the ribosomal-RNA decoding site. Angew. Chem. Int. Ed. 2004, 43, 6735–6738. [Google Scholar]
- Szychowski, J.; Kondo, J.; Zahr, O.; Auclair, K.; Westhof, E.; Hanessian, S.; Keillor, J.W. Inhibition of aminoglycoside-deactivating enzymes APH(3′)-IIIa and AAC(6′)-II by amphiphilic paromomycin O2″-ether analogues. ChemMedChem 2011, 6, 1961–1966. [Google Scholar]
- Baral, B.; Mozafari, M.R. Strategic moves of “superbugs” against available chemical scaffolds: Signaling, regulation, and challenges. ACS Pharmacol. Transl. Sci. 2020, 3, 373–400. [Google Scholar]
- MacNair, C.R.; Tsai, C.N.; Brown, E.D. Creative targeting of the gram-negative outer membrane in antibiotic discovery. Ann. N. Y. Acad. Sci. 2020, 1459, 69–85. [Google Scholar] [PubMed]
- Schmidt, N.W.; Deshayes, S.; Hawker, S.; Blacker, A.; Kasko, A.M.; Wong, G.C.L. Engineering persister-specific antibiotics with synergistic antimicrobial functions. ACS Nano 2014, 8, 8786–8793. [Google Scholar] [PubMed] [Green Version]
- Allison, K.R.; Brynildsen, M.P.; Collins, J.J. Heterogeneous bacterial persisters and engineering approaches to eliminate them. Curr. Opin. Microbiol. 2011, 14, 593–598. [Google Scholar] [PubMed] [Green Version]
- Hurdle, J.G.; O’Neill, A.J.; Chopra, I.; Lee, R.E. Targeting bacterial membrane function: An underexploited mechanism for treating persistent infections. Nat. Rev. Microbiol. 2011, 9, 62–75. [Google Scholar]
- Ganewatta, M.S.; Tang, C. Controlling macromolecular structures towards effective antimicrobial polymers. Polymers 2015, 63, A1–A29. [Google Scholar]
- Steinbuch, K.B.; Fridman, M. Mechanisms of resistance to membrane-disrupting antibiotics in gram-positive and gram-negative bacteria. MedChemComm 2016, 7, 86–102. [Google Scholar]
- Yin, J.; Meng, Q.; Cheng, D.; Fu, J.; Luo, Q.; Liu, Y.; Yu, Z. Mechanisms of bactericidal action and resistance of polymyxins for Gram-positive bacteria. Appl. Microbiol. Biotechnol. 2020, 104, 3771–3780. [Google Scholar]
- Moubareck, C.A. Polymyxins and bacterial membranes: A review of antibacterial activity and mechanisms of resistance. Membranes 2020, 10, 30. [Google Scholar]
- Gellatly, S.L.; Hancock, R.E. Pseudomonas aeruginosa: New insights into pathogenesis and host defenses. Pathog. Dis. 2013, 67, 159–173. [Google Scholar]
- Airoldi, C.; Sommaruga, S.; Merlo, S.; Sperandeo, P.; Cipolla, L.; Polissi, A.; Nicotra, F. Targeting bacterial membranes: Identification of Pseudomonas aeruginosa D-arabinose-5P isomerase and NMR characterisation of its substrate recognition and binding properties. Chembiochem 2011, 12, 719–727. [Google Scholar]
- Putker, F.; Bos, M.P.; Tommassen, J. Transport of lipopolysaccharide to the Gram-negative bacterial cell surface. FEMS Microbiol. Rev. 2015, 39, 985–1002. [Google Scholar] [PubMed] [Green Version]
- Sperandeo, P.; Martorana, A.M.; Polissi, A. Lipopolysaccharide biogenesis and transport at the outer membrane of Gram-negative bacteria. Biochim. Biophys. Acta Mol. Cell. Biol. Lipids 2017, 1862, 1451–1460. [Google Scholar] [PubMed]
- Choi, U.; Lee, C.R. Antimicrobial agents that inhibit the outer membrane assembly machines of Gram-negative bacteria. J. Microbiol. Biotechnol. 2019, 29, 1–10. [Google Scholar] [PubMed]
- Ouberai, M.; El Garch, F.; Bussière, A.; Riou, M.; Alsteens, D.; Lins, L.; Baussanne, I.; Dufrêne, Y.F.; Brasseur, R.; Décout, J.L.; et al. The Pseudomonas aeruginosa membranes: A target for a new amphiphilic aminoglycoside derivative? Biochem. Biophys. Acta Biomembr. 2011, 1808, 1716–1727. [Google Scholar]
- Dong, H.; Zhang, Z.; Tang, X.; Huang, S.; Li, H.; Peng, B.; Dong, C. Structural insights into cardiolipin transfer from the inner membrane to the outer membrane by PbgA in gram-negative bacteria. Sci. Rep. 2016, 6, 10. [Google Scholar]
- Bramkamp, M.; van Baarle, S. Division site selection in rod-shaped bacteria. Curr. Opin. Microbiol. 2009, 12, 683–688. [Google Scholar]
- Renner, L.D.; Weibel, D.B. MinD and MinE interact with anionic phospholipids and regulate division plane formation in Escherichia Coli. J. Biol. Chem. 2012, 287, 38835–38844. [Google Scholar]
- Evans, E.A.; Waugh, R. Osmotic correction to elastic area compressibility measurements on red cell membrane. Biophys. J. 1977, 20, 307–313. [Google Scholar]
- Fosso, Y.; Li, Y.; Garneau-Tsodikova, S. New trends in the use of aminoglycosides. MedChemComm 2014, 5, 1075–1091. [Google Scholar]
- Chandrika, N.T.; Garneau-Tsodikova, S.A. Review of patents (2011–2015) towards combating resistance to and toxicity of aminoglycosides. MedChemComm 2016, 7, 50–68. [Google Scholar]
- Bera, S.; Mondal, D.; Palit, S.; Schweizer, F. Structural modifications of the neomycin class of aminoglycosides. MedChemComm 2016, 7, 1499–1534. [Google Scholar] [CrossRef]
- Aradi, K.; di Giorgio, A.; Duca, M. Recent progresses in aminoglycoside conjugation for RNA targeting: Antimicrobials and beyond. Chem. Eur. J. 2020, 26. [Google Scholar] [CrossRef] [PubMed]
- Zimmermann, L.; Bussière, A.; Ouberai, M.; Baussanne, I.; Jolivalt, C.; Mingeot-Leclercq, M.P.; Décout, J.L. Tuning the antibacterial activity of amphiphilic neamine derivatives and comparison to paromamine homologues. J. Med. Chem. 2013, 56, 7691–7705. [Google Scholar] [CrossRef] [PubMed]
- Zimmermann, L.; Kempf, J.; Briée, F.; Swain, J.; Mingeot-Leclercq, M.P.; Décout, J.L. Broad-spectrum antibacterial amphiphilic aminoglycosides: A new focus on the structure of the lipophilic groups extends the series of active dialkyl NEAs. Eur. J. Med. Chem. 2018, 157, 1512–1525. [Google Scholar] [CrossRef]
- Zimmermann, L.; Das, I.; Désiré, J.; Sautrey, G.; Barros, R.S.V.; El Khoury, M.; Mingeot-Leclercq, M.P.; Décout, J.L. New broad-spectrum antibacterial amphiphilic aminoglycosides active against resistant bacteria: From neamine derivatives to smaller neosamine analogues. J. Med. Chem. 2016, 59, 9350–9369. [Google Scholar] [CrossRef]
- Berkov-Zrihen, Y.; Herzog, I.M.; Benhamou, R.I.; Feldman, M.; Steinbuch, K.B.; Shaul, P.; Lerer, S.; Eldar, A.; Fridman, M. Tobramycin and Nebramine as pseudo-oligosaccharide scaffolds for the development of antimicrobial cationic amphiphiles. Chem. Eur. J. 2015, 21, 4340–4349. [Google Scholar] [CrossRef]
- Benhamou, R.I.; Shaul, P.; Herzog, I.M.; Fridman, M. Di-N-methylation of anti-Gram-positive aminoglycoside-derived membrane disruptors improves antimicrobial potency and broadens spectrum to Gram-negative bacteria. Angew. Chem. Int. Ed. Engl. 2015, 54, 13617–13621. [Google Scholar] [CrossRef]
- Riguet, E.; Désiré, J.; Bailly, C.; Décout, J.L. A route for preparing new NEA derivatives targeting HIV-1 TAR RNA. Tetrahedron 2004, 60, 8053–8064. [Google Scholar] [CrossRef]
- Riguet, E.; Désiré, J.; Boden, O.; Ludwig, V.; Göbel, M.; Bailly, C.; Décout, J.L. NEA dimers targeting the HIV-1 TAR RNA. Bioorg. Med. Chem. Lett. 2005, 15, 4651–4655. [Google Scholar] [CrossRef]
- Jackowski, O.; Bussière, A.; Vanhaverbeke, C.; Baussanne, I.; Peyrin, E.; Mingeot-Leclercq, M.P.; Décout, J.L. Major increases of the reactivity and selectivity in aminoglycoside O-alkylation due to the presence of fluoride ions. Tetrahedron 2012, 68, 737–746. [Google Scholar] [CrossRef]
- Sautrey, G.; Zimmermann, L.; Deleu, M.; Delbar, A.; Souza, M.L.; Jeannot, K.; Van, B.; Buyck, J.M.; Décout, J.L.; Mingeot-Leclercq, M.P. New amphiphilic NEA derivatives active against resistant Pseudomonas aeruginosa and their interactions with lipopolysaccharides. Antimicrob. Agents Chemother. 2014, 58, 4420–4430. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Khan, F.; Nguyen Pham, D.T.; Kim, Y.M. Alternative strategies for the application of aminoglycoside antibiotics against the biofilm-forming human pathogenic bacteria. Appl. Microbiol. Biotechn. 2020, 104, 1955–1976. [Google Scholar] [CrossRef] [PubMed]
- Steinbuch, K.B.; Benhamou, R.I.; Levin, L.; Stein, R.; Fridman, M. Increased degree of unsaturation in the lipid of antifungal cationic amphiphiles facilitates selective fungal cell disruption. ACS Infect. Dis. 2018, 4, 825–836. [Google Scholar] [CrossRef] [PubMed]
- Kusnlr, J.; Barna, K. Fluorimetric determination of some basic antibiotics at very low concentrations. Z. Anal. Chem. 1974, 271, 288. [Google Scholar] [CrossRef]
- El-Shabrawy, Y. Fluorimetric determination of aminoglycoside antibiotics in pharmaceutical preparations and biological fluids. Spectrosc. Lett. 2002, 35, 99–109. [Google Scholar] [CrossRef]
- Tekkeli, S.E.K.; Önal, A.; Sagirli, A.O. Spectrofluorimetric determination of tobramycin in human serum and pharmaceutical preparations by derivatization with fluorescamine. Luminescence 2012, 29, 87–91. [Google Scholar] [CrossRef]
- Raetz, C.R.; Whitfield, C. Lipopolysaccharide Endotoxins. Annu. Rev. Biochem. 2002, 71, 635–700. [Google Scholar] [CrossRef] [Green Version]
- Raetz, C.R.H.; Reynolds, C.M.; Trent, M.S.; Bishop Russell, E. Lipid A modification systems in gram-negative bacteria. Annu. Rev. Biochem. 2007, 76, 295–329. [Google Scholar] [CrossRef] [Green Version]
- Wang, X.; Quinn, P.J. Lipopolysaccharide: Biosynthetic pathway and structure modification. Prog. Lipid Res. 2010, 49, 97–107. [Google Scholar] [CrossRef]
- Bohl, T.E.; Aihara, H. Current progress in the structural and biochemical characterization of proteins involved in the assembly of lipopolysaccharide. Int. J. Microbiol. 2018, 2018, 5319146. [Google Scholar] [CrossRef] [Green Version]
- Qiao, S.; Luo, Q.; Zhao, Y.; Zhang, X.C.; Huang, Y. Structural basis for lipopolysaccharide insertion in the bacterial outer membrane. Nature 2014, 511, 108–111. [Google Scholar] [CrossRef] [PubMed]
- Lam, N.H.; Ha, B.Y. Surface-lattice model describes electrostatic interactions of ions and polycations with bacterial lipopolysaccharides: Ion valence and polycation’s excluded area. Langmuir 2014, 30, 13631–13640. [Google Scholar] [CrossRef] [PubMed]
- Neville, F.; Hodges, C.S.; Liu, C.; Konovalov, O.; Gidalevitz, D. In situ characterization of lipid A interaction with antimicrobial peptides using surface X-ray scattering. Biochim. Biophys. Act. 2006, 1758, 232–240. [Google Scholar] [CrossRef] [Green Version]
- Li, A.; Schertzer, J.W.; Yong, X. Molecular dynamics modeling of pseudomonas aeruginosa outer membranes. Phys. Chem. Chem. Phys. 2018, 20, 23635–23648. [Google Scholar] [CrossRef] [PubMed]
- Brandenburg, K.; Funari, S.S.; Koch, M.H.; Seydel, U. Investigation into the acyl chain packing of endotoxins and phospholipids under near physiological conditions by WAXS and FTIR spectroscopy. J. Struct. Biol. 1999, 128, 175–186. [Google Scholar] [CrossRef] [PubMed]
- Dias, R.P.; Lin, L.; Soares, T.A.; Alexov, E. Modeling the electrostatic potential of asymmetric lipopolysaccharide membranes: The MEMPOT algorithm Implemented in DelPhi. J. Comput. Chem. 2014, 35, 1418–1429. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brandenburg, K.; Andra, J.; Muller, M.; Koch, M.H.; Garidel, P. physicochemical properties of bacterial glycopolymers in relation to bioactivity. Carbohydr. Res. 2003, 338, 2477–2489. [Google Scholar] [CrossRef] [PubMed]
- Parasassi, T.; Gratton, E.; Yu, W.M.; Wilson, P.; Levi, M. Two-photon fluorescence microscopy of Laurdan generalized polarization domains in model and natural membranes. Biophys. J. 1997, 72, 2413–2429. [Google Scholar] [CrossRef] [Green Version]
- Kathi Scheinpflug, K.; Krylova, O.; Strahl, H. Measurement of cell membrane fluidity by laurdan GP: Fluorescence spectroscopy and microscopy. Methods Mol. Biol. 2017, 1520, 159–174. [Google Scholar]
- Pristovsek, P.; Kidric, J. Solution structure of polymyxins B and E and effect of binding to lipopolysaccharide: An NMR and molecular modeling study. J. Med. Chem. 1999, 42, 4604–4613. [Google Scholar] [CrossRef]
- Velkov, T.; Thompson, P.E.; Nation, R.L.; Li, J. Structure-activity relationships of polymyxin antibiotics. J. Med. Chem. 2010, 53, 1898–1916. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Beveridge, T.J. Structures of gram-negative cell walls and their derived membrane vesicles. J. Bacteriol. 1999, 181, 4725–4733. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Seydel, U.; Oikawa, M.; Fukase, K.; Kusumoto, S.; Brandenburg, K. Intrinsic conformation of lipid A is responsible for agonistic and antagonistic activity. Eur. J. Biochem. 2000, 267, 3032–3039. [Google Scholar] [CrossRef] [PubMed]
- Mashburn-Warren, L.; Howe, J.; Garidel, P.; Richter, W.; Steiniger, F.; Roessle, M.; Brandenburg, K.; Whiteley, M. Interaction of quorum signals with outer membrane lipids: Insights into prokaryotic membrane vesicle formation. Mol. Microbiol. 2008, 69, 491–502. [Google Scholar] [CrossRef] [Green Version]
- Howe, J.; Hammer, M.; Alexander, C.; Rossle, M.; Fournier, K.; Mach, J.P.; Waelli, T.; Gorczynski, R.M.; Ulmer, A.J.; Zahringer, U.; et al. Biophysical characterization of the interaction of endotoxins with hemoglobins. Med. Chem. 2007, 3, 13–20. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dalebroux, Z.D.; Matamouros, S.; Whittington, D.; Bishop, R.E.; Miller, S.I. PhoPQ Regulates acidic glycerophospholipid content of the Salmonella typhimurium outer membrane. Proc. Natl. Acad. Sci. USA 2014, 111, 1963–1968. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Helander, I.M.; Mattila-Sandholm, T. Fluorometric assessment of Gram-negative bacterial permeabilization. J. Appl. Microbiol. 2000, 88, 213–219. [Google Scholar] [CrossRef]
- Olofsson, G.; Sparr, E. Ionization constants pKa of cardiolipin. PLoS ONE 2013, 8, e73040. [Google Scholar] [CrossRef] [Green Version]
- Boeris, P.S.; Domenech, C.E.; Lucchesi, G.I. Modification of phospholipid composition in pseudomonas putida a ATCC 12633 induced by contact with tetradecyltrimethylammonium. J. Appl. Microbiol. 2007, 103, 1048–1054. [Google Scholar] [CrossRef]
- Lopez, G.A.; Heredia, R.M.; Boeris, P.S.; Lucchesi, G.I. Content of cardiolipin of the membrane and sensitivity to cationic surfactants in Pseudomonas putida. J. Appl. Microbiol. 2016, 121, 1004–1014. [Google Scholar] [CrossRef] [Green Version]
- Kaneko, H.; Takami, H.; Inoue, A.; Horikoshi, K. Effects of hydrostatic pressure and temperature on growth and lipid composition of the inner membrane of Barotolerant Pseudomonas Sp. BT1 isolated from the deep-sea. Biosci. Biotechnol. Biochem. 2000, 64, 72–79. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Broniatowski, M.; Mastalerz, P.; Flasinski, M. Studies of the interactions of ursane-type bioactive terpenes with the model of escherichia coli inner membrane-langmuir monolayer approach. Biochim. Biophys. Acta 2015, 1848, 469–476. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dalebroux, Z.D.; Edrozo, M.B.; Pfuetzner, R.A.; Ressl, S.; Kulasekara, B.R.; Blanc, M.P.; Miller, S.I. Delivery of cardiolipins to the Salmonella outer membrane is necessary for survival within host tissues and virulence. Cell. Host Microbe 2015, 17, 441–451. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ghorbal, S.K.; Chatti, A.; Sethom, M.M.; Maalej, L.; Mihoub, M.; Kefacha, S.; Feki, M.; Landoulsi, A.; Hassen, A. Changes in membrane fatty acid composition of Pseudomonas aeruginosa in response to UV-C radiations. Curr. Microbiol. 2013, 67, 112–117. [Google Scholar] [CrossRef] [PubMed]
- Pizzuto, M.; Lonez, C.; Baroja-Mazo, A.; Martinez-Banaclocha, H.; Tourlomousis, P.; Gangloff, M.; Pelegrin, P.; Ruysschaert, J.M.; Gay, N.J.; Bryant, C.E. Saturation of acyl chains converts cardiolipin from an antagonist to an activator of toll-like receptor-4. Cell. Mol. Life Sci. 2019, 76, 3679–3680. [Google Scholar] [CrossRef] [Green Version]
- Garrett, T.A.; O’Neill, A.C.; Hopson, M.L. Quantification of cardiolipin molecular species in escherichia coli lipid extracts using liquid chromatography/electrospray ionization mass spectrometry. Rapid. Commun. Mass Spectrom. 2012, 26, 2267–2274. [Google Scholar] [CrossRef]
- Oemer, G.; Lackner, K.; Muigg, K.; Krumschnabel, G.; Watschinger, K.; Sailer, S.; Lindner, H.; Gnaiger, E.; Wortmann, S.B.; Werner, E.R.; et al. Molecular structural diversity of mitochondrial cardiolipins. Proc. Natl. Acad. Sci. USA 2018, 115, 4158–4163. [Google Scholar] [CrossRef] [Green Version]
- Prossnigg, F.; Hickel, A.; Pabst, G.; Lohner, K. Packing behaviour of two predominant anionic phospholipids of bacterial cytoplasmic membranes. Biophys. Chem. 2010, 150, 129–135. [Google Scholar] [CrossRef]
- Pluschke, G.; Overath, P. Function of phospholipids in Escherichia coli. Influence of changes in polar head group composition on the lipid phase transition and characterization of a mutant containing only saturated phospholipid acyl chains. J. Biol. Chem. 1981, 256, 3207–3212. [Google Scholar]
- Nichols-Smith, S.; Teh, S.Y.; Kuhl, T.L. Thermodynamic and mechanical properties of model mitochondrial membranes. Biochim. Biophys. Acta 2004, 1663, 82–88. [Google Scholar] [CrossRef]
- Dowhan, W. Molecular basis for membrane phospholipid diversity: Why are there so many lipids? Annu. Rev. Biochem. 1997, 66, 199–232. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kozlovsky, Y.; Chernomordik, L.V.; Kozlov, M.M. Lipid intermediates in membrane fusion: Formation, structure, and decay of hemifusion diaphragm. Biophys. J. 2002, 83, 2634–2651. [Google Scholar] [CrossRef] [Green Version]
- Epand, R.F.; Martinou, J.C.; Fornallaz-Mulhauser, M.; Hughes, D.W.; Epand, R.M. The apoptotic protein tBid promotes leakage by altering membrane curvature. J. Biol. Chem. 2002, 277, 32632–32639. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Beales, P.A.; Bergstrom, C.L.; Geerts, N.; Groves, J.T.; Vanderlick, T.K. Single vesicle observations of the cardiolipin-cytochrome C interaction: Induction of membrane morphology changes. Langmuir 2011, 27, 6107–6115. [Google Scholar] [CrossRef] [Green Version]
- Rogasevskaia, T.P.; Coorssen, J.R. A new approach to the molecular analysis of docking, priming, and regulated membrane fusion. J. Chem. Biol. 2011, 4, 117–136. [Google Scholar] [CrossRef] [Green Version]
- Epand, R.M.; Epand, R.F. Lipid domains in bacterial membranes and the action of antimicrobial agents. Biochim. Biophys. Acta 2009, 1788, 289–294. [Google Scholar] [CrossRef] [Green Version]
- Romantsov, T.; Gonzalez, K.; Sahtout, N.; Culham, D.E.; Coumoundouros, C.; Garner, J.; Kerr, C.H.; Chang, L.; Turner, R.J.; Wood, J.M. Cardiolipin synthase A colocalizes with cardiolipin and osmosensing transporter ProP at the poles of Escherichia coli cells. Mol. Microbiol. 2018, 107, 623–638. [Google Scholar] [CrossRef] [Green Version]
- Sautrey, G.; El Koury, M.; Dos Santos, A.G.; Zimmermann, L.; Deleu, M.; Lins, L.; Décout, J.L.; Mingeot-Leclercq, M.P. Negatively charged lipids as a potential target for new amphiphilic aminoglycoside antibiotics: A biophysical study. J. Biol. Chem. 2016, 291, 13864–13874. [Google Scholar] [CrossRef] [Green Version]
- El Khoury, M.; Swain, J.; Sautrey, G.; Zimmermann, L.; Van der Smissen, P.; Décout, J.L.; Mingeot-Leclercq, M.P. Targeting bacterial cardiolipin enriched microdomains: An antimicrobial strategy used by amphiphilic aminoglycoside antibiotics. Sci. Rep. 2017, 7, 12. [Google Scholar] [CrossRef] [Green Version]
- Van den Brink-van der Laan, E.; Boots, J.W.P.; Spelbrink, R.E.J.; Kool, G.M.; Breukink, E.; Killian, J.A.; de Kruijff, B. Membrane interaction of the glycosyltransferase MurG: A special role for cardiolipin. J. Bacteriol. 2003, 185, 3773–3779. [Google Scholar] [CrossRef] [Green Version]
- Ugarte-Uribe, B.; Müller, H.M.; Otsuki, M.; Nickel, W.; García-Sáez, A.J. Dynamin-related protein 1 (Drp1) promotes structural intermediates of membrane division. J. Biol. Chem. 2014, 289, 30645–30656. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Swain, J.; El Khoury, M.; Kempf, J.; Briée, F.; van der Smissen, P.; Décout, J.L.; Mingeot-Leclercq, M.P. Effect of cardiolipin on the antimicrobial activity of new amphiphilic aminoglycoside derivative on P. aeruginosa. PLoS ONE 2018, 13, e0201752. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roszkowiak, J.; Jajor, P.; Guła, G.; Gubernator, J.; Żak, A.; Drulis-Kawa, Z.; Augustyniak, D. Interspecies outer membrane vesicles (OMVs) modulate the sensitivity of pathogenic bacteria and pathogenic yeasts to cationic peptides and serum complement. Int. J. Mol. Sci. 2019, 20, 5577. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Swain, J.; El Khoury, M.; Flament, A.; Dezanet, C.; Briée, F.; van der Smissen, P.; Decout, J.L.; Mingeot-Leclercq, M.P. Antimicrobial activity of amphiphilic neamine derivatives: Understanding the mechanism of action on gram-positive bacteria. Biochem. Biophys. Acta Biomembr. 2019, 1861, 10. [Google Scholar] [CrossRef] [PubMed]
- Libardo, M.D.; Cervantes, J.L.; Salazar, J.C.; Angeles-Boza, A.M. Improved bioactivity of antimicrobial peptides by addition of amino-terminal copper and nickel (ATCUN) binding motifs. ChemMedChem. 2014, 9, 1892–1901. [Google Scholar] [CrossRef] [Green Version]
- Libardo, M.D.J.; Nagella, S.; Lugo, A.; Pierce, S.; Angeles-Boza, A.M. Copper binding tripeptide motif increases potency of the antimicrobial peptide anoplin via reactive oxygen species generation. Biochem. Biophys. Res. Commun. 2015, 456, 446–451. [Google Scholar] [CrossRef]
- Joyner, J.C.; Hodnick, W.F.; Cowan, T.D.; Boyd, R.; Cowan, J.A. Antimicrobial metallopeptides with broad nuclease and ribonuclease activity. Chem. Commun. 2013, 49, 2118–2120. [Google Scholar] [CrossRef]
- Allam, A.; Maigre, L.; Alves de Sousa, R.; Dumont, E.; Vergalli, J.; Pages, J.M.; Artaud, I. New amphiphilic neamine conjugates bearing a metal binding motif active against MDR E. aerogenes gram-negative bacteria. Eur. J. Med. Chem. 2017, 12, 748–756. [Google Scholar] [CrossRef]
- Story, S.; Skriba, M.J.; Maiti, K.; Ranjan, N.; Degtyareva, N.N.; Green, K.D.; Khodaverdian, V.; Oyelere, A.K.; Garneau-Tsodikova, S.; Arya, D.P.; et al. Synthesis, antimicrobial activity, attenuation of aminoglycoside resistance in MRSA, and ribosomal A-site binding of pyrene-neomycin conjugates. Eur. J. Med. Chem. 2019, 163, 381–393. [Google Scholar] [CrossRef]
- Domalaon, R.; Idowu, T.; Zhanel, G.G.; Schweizer, F. Antibiotic hybrids: The next generation of agents and adjuvants against gram-negative pathogens? Clin. Microb. Rev. 2018, 31, 45. [Google Scholar] [CrossRef] [Green Version]
- Gupta, V.; Datta, P. Next-generation strategy for treating drug resistant bacteria: Antibiotic hybrids. Indian J. Med. Res. 2019, 7, 97–106. [Google Scholar]
- Schweizer, F. Enhancing uptake of antibiotics into gram-negative bacteria using nonribosome-targeting aminoglycoside-based adjuvants. Future Med. Chem. 2019, 11, 1519–1522. [Google Scholar] [CrossRef] [PubMed]
- Bera, S.; Zhanel, G.; Schweizer, F. Synthesis and antibacterial activity of amphiphilic lysine-ligated neomycin B conjugates. Carbohydr. Res. 2011, 346, 560–568. [Google Scholar]
- Lyu, Y.; Yang, X.; Goswami, S.; Gorityala, B.K.; Idowu, T.; Domalaon, R.; Zhanel, G.G.; Shan, A.; Schweizer, F. Amphiphilic tobramycin-lysine conjugates sensitize multidrug resistant gram-negative bacteria to rifampicin and minocycline. J. Med. Chem. 2017, 60, 3684–3702. [Google Scholar] [CrossRef] [PubMed]
- Lyu, Y.; Domalaon, R.; Yang, X.; Schweizer, F. Amphiphilic lysine conjugated to tobramycin synergizes legacy antibiotics against wild-type and multidrug-resistant Pseudomonas aeruginosa. Peptide Sci. 2019, 111, 7. [Google Scholar] [CrossRef]
- Yang, X.; Goswami, S.; Gorityala, B.K.; Domalaon, R.; Lyu, Y.; Kumar, A.; Zhanel, G.G.; Schweizer, F. A Tobramycin vector enhances synergy and efficacy of efflux pump inhibitors against multidrug-resistant Gram-negative bacteria. J. Med. Chem. 2017, 60, 3913–3932. [Google Scholar] [CrossRef]
- Yang, X.; Domalaon, R.; Lyu, Y.; Zhanel, G.G.; Schweizer, F. Tobramycin-linked efflux pump inhibitor conjugates synergize fluoroquinolones, rifampicin and fosfomycin against multidrug-resistant Pseudomonas aeruginosa. J. Clin. Med. 2018, 7, 158. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gorityala, B.K.; Guchhait, G.; Goswami, S.; Fernando, D.M.; Kumar, A.; Zhanel, G.G.; Schweizer, F. Hybrid antibiotic overcomes resistance in P. aeruginosa by enhancing outer membrane penetration and reducing efflux. J. Med. Chem. 2016, 59, 8441–8455. [Google Scholar]
- Gorityala, B.K.; Guchhait, G.; Fernando, D.M.; Deo, S.; McKenna, S.A.; Zhanel, G.G.; Kumar, A.; Schweizer, F. Adjuvants based on hybrid antibiotics overcome resistance in Pseudomonas aeruginosa and enhance fluoroquinolone efficacy. Angew. Chem. Int. Ed. Engl. 2016, 55, 555–559. [Google Scholar] [CrossRef]
- Domalaon, R.; Ammeter, D.; Brizuela, M.; Gorityala, B.K.; Zhanel, G.G.; Schweizer, F. Repurposed antimicrobial combination therapy: Tobramycin-Ciprofloxacin hybrid augments activity of the anticancer drug Mitomycin C against Multidrug-Resistant Gram-Negative bacteria. Front. Microbiol. 2019, 10, 9. [Google Scholar] [CrossRef] [Green Version]
- Yang, X.; Ammeter, D.; Idowu, T.; Domalaon, R.; Brizuela, M.; Okunnu, O.; Bi, L.; Guerrero, Y.A.; Zhanel, G.G.; Kumar, A.; et al. Amphiphilic nebramine-based hybrids rescue legacy antibiotics from intrinsic resistance in multidrug-resistant gram-negative bacilli. Eur. J. Med. Chem. 2019, 175, 187–200. [Google Scholar] [CrossRef] [PubMed]
- Domalaon, R.; Yang, X.; Lyu, Y.; Zhanel, G.G.; Schweizer, F. Polymyxin B-3-tobramycin hybrids with Pseudomonas aeruginosa-selective antibacterial activity and strong potentiation of rifampicin, minocycline, and vancomycin. ACS Infect. Dis. 2017, 3, 941–954. [Google Scholar] [CrossRef] [PubMed]
- Idowu, T.; Arthur, G.; Zhanel, G.G.; Schweizer, F. Heterodimeric rifampicin-tobramycin conjugates break intrinsic resistance of Pseudomonas aeruginosa to doxycycline and chloramphenicol in vitro and in a Galleria mellonella in vivo model. Eur. J. Med. Chem. 2019, 174, 16–32. [Google Scholar] [CrossRef] [PubMed]
- Idowu, T.; Ammeter, D.; Arthur, G.; Zhanel, G.G.; Schweizer, F. Potentiation of β-lactam antibiotics and β -lactam/β-lactamase inhibitor combinations against MDR and XDR Pseudomonas aeruginosa using non-ribosomal Tobramycin-Cyclam conjugates. J. Antimicrob. Chemother. 2019, 74, 2640–2648. [Google Scholar] [CrossRef] [PubMed]
- Ammeter, D.; Idowu, T.; Zhanel, G.G.; Schweizer, F. Development of a nebramine-cyclam conjugate as an antibacterial adjuvant to potentiate beta-lactam antibiotics against multidrug-resistant P. aeruginosa. J. Antibiot. 2019, 72, 816–826. [Google Scholar] [CrossRef]
- Singh, I.; Priyam, A.; Jha, D.; Dhawan, G.; Gautam, H.K.; Kumar, P. Polydopamine-aminoglycoside nanoconjugates: Synthesis, characterization, antimicrobial evaluation and cytocompatibility. Mater. Sci. Eng. C 2020, 107, 11. [Google Scholar] [CrossRef]
- Torrado, J.J.; Espada, R.; Ballesteros, M.P.; Torrado-Santiago, S. Amphotericin B formulations and drug targeting. J. Pharm. Sci. 2008, 97, 2405–2425. [Google Scholar]
- Barker, W.T.; Martin, S.E.; Chandler, C.E.; Nguyen, T.V.; Harris, T.L.; Goodell, C.; Melander, R.J.; Doi, Y.; Ernst, R.K.; Melander, C. Small molecule adjuvants that suppress both chromosomal and mcr-1 encoded colistin-resistance and amplify colistin efficacy in polymyxin-susceptible bacteria. Bioorg. Med. Chem. 2017, 25, 5749–5753. [Google Scholar] [CrossRef]
- Kathayat, D.; Antony, L.; Deblais, L.; Helmy, Y.A.S.; Caria, J.; Rajashekara, G. Small molecule adjuvants potentiate Colistin activity and attenuate resistance development in Escherichia coli by affecting pmrAB system. Infect. Drug Resist. 2020, 13, 2205–2222. [Google Scholar] [CrossRef]
- Howard, K.C.; Dennis, E.K.; Watt, D.S.; Garneau-Tsodikova, S. A comprehensive overview of the medicinal chemistry of antifungal drugs including: Perspectives and promise. Chem. Soc. Rev. 2020, 49, 2426–2480. [Google Scholar] [CrossRef]
- Chang, C.W.T.; Takemoto, J.Y. Antifungal amphiphilic aminoglycosides. Med. Chem. Commun. 2014, 5, 1048–1057. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chang, C.W.T.; Fosso, M.; Kawasaki, Y.; Shrestha, S.; Bensaci, M.F.; Wang, J.; Evans, C.K.; Takemoto, J.Y. Divergent synthesis of three classes of antifungal amphiphilic kanamycin derivatives. J. Antibiot. 2010, 63, 667–672. [Google Scholar] [CrossRef] [PubMed] [Green Version]
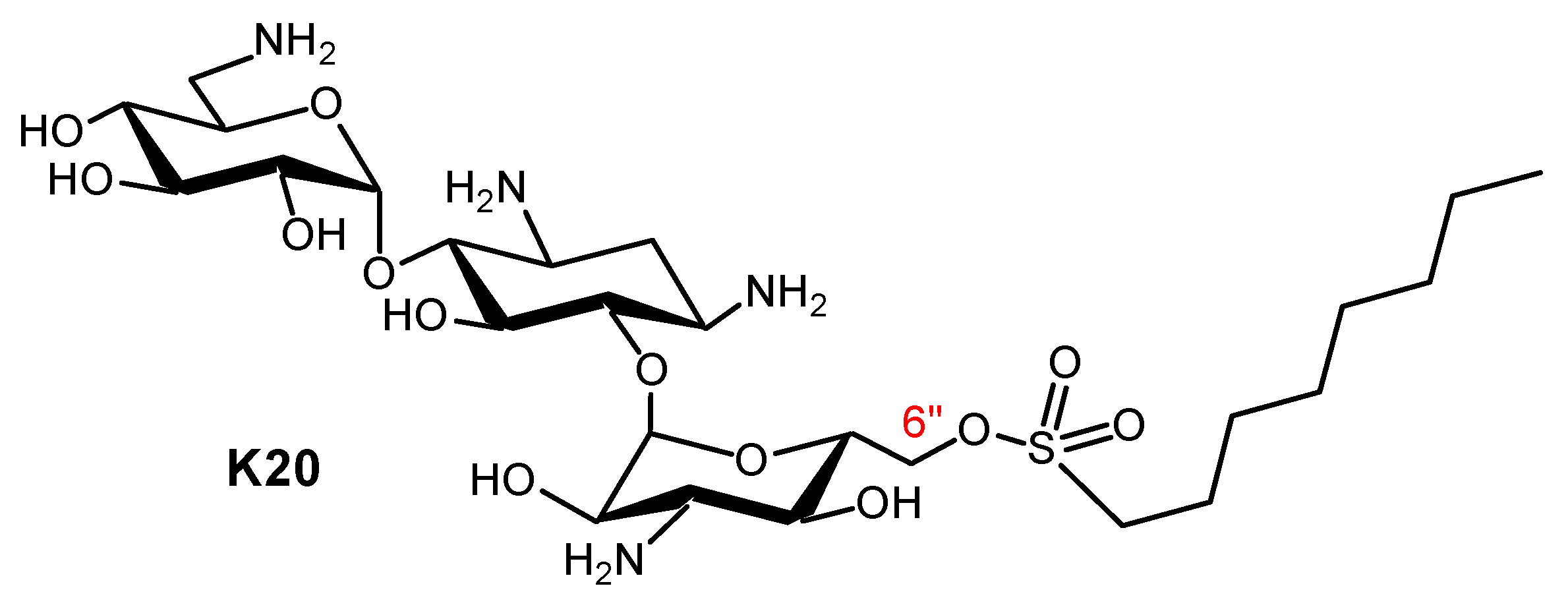
- Takemoto, J.Y.; Wegulo, S.N.; Yuen, G.Y.; Stevens, J.A.; Jochum, C.C.; Chang, C.W.T.; Kawasaki, Y.; Miller, G.W. Suppression of wheat Fusarium head blight by novel amphiphilic aminoglycoside fungicide K20. Fungal Biol. 2017, 122, 465–470. [Google Scholar] [CrossRef] [PubMed]
- Subedi, Y.P.; AlFindee, M.N.; Takemoto, J.Y.; Chang, C.W.T. Antifungal amphiphilic kanamycins: New life for an old drug. MedChemComm 2018, 9, 909–919. [Google Scholar] [CrossRef]
- Subedi, Y.P.; Roberts, P.; Grilley, M.; Takemoto, J.Y.; Chang, C.T. Development of fungal selective amphiphilic kanamycin: Cost-effective synthesis and use of fluorescent analogs for mode of action investigation. ACS Infect. Dis. 2019, 5, 473–483. [Google Scholar] [CrossRef]
- Alfindee, M.N.; Subedi, Y.P.; Grilley, M.M.; Takemoto, J.Y.; Chang, C.W.T. Antifungal activities of 4″,6″-disubstituted amphiphilic kanamycins. Molecules 2019, 24, 1882. [Google Scholar] [CrossRef] [Green Version]
- Subedi, Y.P.; Pandey, U.; Alfindee, M.N.; Montgomery, H.; Roberts, P.; Wight, J.; Nichols, G.; Grilley, M.; Takemoto, J.Y.; Chang, C.W.T.; et al. Scalable and cost-effective tosylation-mediated synthesis of antifungal and fungal diagnostic 6″-Modified amphiphilic kanamycins. Eur. J. Med. Chem. 2019, 182, 8. [Google Scholar] [CrossRef]
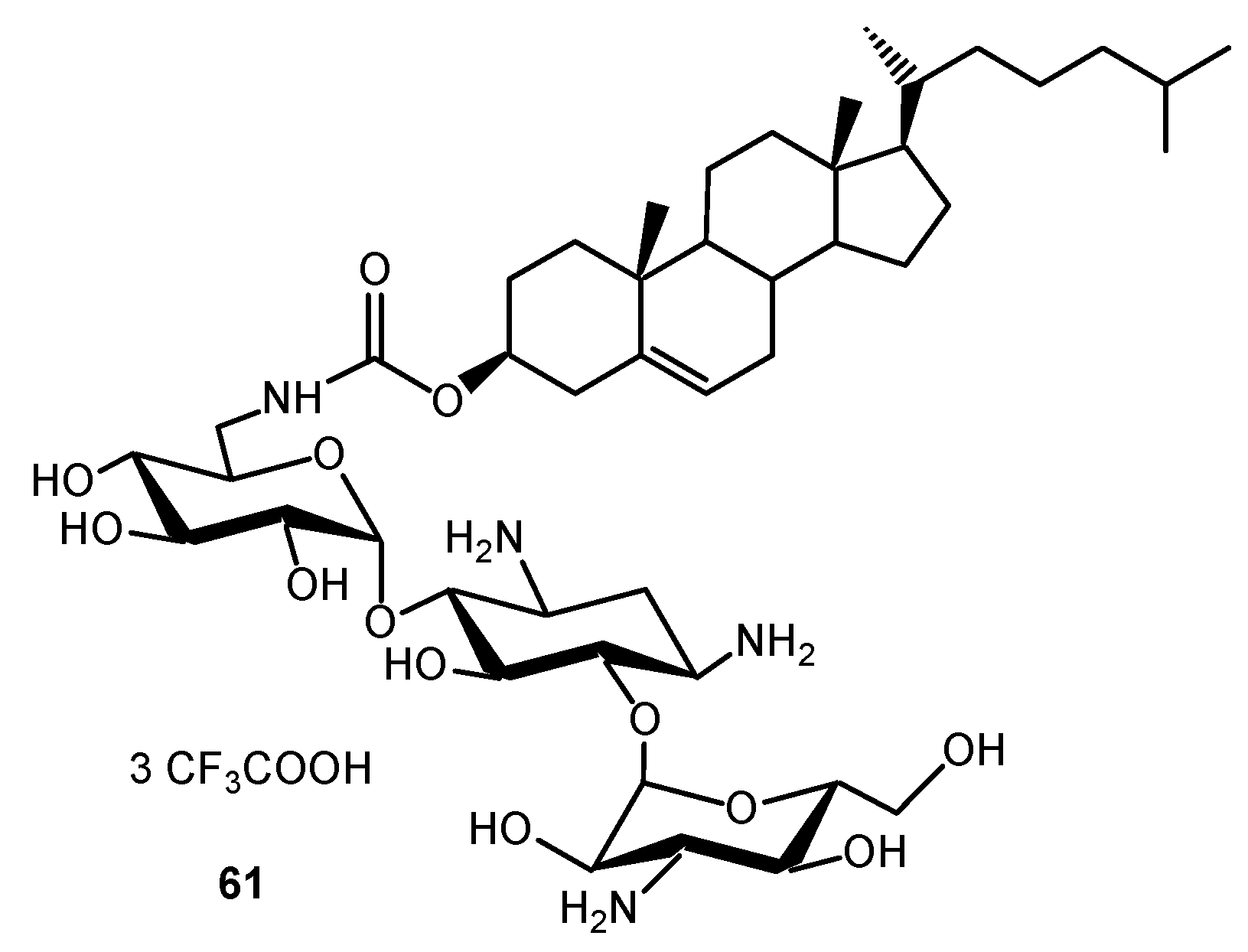
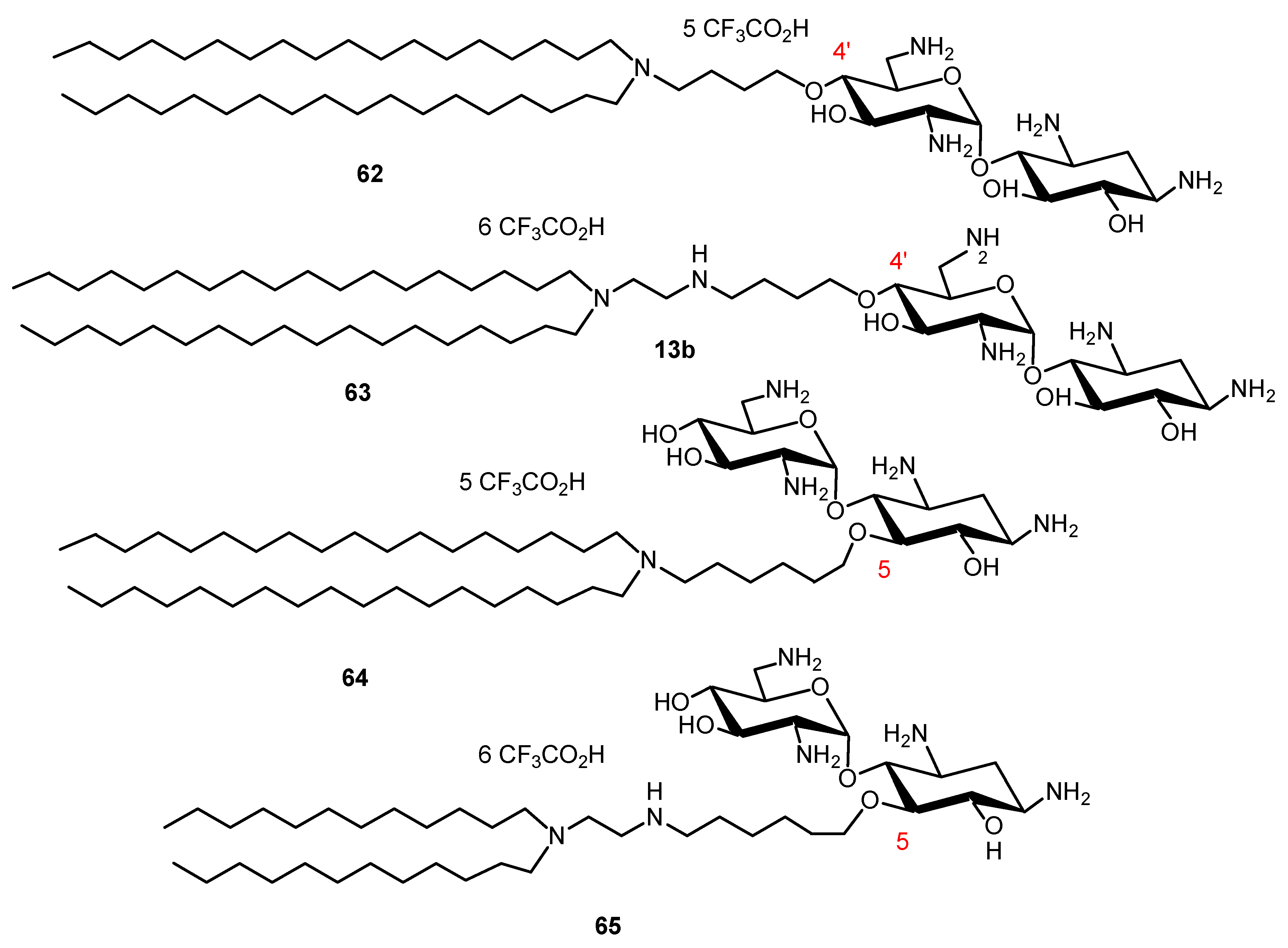
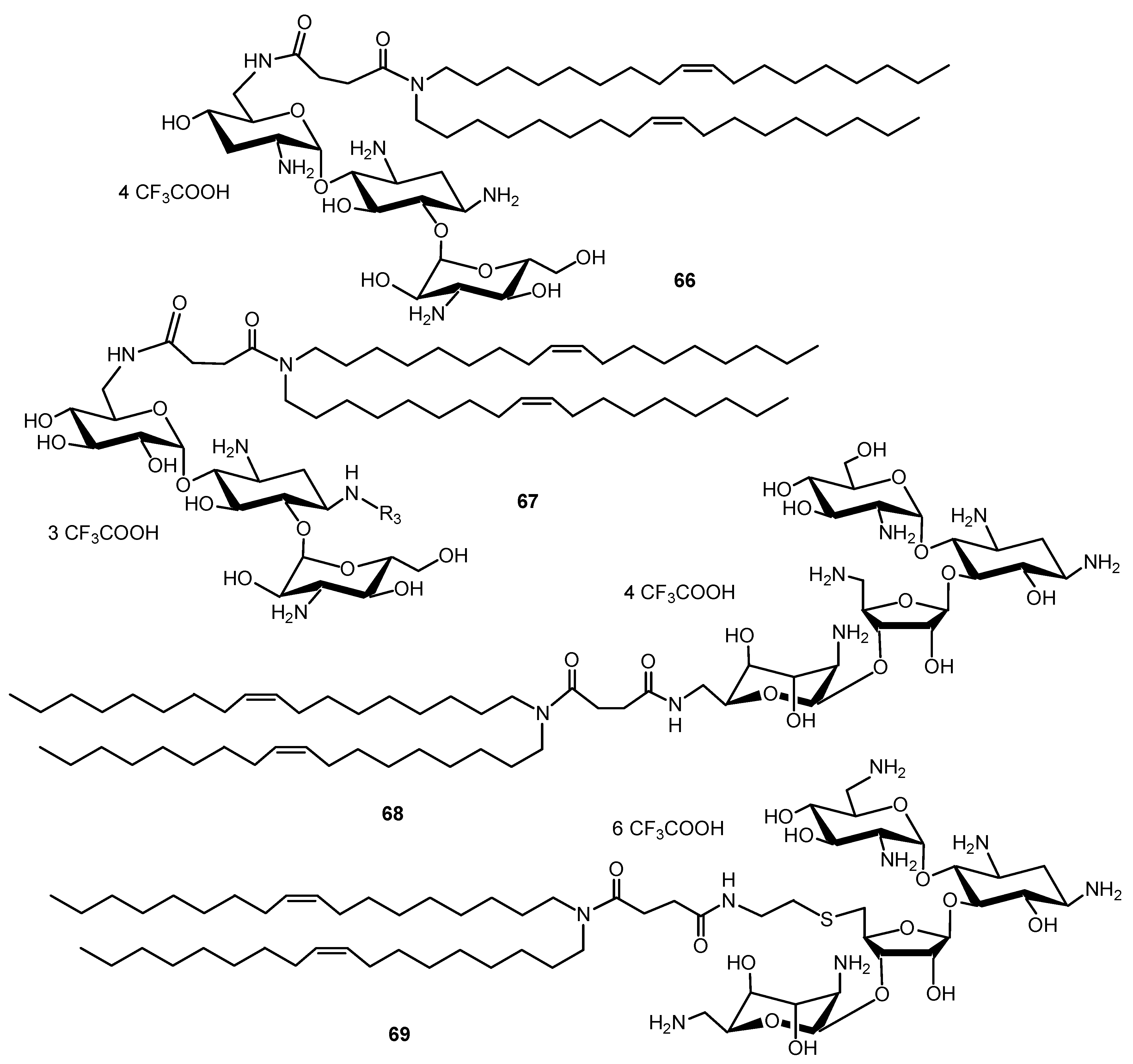
- Bellucci, M.C.; Volonterio, A. Aminoglycosides: From antibiotics to building blocks for the synthesis and development of gene delivery vehicles. Antibiotics 2020, 9, 504. [Google Scholar] [CrossRef]
- Belmont, P.; Aissaoui, A.; Hauchecorne, M.; Oudrhiri, N.; Petit, L.; Vigneron, J.P.; Lehn, J.M.; Lehn, P. Aminoglycoside-derived cationic lipids as efficient vectors for gene transfection in vitro and in vivo. J. Gene Med. 2002, 4, 517–526. [Google Scholar] [CrossRef]
- Sainlos, M.; Belmont, P.; Vigneron, J.P.; Lehn, P.; Lehn, J.M. Aminoglycoside-derived cationic lipids for gene transfection: Synthesis of kanamycin A derivatives. Eur. J. Org. Chem. 2003, 15, 2764–2774. [Google Scholar] [CrossRef]
- Sainlos, M.; Hauchecorne, M.; Oudrhiri, N.; Zertal-Zidani, S.; Aissaoui, A.; Vigneron, J.P.; Lehn, J.M.; Lehn, P. Kanamycin A-derived cationic lipids as vectors for gene transfection. ChemBioChem 2005, 6, 1023–1033. [Google Scholar] [CrossRef] [PubMed]
- Mevel, M.; Sainlos, M.; Chatin, B.; Oudrhiri, N.; Hauchecorne, M.; Lambert, O.; Vigneron, J.P.; Lehn, P.; Pitard, B.; Lehn, J.M. Paromomycin and neomycin B derived cationic lipids: Synthesis and transfection studies. J. Control. Release 2012, 158, 461–469. [Google Scholar] [CrossRef] [PubMed]
- Le Gall, T.; Baussanne, I.; Halder, S.; Carmoy, N.; Montier, T.; Lehn, P.; Décout, J.L. Synthesis and transfection properties of a series of lipidic neamine derivatives. Bioconjugate Chem. 2009, 20, 2032–2046. [Google Scholar] [CrossRef] [PubMed]
- Desigaux, L.; Sainlos, M.; Lambert, O.; Chèvre, R.; Letrou-Bonneval, E.; Vigneron, J.P.; Lehn, P.; Lehn, J.M.; Pitard, B. Self-assembled lamellar complexes of siRNA with lipidic aminoglycoside derivatives promote efficient siRNA delivery and interference. Proc. Natl. Acad. Sci. USA 2007, 104, 16534–16539. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bono, N.; Pennetta, C.; Sganappa, A.; Giupponi, E.; Sansone, F.; Volonterio, A.; Candiani, G. Design and synthesis of biologically active cationic amphiphiles built on the calix [4]arene scaffold. Int. J. Pharm. 2018, 549, 436–445. [Google Scholar] [CrossRef] [PubMed]
- Irudayasamy, C.; Xi, H.; Arya, D.P. Sequence specific targeting of RNA with an oligonucleotide-neomycin conjugate. Bioconjugate Chem. 2007, 18, 160–169. [Google Scholar]
- Ketomäki, K.; Virta, P. Synthesis of aminoglycoside conjugates of 2′-O-methyl oligoribonucleotides. Bioconjugate Chem. 2008, 19, 766–777. [Google Scholar] [CrossRef]
- Kiviniemi, A.; Virta, P.; Lönnberg, H. Utilization of intrachain 4′-C-azidomethylthymidine for preparation of oligodeoxyribonucleotide conjugates by click chemistry in solution and on a solid support. Bioconjugate Chem. 2008, 19, 1726–1734. [Google Scholar] [CrossRef]
- Kiviniemi, A.; Virta, P.; Lönnberg, H. Solid supported synthesis and click conjugation of 4′-C-alkyne functionalized oligodeoxyribonucleotides. Bioconjugate Chem. 2010, 21, 1890–1901. [Google Scholar] [CrossRef]
- Kiviniemi, A.; Virta, P. Synthesis of aminoglycoside-3′- conjugates of 2′-O-methyl oligoribonucleotides and their invasion to a 19F labeled HIV-1 TAR model. Bioconjugate Chem. 2011, 22, 1559–1566. [Google Scholar] [CrossRef]
- Mei, H.; Xing, L.; Cai, L.; Hong-Wei, J.; Zhao, P.; Yang, Z.J.; Zhang, L.R.; Zhang, L.H. Studies on the synthesis of neamine−dinucleosides and neamine−PNA conjugates and their interaction with RNA. Bioorg. Med. Chem. Lett. 2008, 18, 5355–5358. [Google Scholar] [CrossRef] [PubMed]
- Nielsen, P.E.; Egholm, M.; Berg, R.H.; Buchardt, O. Sequence selective recognition of DNA by strand displacement with a thymine-substituted polyamide. Science 1991, 254, 1497–1500. [Google Scholar] [CrossRef] [PubMed]
- Ghosal, A.; Nielsen, P. Potent antibacterial antisense peptide–peptide nucleic acid conjugates against Pseudomonas aeruginosa. Nucleic Acid Ther. 2012, 22, 323–334. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wojciechowska, M.; Rownicki, M.; Mieczkowski, A.; Miszkiewicz, J.; Trylska, J. Antibacterial peptide nucleic acids-facts and perspectives. Molecules 2020, 25, 559. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Riguet, E.; Tripathi, S.; Chaubey, B.; Désiré, J.; Pandey, V.N.; Décout, J.L. A peptide nucleic acid−neamine conjugate that targets and cleaves HIV-1 TAR RNA inhibits viral replication. J. Med. Chem. 2004, 47, 4806–4809. [Google Scholar] [CrossRef]
- Charles, I.; Arya, D.P. Synthesis of neomycin−DNA/peptide nucleic acid conjugates. J. Carbohydr. Chem. 2005, 24, 145–160. [Google Scholar] [CrossRef]
- Soonsil, H.; Hyun, L.K.; Jaehoon, Y. A strategy for the design of selective RNA binding agents. Preparation and RRE RNA binding affinities of a neomycin−peptide nucleic acid heteroconjugate library. Bioorg. Med. Chem. Lett. 2006, 16, 4757–4759. [Google Scholar]
- Chaubey, B.; Tripathi, S.; Désiré, J.; Baussanne, I.; Décout, J.L.; Pandey, V.N. Mechanism of RNA cleavage catalyzed by sequence specific polyamide nucleic acid−neamine conjugate. Oligonucleotides 2007, 17, 302–313. [Google Scholar] [CrossRef]
- Alguacil, J.; Defaus, S.; Claudio, A.; Trapote, A.; Masides, M.; Robles, J. A straigtforward preparation of aminoglycoside−dinucleotide and−diPNA conjugates via Click ligation assisted by microwaves. Eur. J. Org. Chem. 2010, 20, 3102–3109. [Google Scholar] [CrossRef]
- Das, I.; Désiré, J.; Manvar, D.; Baussanne, I.; Décout, J.L. A peptide nucleic acid-aminosugar conjugate targeting transactivation response element of HIV-1 RNA genome shows a high bioavailability in human cells and strongly inhibits Tat-mediated transactivation of HIV-1 transcription. J. Med. Chem. 2012, 55, 6021–6032. [Google Scholar] [CrossRef]
- Devi, G.; Yuan, Z.; Lu, Y.; Zhao, Y.; Chen, G. Incorporation of thio-pseudoisocytosine into triplex-forming peptide nucleic acids for enhanced recognition of RNA duplexes. Nucleic Acids Res. 2014, 42, 4008–4018. [Google Scholar] [CrossRef] [PubMed]
- Kesy, J.; Patil, K.; Subaschandrabose, R.; Shu, Z.; Yong, H.Y.; Zimmermann, L.; Ong, A.A.L.; Toh, D.F.K.; Krishna, M.S.; Yang, L.; et al. A Short chemically-modified dsRNA-binding PNA (dbPNA) inhibits influenza viral replication by targeting viral RNA panhandle structure. Bioconjugate Chem. 2019, 30, 931–943. [Google Scholar] [CrossRef] [PubMed]
- Ong, A.A.L.; Tan, J.; Bhadra, M.; Dezanet, C.; Patil, K.M.; Chong, M.S.; Kierzek, R.; Decout, J.L.; Roca, X.; Chen, G.; et al. RNA secondary structure-based design of antisense peptide nucleic acids for modulating disease-associated aberrant tau pre-mRNA alternative splicing. Molecules 2019, 24, 3020. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ying, L.; Zhu, H.; Fosso, M.Y.; Garneau-Tsodikova, S.; Fredrick, K. Modified aminoglycosides bind nucleic acids in high-molecular-weight complexes. Antibiotics 2020, 9, 93. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Greenberg, M.M. The formamidopyrimidines: Purine lesions formed in competition with 8-oxopurines from oxidative stress. Accounts Chem. Res. 2012, 45, 588–597. [Google Scholar] [CrossRef] [Green Version]
- Perigolo, M.; Constant, J.F.; Peuchmaur, M.; Pitta, I.; Décout, J.L. Antibiotic drugs aminoglycosides cleave DNA at abasic sites: Shedding new light on their toxicity? Chem. Res. Tox. 2013, 26, 1710–1719. [Google Scholar] [CrossRef]
- Deka, S.R.; Yadav, S.; Mahato, M.; Sharma, A.K. Azobenzene-aminoglycoside: Self-assembled smart amphiphilic nanostructures for drug delivery. Colloid Surface B 2015, 135, 150–157. [Google Scholar] [CrossRef]
- Miryala, B.; Godeshala, S.; Grandhi, T.S.P.; Christensen, M.D.; Tian, Y.; Rege, K. Aminoglycoside-derived amphiphilic nanoparticles for molecular delivery. Colloid Surface B 2016, 146, 924–937. [Google Scholar] [CrossRef]
- AlFindee, M.N.; Subedi, Y.P.; Fiori, M.C.; Krishnan, S.; Kjellgren, A.; Altenberg, G.A.; Chang, C.W.T. Inhibition of connexin hemichannels by new amphiphilic aminoglycosides without antibiotic activity. ACS Med. Chem. Lett. 2018, 9, 697–701. [Google Scholar] [CrossRef]




















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
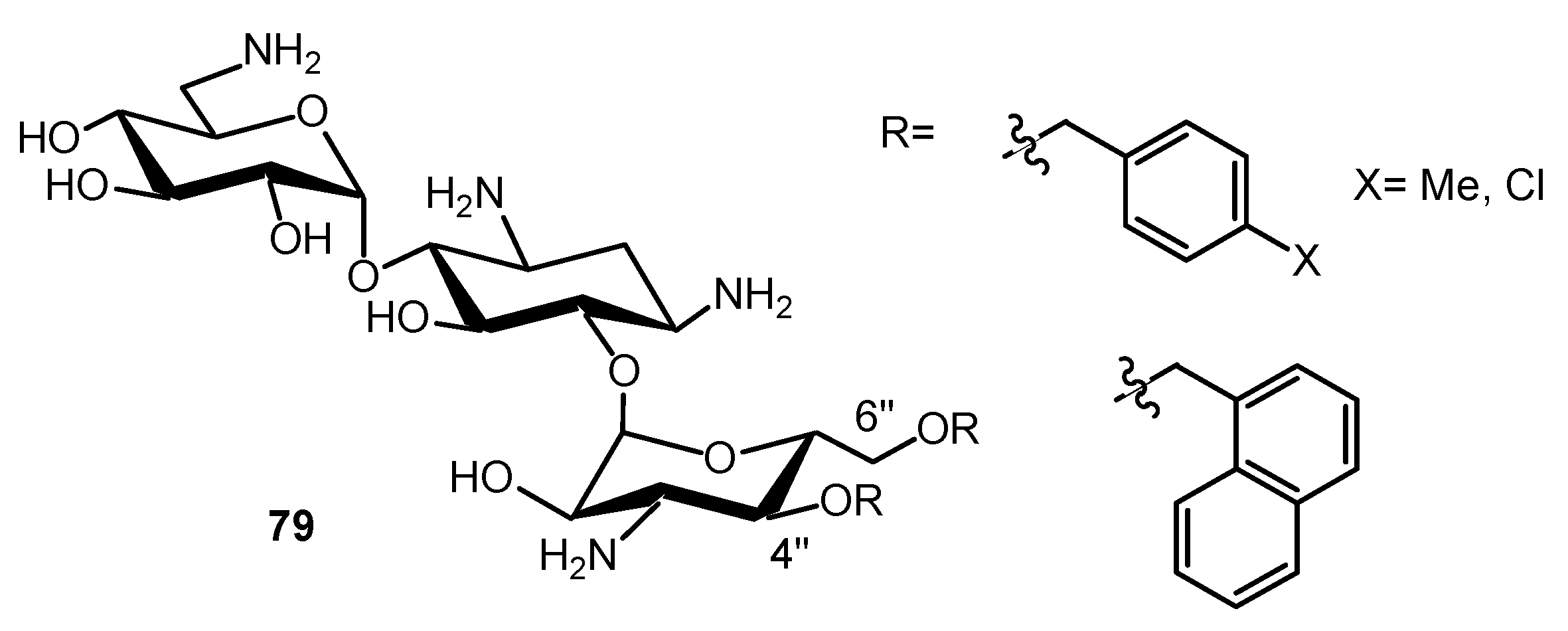{kind=link}
| AGs | Lipophilicity Expressed as clogP | MIC µg/mL | ||||||
|---|---|---|---|---|---|---|---|---|
| S. aureus | P. aeruginosa | |||||||
| ATCC 25923 | SA-1 Pump NorA | ATCC 33592 HA-MRSA | ATCC 27853 | Psa. FO3 a | PA22 b | PA406 c | ||
| NEO 1 | −29.9 | 1–2 | 0.5–1 | >128 | 64 | 128 | 32–64 | 2–4 |
| NEA 6 | −19.4 | 16–32 | 8 | >128 | >128 | >128 | >128 | 64 |
| 3′,6-diNn 24 | −11.9 | 1 | 1 | 2–4 | 2–4 | 4–8 | 4 | 2–4 |
| 3′,6-di2NP 26 | −11.4 | 2 | 2 | 2 | 8-16 | 16 | 16 | 2–4 |
| 3′,6-diOc 39 | −12.7 | 1 | 1 | 2 | 2 | 8 | 8 | 2 |
| AAG | Lipophilicity Expressed as clogP | Viability % | |
|---|---|---|---|
| 10 µM | 30 µM | ||
| NEO 1 | −29.9 | 87.3 (10) | 69.8 (2) |
| NEA 6 | −19.4 | 94.8 (9) | 84.4 (2) |
| 3′,6-diNn 24 | −11.9 | 86.7 (9) | 67.4 (3) |
| 3′,6-di2NP 26 | −11.4 | 91.1 (13) | 89.5 (2) |
| 3′,6-diOc 39 | −12.7 | 91.3 (4) | 65.1 (3) |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dezanet, C.; Kempf, J.; Mingeot-Leclercq, M.-P.; Décout, J.-L. Amphiphilic Aminoglycosides as Medicinal Agents. Int. J. Mol. Sci. 2020, 21, 7411. https://doi.org/10.3390/ijms21197411
Dezanet C, Kempf J, Mingeot-Leclercq M-P, Décout J-L. Amphiphilic Aminoglycosides as Medicinal Agents. International Journal of Molecular Sciences. 2020; 21(19):7411. https://doi.org/10.3390/ijms21197411
Chicago/Turabian StyleDezanet, Clément, Julie Kempf, Marie-Paule Mingeot-Leclercq, and Jean-Luc Décout. 2020. "Amphiphilic Aminoglycosides as Medicinal Agents" International Journal of Molecular Sciences 21, no. 19: 7411. https://doi.org/10.3390/ijms21197411
APA StyleDezanet, C., Kempf, J., Mingeot-Leclercq, M. -P., & Décout, J. -L. (2020). Amphiphilic Aminoglycosides as Medicinal Agents. International Journal of Molecular Sciences, 21(19), 7411. https://doi.org/10.3390/ijms21197411