Regulation of CFTR Biogenesis by the Proteostatic Network and Pharmacological Modulators

{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Cystic Fibrosis
1.1. Pathology
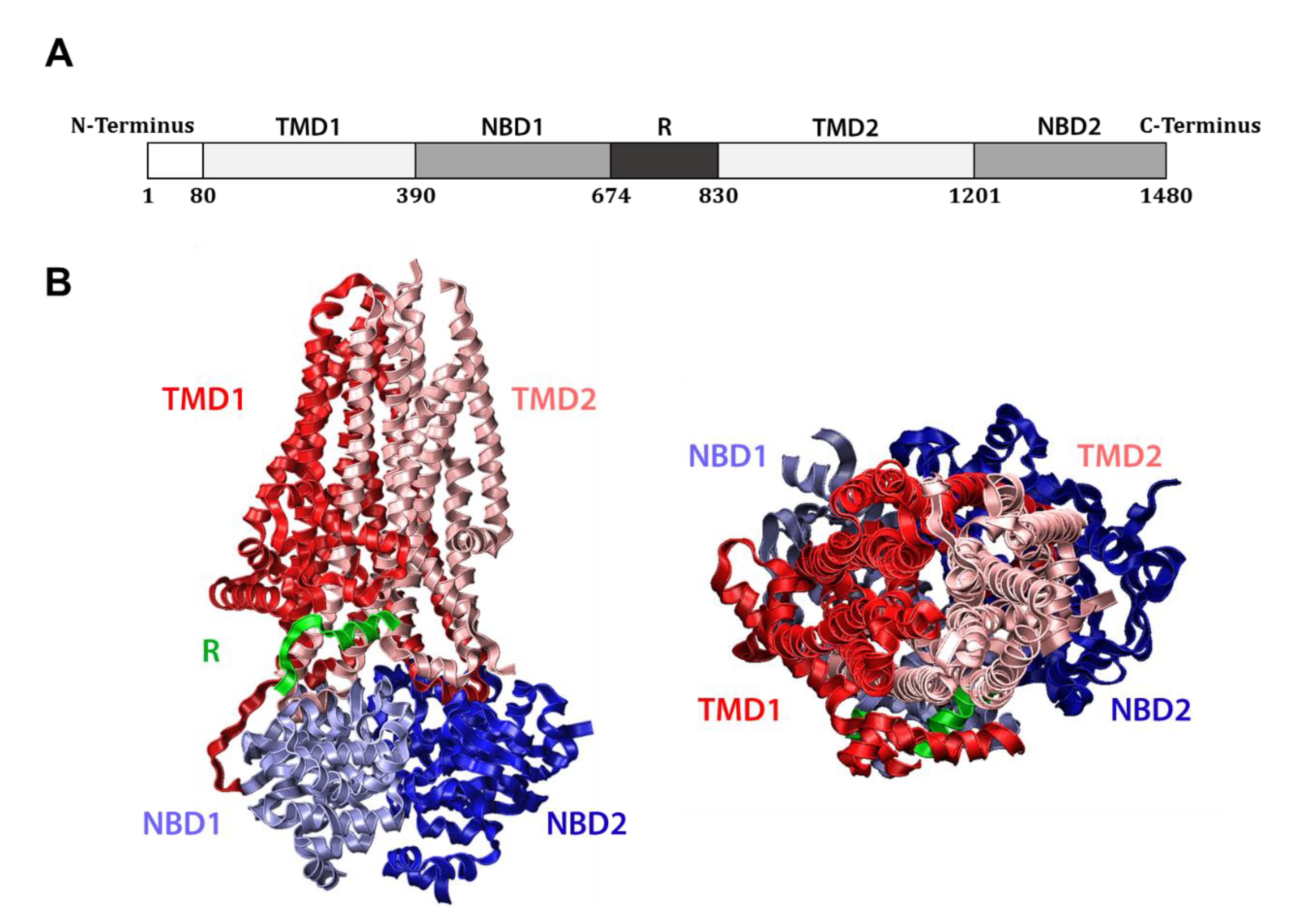
1.2. CFTR Structure and Function
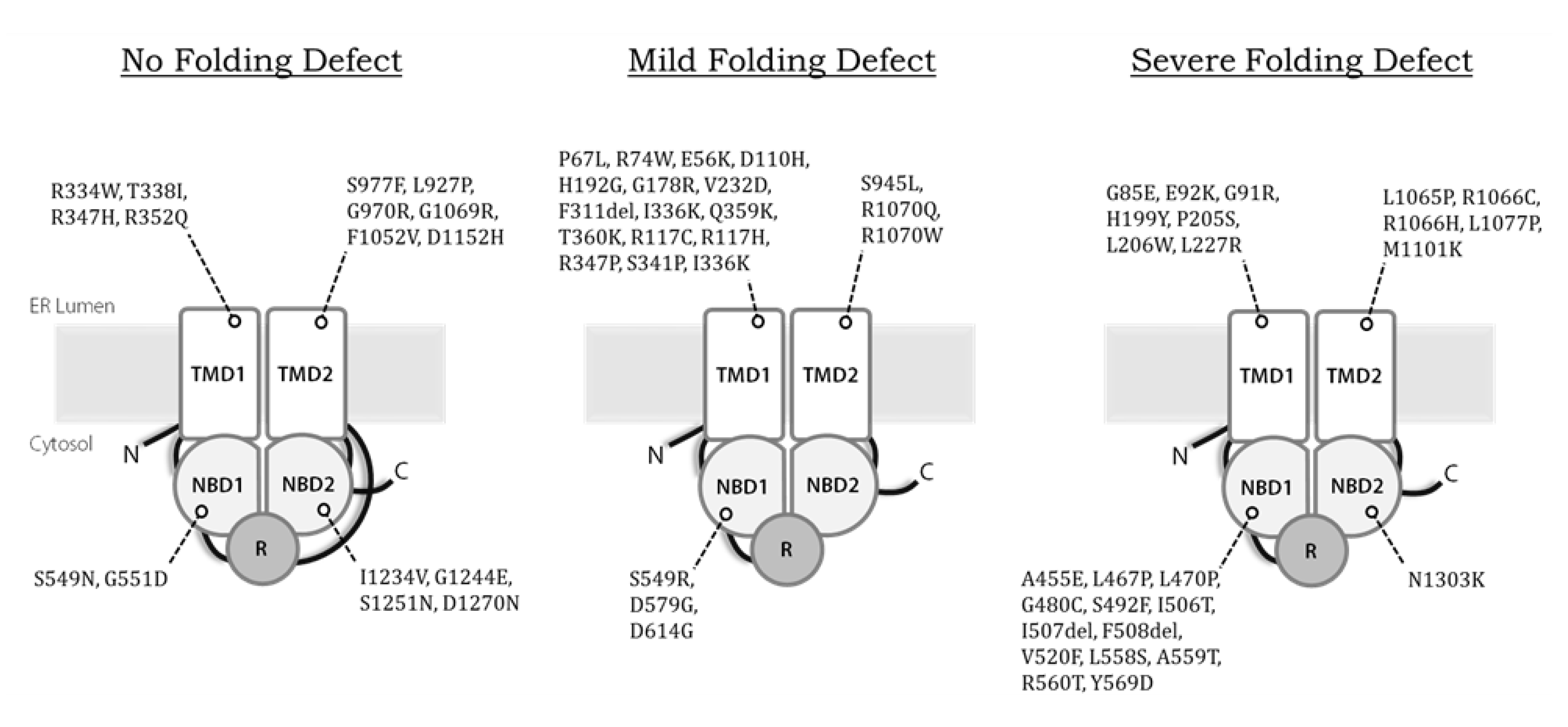
1.3. Inheritance of Dysfunctional CFTR Variants Causes CF
2. CFTR is Subject to Multiple Protein Quality Control Checkpoints
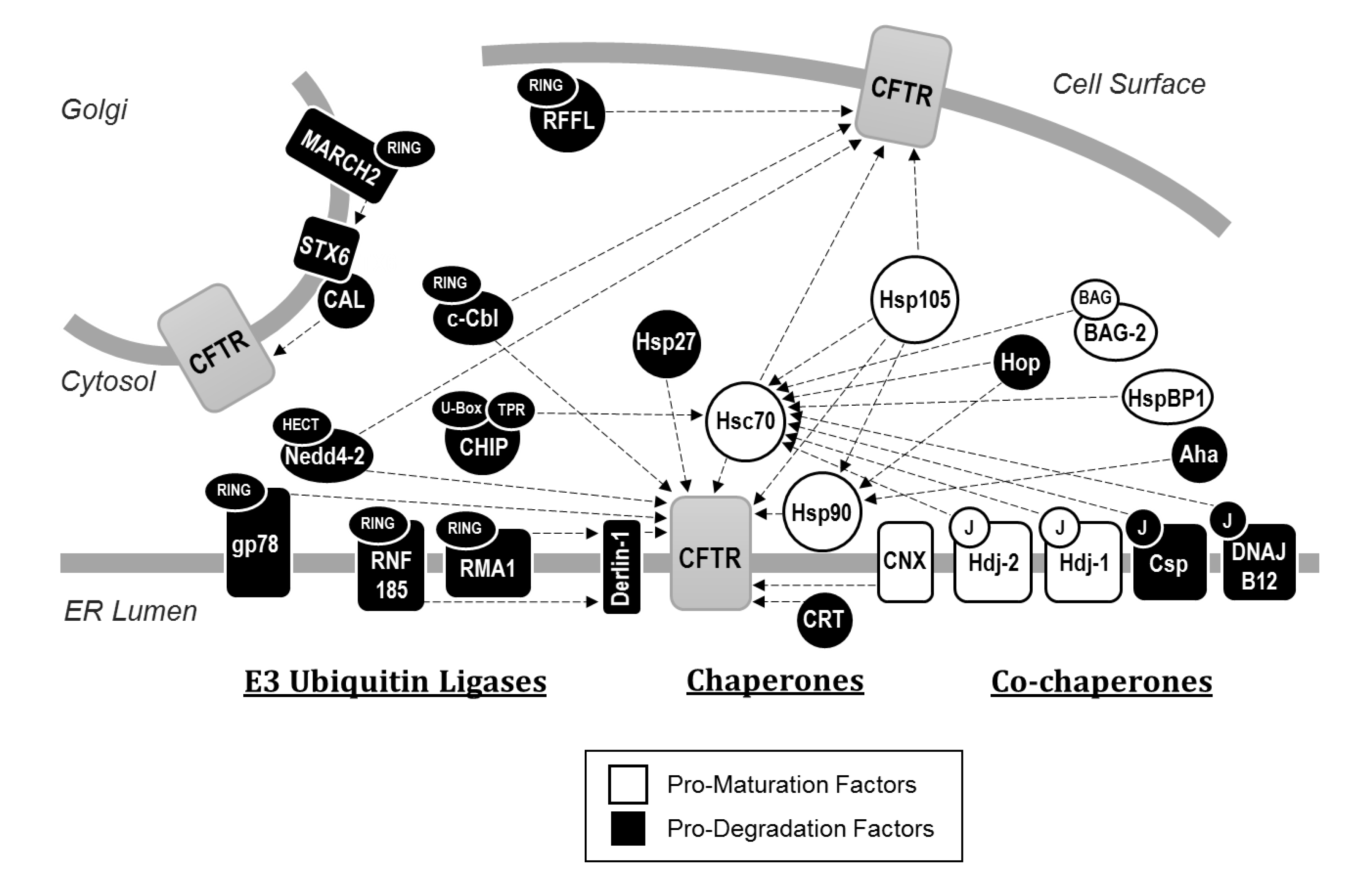
2.1. Protein Quality Control at the ER: The Roles of Molecular Chaperones and the Proteostatic Network
2.2. The Targeting of CFTR for Endoplasmic Reticulum Associated Degradation (ERAD)
2.3. Trafficking of CFTR from the ER
2.4. Post-ER Quality Control: Targeting of Plasma Membrane and Endosomal CFTR for Lysosomal Degradation
3. Pharmacological Interventions in Cystic Fibrosis
3.1. Correction of CFTR Misfolding Promotes Forward Trafficking
3.2. Potentiation of Channel Conductance
3.3. Compounds Currently in Clinical Use
4. The Path Forward in Cystic Fibrosis
Funding
Conflicts of Interest
References
- Anderson, D.H. Cystic Fibrosis of the Pancreas and its Relation to Celiac Disease: A Clinical and Pathologic Study. Am. J. Dis. Child. 1938, 56, 344–399. [Google Scholar] [CrossRef]
- Singh, V.K.; Schwarzenberg, S.J. Pancreatic insufficiency in Cystic Fibrosis. J. Cyst. Fibros. 2017, 16, S70–S78. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Anderson, D.H.; Hodges, R.G. Genetics of Cystic Fibrosis of the Pancreas with a Consideration of Etiology. Am. J. Dis. Child. 1946, 72, 62–80. [Google Scholar] [CrossRef] [PubMed]
- Verkman, A.S.; Song, Y.; Thiagarajah, J.R. Role of airway surface liquid and submucosal glands in cystic fibrosis lung disease. Am. J. Physiol. Cell Physiol. 2003, 284, C2–C15. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Haq, I.J.; Gray, M.A.; Garnett, J.P.; Ward, C.; Brodlie, M. Airway surface liquid homeostasis in cystic fibrosis: Pathophysiology and therapeutic targets. Thorax 2016, 71, 284–287. [Google Scholar] [CrossRef] [Green Version]
- Frayman, K.B.; Armstrong, D.S.; Grimwood, K.; Ranganathan, S.C. The airway microbiota in early cystic fibrosis lung disease. Pediatr. Pulmonol. 2017, 52, 1384–1404. [Google Scholar] [CrossRef]
- Cantin, A.M.; Hartl, D.; Konstan, M.W.; Chmiel, J.F. Inflammation in cystic fibrosis lung disease: Pathogenesis and therapy. J. Cyst. Fibros. 2015, 14, 419–430. [Google Scholar] [CrossRef] [Green Version]
- Szczesniak, R.; Heltshe, S.L.; Stanojevic, S.; Mayer-Hamblett, N. Use of FEV1 in cystic fibrosis epidemiologic studies and clinical trials: A statistical perspective for the clinical researcher. J. Cyst. Fibros. 2017, 16, 318–326. [Google Scholar] [CrossRef]
- Furukawa, B.S.; Flume, P.A. Nontuberculous Mycobacteria in Cystic Fibrosis. Semin. Respir. Crit. Care Med. 2018, 39, 383–391. [Google Scholar]
- Hector, A.; Kirn, T.; Ralhan, A.; Graepler-Mainka, U.; Berenbrinker, S.; Riethmueller, J.; Hogardt, M.; Wagner, M.; Pfleger, A.; Autenrieth, I.; et al. Microbial colonization and lung function in adolescents with cystic fibrosis. J. Cyst. Fibros. 2016, 15, 340–349. [Google Scholar] [CrossRef] [Green Version]
- Rosen, B.H.; Evans, T.I.A.; Moll, S.R.; Gray, J.S.; Liang, B.; Sun, X.; Zhang, Y.; Jensen-Cody, C.W.; Swatek, A.M.; Zhou, W.; et al. Infection is Not Required for Mucoinflammatory Lung Disease in CFTR-knockout Ferrets. Am. J. Respir. Crit. Care Med. 2018, 197, 1308–1318. [Google Scholar] [CrossRef] [PubMed]
- Mogayzel, P.J., Jr.; Naureckas, E.T.; Robinson, K.A.; Brady, C.; Guill, M.; Lahiri, T.; Lubsch, L.; Matsui, J.; Oermann, C.M.; Ratjen, F.; et al. Cystic Fibrosis Foundation pulmonary guideline. Pharmacologic approaches to prevention and eradication of initial Pseudomonas aeruginosa infection. Ann. Am. Thorac. Soc. 2014, 11, 1640–1650. [Google Scholar] [CrossRef] [PubMed]
- McIlwaine, M.; Button, B.; Nevitt, S.J. Positive expiratory pressure physiotherapy for airway clearance in people with cystic fibrosis. Cochrane Database Syst. Rev. 2019, 2019, 11. [Google Scholar] [CrossRef] [PubMed]
- Grosse-Onnebrink, J.; Mellies, U.; Olivier, M.; Werner, C.; Stehling, F. Chest physiotherapy can affect the lung clearance index in cystic fibrosis patients. Pediatr. Pulmonol. 2017, 52, 625–631. [Google Scholar] [CrossRef]
- Cystic Fibrosis Foundation. Patient Registry 2018 Annual Data Report; Cystic Fibrosis Foundation: Bethesda, MD, USA, 2019. [Google Scholar]
- Maqbool, A.; Pauwels, A. Cystic Fibrosis and gastroesophageal reflux disease. J. Cyst. Fibros. 2017, 16, S2–S13. [Google Scholar] [CrossRef] [Green Version]
- Kelsey, R.; Manderson Koivula, F.N.; McClenaghan, N.H.; Kelly, C. Cystic Fibrosis-Related Diabetes: Pathophysiology and Therapeutic Challenges. Clin. Med. Insights Endocrinol. Diabetes 2019, 12, 1–7. [Google Scholar] [CrossRef] [Green Version]
- Moheet, A.; Moran, A. Pharmacological management of cystic fibrosis related diabetes. Expert Rev. Clin. Pharm. 2018, 11, 185–191. [Google Scholar] [CrossRef]
- Kamal, N.; Surana, P.; Koh, C. Liver disease in patients with cystic fibrosis. Curr. Opin. Gastroenterol. 2018, 34, 146–151. [Google Scholar] [CrossRef]
- Chillon, M.; Casals, T.; Mercier, B.; Bassas, L.; Lissens, W.; Silber, S.; Romey, M.C.; Ruiz-Romero, J.; Verlingue, C.; Claustres, M. Mutations in the cystic fibrosis gene in patients with congenital absence of the vas deferens. N. Engl. J. Med. 1995, 332, 1475–1480. [Google Scholar] [CrossRef] [Green Version]
- Bernardino, R.L.; Jesus, T.T.; Martins, A.D.; Sousa, M.; Barros, A.; Cavaco, J.E.; Socorro, S.; Alves, M.G.; Oliveira, P.F. Molecular basis of bicarbonate membrane transport in the male reproductive tract. Curr. Med. Chem. 2013, 20, 4037–4049. [Google Scholar] [CrossRef]
- Quittner, A.L.; Goldbeck, L.; Abbott, J.; Duff, A.; Lambrecht, P.; Sole, A.; Tibosch, M.M.; Bergsten Brucefors, A.; Yuksel, H.; Catastini, P.; et al. Prevalence of depression and anxiety in patients with cystic fibrosis and parent caregivers: Results of The International Depression Epidemiological Study across nine countries. Thorax 2014, 69, 1090–1097. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Smith, B.A.; Georgiopoulos, A.M.; Quittner, A.L. Maintaining mental health and function for the long run in cystic fibrosis. Pediatr. Pulmonol. 2016, 51, S71–S78. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kerem, B.S.; Zielenski, J.; Markiewicz, D.; Bozon, D.; Gazit, E.; Yahav, J.; Kennedy, D.; Riordan, J.R.; Collins, F.S.; Rommens, J.M. Identification of mutations in regions corresponding to the two putative nucleotide (ATP)-binding folds of the cystic fibrosis gene. Proc. Natl. Acad. Sci. USA 1990, 87, 8447–8451. [Google Scholar] [CrossRef] [Green Version]
- Riordan, J.R.; Rommens, J.M.; Kerem, B.; Alon, N.; Rozmahel, R.; Grzelczak, Z.; Zielenski, J.; Lok, S.; Plavsic, N.; Chou, J.L. Identification of the cystic fibrosis gene: Cloning and characterization of complementary DNA. Science 1989, 245, 1066–1073. [Google Scholar] [CrossRef] [PubMed]
- Mornon, J.P.; Lehn, P.; Callebaut, I. Atomic model of human cystic fibrosis transmembrane conductance regulator: Membrane-spanning domains and coupling interfaces. Cell Mol. Life Sci. 2008, 65, 2594–2612. [Google Scholar] [CrossRef] [PubMed]
- Cheng, S.H.; Rich, D.P.; Marshall, J.; Gregory, R.J.; Welsh, M.J.; Smith, A.E. Phosphorylation of the R domain by cAMP-dependent protein kinase regulates the CFTR chloride channel. Cell 1991, 66, 1027–1036. [Google Scholar] [CrossRef]
- Liu, F.; Zhang, Z.; Csanady, L.; Gadsby, D.C.; Chen, J. Molecular Structure of the Human CFTR Ion Channel. Cell 2017, 169, 85–95. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Z.; Chen, J. Atomic Structure of the Cystic Fibrosis Transmembrane Conductance Regulator. Cell 2016, 167, 1586–1597. [Google Scholar] [CrossRef] [Green Version]
- Moran, O. On the structural organization of the intracellular domains of CFTR. Int. J. Biochem. Cell Biol. 2014, 52, 7–14. [Google Scholar] [CrossRef]
- Hwang, T.C.; Nagel, G.; Nairn, A.C.; Gadsby, D.C. Regulation of the gating of cystic fibrosis transmembrane conductance regulator C1 channels by phosphorylation and ATP hydrolysis. Proc. Natl. Acad. Sci. USA 1994, 91, 4698–4702. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Z.; Liu, F.; Chen, J. Molecular structure of the ATP-bound, phosphorylated human CFTR. Proc. Natl. Acad. Sci. USA 2018, 115, 12757–12762. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fay, J.F.; Aleksandrov, L.A.; Jensen, T.J.; Cui, L.L.; Kousouros, J.N.; He, L.; Aleksandrov, A.A.; Gingerich, D.S.; Riordan, J.R.; Chen, J.Z. Cryo-EM Visualization of an Active High Open Probability CFTR Anion Channel. Biochemistry 2018, 57, 6234–6246. [Google Scholar] [CrossRef] [PubMed]
- Sheppard, D.N.; Welsh, M.J. Structure and function of the CFTR chloride channel. Physiol. Rev. 1999, 79, S23–S45. [Google Scholar] [CrossRef] [PubMed]
- Locher, K.P. Mechanistic diversity in ATP-binding cassette (ABC) transporters. Nat. Struct. Mol. Biol. 2016, 23, 487–493. [Google Scholar] [CrossRef] [Green Version]
- Shah, V.S.; Meyerholz, D.K.; Tang, X.X.; Reznikov, L.; Abou Alaiwa, M.; Ernst, S.E.; Karp, P.H.; Wohlford-Lenane, C.L.; Heilmann, K.P.; Leidinger, M.R.; et al. Airway acidification initiates host defense abnormalities in cystic fibrosis mice. Science 2016, 351, 503–507. [Google Scholar] [CrossRef] [Green Version]
- Pezzulo, A.A.; Tang, X.X.; Hoegger, M.J.; Abou Alaiwa, M.H.; Ramachandran, S.; Moninger, T.O.; Karp, P.H.; Wohlford-Lenane, C.L.; Haagsman, H.P.; van Eijk, M.; et al. Reduced airway surface pH impairs bacterial killing in the porcine cystic fibrosis lung. Nature 2012, 487, 109–113. [Google Scholar] [CrossRef]
- Ram, S.J.; Kirk, K.L. Cl- permeability of human sweat duct cells monitored with fluorescence-digital imaging microscopy: Evidence for reduced plasma membrane Cl- permeability in cystic fibrosis. Proc. Natl. Acad. Sci. USA 1989, 86, 10166–10170. [Google Scholar] [CrossRef] [Green Version]
- Di Sant’Agnese, P.A.; Darling, R.C.; Perara, G.A.; Shea, A. Abnormal electrolyte composition of sweat in cystic fibrosis of the pancreas. AMA Am. J. Dis. Child. 1953, 86, 618–619. [Google Scholar]
- LeGrys, V.A.; Moon, T.C.; Laux, J.; Rock, M.J.; Accurso, F. Analytical and biological variation in repeated sweat chloride concentrations in clinical trials for CFTR modulator therapy. J. Cyst. Fibros. 2018, 17, 43–49. [Google Scholar] [CrossRef] [Green Version]
- Farrell, P.; Ferec, C.; Macek, M.; Frischer, T.; Renner, S.; Riss, K.; Barton, D.; Repetto, T.; Tzetis, M.; Giteau, K.; et al. Estimating the age of p.(Phe508del) with family studies of geographically distinct European populations and the early spread of cystic fibrosis. Eur. J. Hum. Genet 2018, 26, 1832–1839. [Google Scholar] [CrossRef] [Green Version]
- Gabriel, S.E.; Brigman, K.N.; Koller, B.H.; Boucher, R.C.; Stutts, M.J. Cystic fibrosis heterozygote resistance to cholera toxin in the cystic fibrosis mouse model. Science 1994, 266, 107–109. [Google Scholar] [CrossRef] [PubMed]
- Welsh, M.J.; Smith, A.E. Molecular mechanisms of CFTR chloride channel dysfunction in cystic fibrosis. Cell 1993, 73, 1251–1254. [Google Scholar] [CrossRef]
- Sosnay, P.R.; Siklosi, K.R.; Van Goor, F.; Kaniecki, K.; Yu, H.; Sharma, N.; Ramalho, A.S.; Amaral, M.D.; Dorfman, R.; Zielenski, J.; et al. Defining the disease liability of variants in the cystic fibrosis transmembrane conductance regulator gene. Nat. Genet. 2013, 45, 1160–1167. [Google Scholar] [CrossRef] [Green Version]
- Thomas, P.J.; Shenbagamurthi, P.; Sondek, J.; Hullihen, J.M.; Pedersen, P.L. The cystic fibrosis transmembrane conductance regulator. Effects of the most common cystic fibrosis-causing mutation on the secondary structure and stability of a synthetic peptide. J. Biol. Chem. 1992, 267, 5727–5730. [Google Scholar] [PubMed]
- Qu, B.H.; Strickland, E.H.; Thomas, P.J. Localization and suppression of a kinetic defect in cystic fibrosis transmembrane conductance regulator folding. J. Biol. Chem. 1997, 272, 15739–15744. [Google Scholar] [CrossRef] [Green Version]
- Zhang, F.; Kartner, N.; Lukacs, G.L. Limited proteolysis as a probe for arrested conformational maturation of delta F508 CFTR. Nat. Struct. Biol. 1998, 5, 180–183. [Google Scholar] [CrossRef]
- Sharma, M.; Benharouga, M.; Hu, W.; Lukacs, G.L. Conformational and temperature-sensitive stability defects of the delta F508 cystic fibrosis transmembrane conductance regulator in post-endoplasmic reticulum compartments. J. Biol. Chem. 2001, 276, 8942–8950. [Google Scholar] [CrossRef] [Green Version]
- Younger, J.M.; Chen, L.; Ren, H.Y.; Rosser, M.F.; Turnbull, E.L.; Fan, C.Y.; Patterson, C.; Cyr, D.M. Sequential quality-control checkpoints triage misfolded cystic fibrosis transmembrane conductance regulator. Cell 2006, 126, 571–582. [Google Scholar] [CrossRef] [Green Version]
- Hoelen, H.; Kleizen, B.; Schmidt, A.; Richardson, J.; Charitou, P.; Thomas, P.J.; Braakman, I. The primary folding defect and rescue of ΔF508 CFTR emerge during translation of the mutant domain. PLoS ONE 2010, 5, e15458. [Google Scholar] [CrossRef] [Green Version]
- Yu, H.; Burton, B.; Huang, C.J.; Worley, J.; Cao, D.; Johnson, J.P., Jr.; Urrutia, A.; Joubran, J.; Seepersaud, S.; Sussky, K.; et al. Ivacaftor potentiation of multiple CFTR channels with gating mutations. J. Cyst. Fibros. 2012, 11, 237–245. [Google Scholar] [CrossRef] [Green Version]
- Sheppard, D.N.; Ostedgaard, L.S.; Winter, M.C.; Welsh, M.J. Mechanism of dysfunction of two nucleotide binding domain mutations in cystic fibrosis transmembrane conductance regulator that are associated with pancreatic sufficiency. EMBO J. 1995, 14, 876–883. [Google Scholar] [CrossRef] [PubMed]
- Veit, G.; Avramescu, R.G.; Chiang, A.N.; Houck, S.A.; Cai, Z.; Peters, K.W.; Hong, J.S.; Pollard, H.B.; Guggino, W.B.; Balch, W.E.; et al. From CFTR biology toward combinatorial pharmacotherapy: Expanded classification of cystic fibrosis mutations. Mol. Biol. Cell 2016, 27, 424–433. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van Goor, F.; Hadida, S.; Grootenhuis, P.D.; Burton, B.; Cao, D.; Neuberger, T.; Turnbull, A.; Singh, A.; Joubran, J.; Hazlewood, A.; et al. Rescue of CF airway epithelial cell function in vitro by a CFTR potentiator, VX-770. Proc. Natl. Acad. Sci. USA 2009, 106, 18825–18830. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dalemans, W.; Barbry, P.; Champigny, G.; Jallat, S.; Dott, K.; Dreyer, D.; Crystal, R.G.; Pavirani, A.; Lecocq, J.P.; Lazdunski, M. Altered chloride ion channel kinetics associated with the delta F508 cystic fibrosis mutation. Nature 1991, 354, 526–528. [Google Scholar] [CrossRef]
- Hwang, T.C.; Wang, F.; Yang, I.C.; Reenstra, W.W. Genistein potentiates wild-type and delta F508-CFTR channel activity. Am. J. Physiol. 1997, 273, C988–C998. [Google Scholar] [CrossRef]
- Ward, C.L.; Kopito, R.R. Intracellular turnover of cystic fibrosis transmembrane conductance regulator. Inefficient processing and rapid degradation of wild-type and mutant proteins. J. Biol. Chem. 1994, 269, 25710–25718. [Google Scholar]
- Kleizen, B.; van Vlijmen, T.; de Jonge, H.R.; Braakman, I. Folding of CFTR is predominantly cotranslational. Mol. Cell 2005, 20, 277–287. [Google Scholar] [CrossRef]
- Rabeh, W.M.; Bossard, F.; Xu, H.; Okiyoneda, T.; Bagdany, M.; Mulvihill, C.M.; Du, K.; di Bernardo, S.; Liu, Y.; Konermann, L.; et al. Correction of both NBD1 energetics and domain interface is required to restore ΔF508 CFTR folding and function. Cell 2012, 148, 150–163. [Google Scholar] [CrossRef] [Green Version]
- Du, K.; Lukacs, G.L. Cooperative assembly and misfolding of CFTR domains in vivo. Mol. Biol. Cell 2009, 20, 1903–1915. [Google Scholar] [CrossRef] [Green Version]
- Brodsky, J.L. The protective and destructive roles played by molecular chaperones during ERAD (endoplasmic-reticulum-associated degradation). Biochem. J. 2007, 404, 353–363. [Google Scholar] [CrossRef]
- Loo, M.A.; Jensen, T.J.; Cui, L.; Hou, Y.; Chang, X.B.; Riordan, J.R. Perturbation of Hsp90 interaction with nascent CFTR prevents its maturation and accelerates its degradation by the proteasome. EMBO J. 1998, 17, 6879–6887. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meacham, G.C.; Lu, Z.; King, S.; Sorscher, E.; Tousson, A.; Cyr, D.M. The Hdj-2/Hsc70 chaperone pair facilitates early steps in CFTR biogenesis. EMBO J. 1999, 18, 1492–1505. [Google Scholar] [CrossRef] [PubMed]
- Scott-Ward, T.S.; Amaral, M.D. Deletion of Phe508 in the first nucleotide-binding domain of the cystic fibrosis transmembrane conductance regulator increases its affinity for the heat shock cognate 70 chaperone. FEBS J. 2009, 276, 7097–7109. [Google Scholar] [CrossRef] [PubMed]
- Bagdany, M.; Veit, G.; Fukuda, R.; Avramescu, R.G.; Okiyoneda, T.; Baaklini, I.; Singh, J.; Sovak, G.; Xu, H.; Apaja, P.M.; et al. Chaperones rescue the energetic landscape of mutant CFTR at single molecule and in cell. Nat. Commun. 2017, 8, 398. [Google Scholar] [CrossRef]
- Matsumura, Y.; David, L.L.; Skach, W.R. Role of Hsc70 binding cycle in CFTR folding and endoplasmic reticulum-associated degradation. Mol. Biol. Cell 2011, 22, 2797–2809. [Google Scholar] [CrossRef]
- Marozkina, N.V.; Yemen, S.; Borowitz, M.; Liu, L.; Plapp, M.; Sun, F.; Islam, R.; Erdmann-Gilmore, P.; Townsend, R.R.; Lichti, C.F.; et al. Hsp 70/Hsp 90 organizing protein as a nitrosylation target in cystic fibrosis therapy. Proc. Natl. Acad. Sci. USA 2010, 107, 11393–11398. [Google Scholar] [CrossRef] [Green Version]
- Zaman, K.; Sawczak, V.; Zaidi, A.; Butler, M.; Bennett, D.; Getsy, P.; Zeinomar, M.; Greenberg, Z.; Forbes, M.; Rehman, S.; et al. Augmentation of CFTR maturation by S-nitrosoglutathione reductase. Am. J. Physiol. Lung Cell Mol. Physiol. 2016, 310, L263–L270. [Google Scholar] [CrossRef] [Green Version]
- Kabani, M.; McLellan, C.; Raynes, D.A.; Guerriero, V.; Brodsky, J.L. HspBP1, a homologue of the yeast Fes1 and Sls1 proteins, is an Hsc70 nucleotide exchange factor. FEBS Lett. 2002, 531, 339–342. [Google Scholar] [CrossRef] [Green Version]
- Alberti, S.; Bohse, K.; Arndt, V.; Schmitz, A.; Hohfeld, J. The cochaperone HspBP1 inhibits the CHIP ubiquitin ligase and stimulates the maturation of the cystic fibrosis transmembrane conductance regulator. Mol. Biol. Cell 2004, 15, 4003–4010. [Google Scholar] [CrossRef]
- Strickland, E.; Qu, B.H.; Millen, L.; Thomas, P.J. The molecular chaperone Hsc70 assists the in vitro folding of the N-terminal nucleotide-binding domain of the cystic fibrosis transmembrane conductance regulator. J. Biol. Chem. 1997, 272, 25421–25424. [Google Scholar] [CrossRef] [Green Version]
- Farinha, C.M.; Nogueira, P.; Mendes, F.; Penque, D.; Amaral, M.D. The human DnaJ homologue (Hdj)-1/heat-shock protein (Hsp) 40 co-chaperone is required for the in vivo stabilization of the cystic fibrosis transmembrane conductance regulator by Hsp70. Biochem. J. 2002, 366, 797–806. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Younger, J.M.; Ren, H.Y.; Chen, L.; Fan, C.Y.; Fields, A.; Patterson, C.; Cyr, D.M. A foldable CFTR ΔF508 biogenic intermediate accumulates upon inhibition of the Hsc70-CHIP E3 ubiquitin ligase. J. Cell Biol. 2004, 167, 1075–1085. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, H.; Peters, K.W.; Sun, F.; Marino, C.R.; Lang, J.; Burgoyne, R.D.; Frizzell, R.A. Cysteine string protein interacts with and modulates the maturation of the cystic fibrosis transmembrane conductance regulator. J. Biol. Chem. 2002, 277, 28948–28958. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, H.; Schmidt, B.Z.; Sun, F.; Condliffe, S.B.; Butterworth, M.B.; Youker, R.T.; Brodsky, J.L.; Aridor, M.; Frizzell, R.A. Cysteine string protein monitors late steps in cystic fibrosis transmembrane conductance regulator biogenesis. J. Biol. Chem. 2006, 281, 11312–11321. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schmidt, B.Z.; Watts, R.J.; Aridor, M.; Frizzell, R.A. Cysteine string protein promotes proteasomal degradation of the cystic fibrosis transmembrane conductance regulator (CFTR) by increasing its interaction with the C terminus of Hsp70-interacting protein and promoting CFTR ubiquitylation. J. Biol. Chem. 2009, 284, 4168–4178. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yamamoto, Y.H.; Kimura, T.; Momohara, S.; Takeuchi, M.; Tani, T.; Kimata, Y.; Kadokura, H.; Kohno, K. A novel ER J-protein DNAJB12 accelerates ER-associated degradation of membrane proteins including CFTR. Cell Struct. Funct. 2010, 35, 107–116. [Google Scholar] [CrossRef] [Green Version]
- Grove, D.E.; Fan, C.Y.; Ren, H.Y.; Cyr, D.M. The endoplasmic reticulum-associated Hsp40 DNAJB12 and Hsc70 cooperate to facilitate RMA1 E3-dependent degradation of nascent CFTR ΔF508. Mol. Biol. Cell 2011, 22, 301–314. [Google Scholar] [CrossRef]
- Youker, R.T.; Walsh, P.; Beilharz, T.; Lithgow, T.; Brodsky, J.L. Distinct roles for the Hsp40 and Hsp90 molecular chaperones during cystic fibrosis transmembrane conductance regulator degradation in yeast. Mol. Biol. Cell 2004, 15, 4787–4797. [Google Scholar] [CrossRef] [Green Version]
- Yang, Y.; Janich, S.; Cohn, J.A.; Wilson, J.M. The common variant of cystic fibrosis transmembrane conductance regulator is recognized by hsp70 and degraded in a pre-Golgi nonlysosomal compartment. Proc. Natl. Acad. Sci. USA 1993, 90, 9480–9484. [Google Scholar] [CrossRef] [Green Version]
- Mogk, A.; Ruger-Herreros, C.; Bukau, B. Cellular Functions and Mechanisms of Action of Small Heat Shock Proteins. Annu. Rev. Microbiol. 2019, 73, 89–110. [Google Scholar] [CrossRef]
- Ahner, A.; Nakatsukasa, K.; Zhang, H.; Frizzell, R.A.; Brodsky, J.L. Small heat-shock proteins select deltaF508-CFTR for endoplasmic reticulum-associated degradation. Mol. Biol. Cell 2007, 18, 806–814. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ahner, A.; Gong, X.; Schmidt, B.Z.; Peters, K.W.; Rabeh, W.M.; Thibodeau, P.H.; Lukacs, G.L.; Frizzell, R.A. Small heat shock proteins target mutant cystic fibrosis transmembrane conductance regulator for degradation via a small ubiquitin-like modifier-dependent pathway. Mol. Biol. Cell 2013, 24, 74–84. [Google Scholar] [CrossRef] [PubMed]
- Gong, X.; Ahner, A.; Roldan, A.; Lukacs, G.L.; Thibodeau, P.H.; Frizzell, R.A. Non-native Conformers of Cystic Fibrosis Transmembrane Conductance Regulator NBD1 Are Recognized by Hsp27 and Conjugated to SUMO-2 for Degradation. J. Biol. Chem. 2016, 291, 2004–2017. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, X.; Venable, J.; LaPointe, P.; Hutt, D.M.; Koulov, A.V.; Coppinger, J.; Gurkan, C.; Kellner, W.; Matteson, J.; Plutner, H.; et al. Hsp90 cochaperone Aha1 downregulation rescues misfolding of CFTR in cystic fibrosis. Cell 2006, 127, 803–815. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koulov, A.V.; LaPointe, P.; Lu, B.; Razvi, A.; Coppinger, J.; Dong, M.Q.; Matteson, J.; Laister, R.; Arrowsmith, C.; Yates, J.R.; et al. Biological and structural basis for Aha1 regulation of Hsp90 ATPase activity in maintaining proteostasis in the human disease cystic fibrosis. Mol. Biol. Cell 2010, 21, 871–884. [Google Scholar] [CrossRef]
- Hutt, D.M.; Roth, D.M.; Chalfant, M.A.; Youker, R.T.; Matteson, J.; Brodsky, J.L.; Balch, W.E. FK506 binding protein 8 peptidylprolyl isomerase activity manages a late stage of cystic fibrosis transmembrane conductance regulator (CFTR) folding and stability. J. Biol. Chem. 2012, 287, 21914–21925. [Google Scholar] [CrossRef] [Green Version]
- Bracher, A.; Verghese, J. The nucleotide exchange factors of Hsp70 molecular chaperones. Front. Mol. Biosci. 2015, 2, 10. [Google Scholar] [CrossRef]
- Saxena, A.; Banasavadi-Siddegowda, Y.K.; Fan, Y.; Bhattacharya, S.; Roy, G.; Giovannucci, D.R.; Frizzell, R.A.; Wang, X. Human heat shock protein 105/110 kDa (Hsp105/110) regulates biogenesis and quality control of misfolded cystic fibrosis transmembrane conductance regulator at multiple levels. J. Biol. Chem. 2012, 287, 19158–19170. [Google Scholar] [CrossRef] [Green Version]
- Denning, G.M.; Anderson, M.P.; Amara, J.F.; Marshall, J.; Smith, A.E.; Welsh, M.J. Processing of mutant cystic fibrosis transmembrane conductance regulator is temperature-sensitive. Nature 1992, 358, 761–764. [Google Scholar] [CrossRef]
- Pankow, S.; Bamberger, C.; Calzolari, D.; Martinez-Bartolome, S.; Lavallee-Adam, M.; Balch, W.E.; Yates III, J.R. F508 CFTR interactome remodelling promotes rescue of cystic fibrosis. Nature 2015, 528, 510–516. [Google Scholar] [CrossRef] [Green Version]
- Bebök, Z.; Mazzochi, C.; King, S.A.; Hong, J.S.; Sorscher, E.J. The mechanism underlying cystic fibrosis transmembrane conductance regulator transport from the endoplasmic reticulum to the proteasome includes Sec61beta and a cytosolic, deglycosylated intermediary. J. Biol. Chem. 1998, 273, 29873–29878. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oberdorf, J.; Pitonzo, D.; Skach, W.R. An energy-dependent maturation step is required for release of the cystic fibrosis transmembrane conductance regulator from early endoplasmic reticulum biosynthetic machinery. J. Biol. Chem. 2005, 280, 38193–38202. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pitonzo, D.; Yang, Z.; Matsumura, Y.; Johnson, A.E.; Skach, W.R. Sequence-specific retention and regulated integration of a nascent membrane protein by the endoplasmic reticulum Sec61 translocon. Mol. Biol. Cell 2009, 20, 685–698. [Google Scholar] [CrossRef] [PubMed]
- Rapoport, T.A.; Li, L.; Park, E. Structural and Mechanistic Insights into Protein Translocation. Annu. Rev. Cell Dev. Biol. 2017, 33, 369–390. [Google Scholar] [CrossRef]
- Cheng, S.H.; Gregory, R.J.; Marshall, J.; Paul, S.; Souza, D.W.; White, G.A.; O’Riordan, C.R.; Smith, A.E. Defective intracellular transport and processing of CFTR is the molecular basis of most cystic fibrosis. Cell 1990, 63, 827–834. [Google Scholar] [CrossRef]
- Pind, S.; Riordan, J.R.; Williams, D.B. Participation of the endoplasmic reticulum chaperone calnexin (p88, IP90) in the biogenesis of the cystic fibrosis transmembrane conductance regulator. J. Biol. Chem. 1994, 269, 12784–12788. [Google Scholar]
- Harada, K.; Okiyoneda, T.; Hashimoto, Y.; Ueno, K.; Nakamura, K.; Yamahira, K.; Sugahara, T.; Shuto, T.; Wada, I.; Suico, M.A.; et al. Calreticulin negatively regulates the cell surface expression of cystic fibrosis transmembrane conductance regulator. J. Biol. Chem. 2006, 281, 12841–12848. [Google Scholar] [CrossRef] [Green Version]
- Rosser, M.F.; Grove, D.E.; Chen, L.; Cyr, D.M. Assembly and misassembly of cystic fibrosis transmembrane conductance regulator: Folding defects caused by deletion of F508 occur before and after the calnexin-dependent association of membrane spanning domain (MSD) 1 and MSD2. Mol. Biol. Cell 2008, 19, 4570–4579. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Egan, M.E.; Pearson, M.; Weiner, S.A.; Rajendran, V.; Rubin, D.; Glockner-Pagel, J.; Canny, S.; Du, K.; Lukacs, G.L.; Caplan, M.J. Curcumin, a major constituent of turmeric, corrects cystic fibrosis defects. Science 2004, 304, 600–602. [Google Scholar] [CrossRef] [Green Version]
- Okiyoneda, T.; Niibori, A.; Harada, K.; Kohno, T.; Michalak, M.; Duszyk, M.; Wada, I.; Ikawa, M.; Shuto, T.; Suico, M.A.; et al. Role of calnexin in the ER quality control and productive folding of CFTR; differential effect of calnexin knockout on wild-type and ΔF508 CFTR. Biochim. Biophys. Acta 2008, 1783, 1585–1594. [Google Scholar] [CrossRef] [Green Version]
- Farinha, C.M.; Amaral, M.D. Most F508del-CFTR is targeted to degradation at an early folding checkpoint and independently of calnexin. Mol. Cell. Biol. 2005, 25, 5242–5252. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bebök, Z.; Collawn, J.F.; Wakefield, J.; Parker, W.; Li, Y.; Varga, K.; Sorscher, E.J.; Clancy, J.P. Failure of cAMP agonists to activate rescued deltaF508 CFTR in CFBE41o- airway epithelial monolayers. J. Physiol. 2005, 569, 601–615. [Google Scholar] [CrossRef]
- Ostedgaard, L.S.; Rogers, C.S.; Dong, Q.; Randak, C.O.; Vermeer, D.W.; Rokhlina, T.; Karp, P.H.; Welsh, M.J. Processing and function of CFTR-ΔF508 are species-dependent. Proc. Natl. Acad. Sci. USA 2007, 104, 15370–15375. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Varga, K.; Jurkuvenaite, A.; Wakefield, J.; Hong, J.S.; Guimbellot, J.S.; Venglarik, C.J.; Niraj, A.; Mazur, M.; Sorscher, E.J.; Collawn, J.F.; et al. Efficient intracellular processing of the endogenous cystic fibrosis transmembrane conductance regulator in epithelial cell lines. J. Biol. Chem. 2004, 279, 22578–22584. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lukacs, G.L.; Mohamed, A.; Kartner, N.; Chang, X.B.; Riordan, J.R.; Grinstein, S. Conformational maturation of CFTR but not its mutant counterpart (Δ F508) occurs in the endoplasmic reticulum and requires ATP. EMBO J. 1994, 13, 6076–6086. [Google Scholar] [CrossRef]
- Xiong, X.; Chong, E.; Skach, W.R. Evidence that endoplasmic reticulum (ER)-associated degradation of cystic fibrosis transmembrane conductance regulator is linked to retrograde translocation from the ER membrane. J. Biol. Chem. 1999, 274, 2616–2624. [Google Scholar] [CrossRef] [Green Version]
- Sun, F.; Zhang, R.; Gong, X.; Geng, X.; Drain, P.F.; Frizzell, R.A. Derlin-1 promotes the efficient degradation of the cystic fibrosis transmembrane conductance regulator (CFTR) and CFTR folding mutants. J. Biol. Chem. 2006, 281, 36856–36863. [Google Scholar] [CrossRef] [Green Version]
- Wang, B.; Heath-Engel, H.; Zhang, D.; Nguyen, N.; Thomas, D.Y.; Hanrahan, J.W.; Shore, G.C. BAP31 interacts with Sec61 translocons and promotes retrotranslocation of CFTR ΔF508 via the derlin-1 complex. Cell 2008, 133, 1080–1092. [Google Scholar] [CrossRef] [Green Version]
- Baldridge, R.D.; Rapoport, T.A. Autoubiquitination of the Hrd1 Ligase Triggers Protein Retrotranslocation in ERAD. Cell 2016, 166, 394–407. [Google Scholar] [CrossRef] [Green Version]
- Ballar, P.; Ors, A.U.; Yang, H.; Fang, S. Differential regulation of CFTR ΔF508 degradation by ubiquitin ligases gp78 and Hrd1. Int. J. Biochem. Cell Biol. 2010, 42, 167–173. [Google Scholar] [CrossRef]
- Oberdorf, J.; Carlson, E.J.; Skach, W.R. Redundancy of mammalian proteasome beta subunit function during endoplasmic reticulum associated degradation. Biochemistry 2001, 40, 13397–13405. [Google Scholar] [CrossRef] [PubMed]
- Varshavsky, A. The ubiquitin system, an immense realm. Annu. Rev. Biochem. 2012, 81, 167–176. [Google Scholar] [CrossRef] [PubMed]
- Preston, G.M.; Brodsky, J.L. The evolving role of ubiquitin modification in endoplasmic reticulum-associated degradation. Biochem. J. 2017, 474, 445–469. [Google Scholar] [CrossRef] [Green Version]
- Komander, D.; Rape, M. The Ubiquitin Code. Annu. Rev. Biochem. 2012, 81, 203–229. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leto, D.E.; Morgens, D.W.; Zhang, L.; Walczak, C.P.; Elias, J.E.; Bassik, M.C.; Kopito, R.R. Genome-wide CRISPR Analysis Identifies Substrate-Specific Conjugation Modules in ER-Associated Degradation. Mol. Cell 2019, 73, 377–389. [Google Scholar] [CrossRef] [Green Version]
- Lee, S.; Henderson, M.J.; Schiffhauer, E.; Despanie, J.; Henry, K.; Kang, P.W.; Walker, D.; McClure, M.L.; Wilson, L.; Sorscher, E.J.; et al. Interference with ubiquitination in CFTR modifies stability of core glycosylated and cell surface pools. Mol. Cell Biol. 2014, 34, 2554–2565. [Google Scholar] [CrossRef] [Green Version]
- Li, W.; Bengtson, M.H.; Ulbrich, A.; Matsuda, A.; Reddy, V.A.; Orth, A.; Chanda, S.K.; Batalov, S.; Joazeiro, C.A. Genome-wide and functional annotation of human E3 ubiquitin ligases identifies MULAN, a mitochondrial E3 that regulates the organelle’s dynamics and signaling. PLoS ONE 2008, 3, e1487. [Google Scholar] [CrossRef]
- Sato, S.; Ward, C.L.; Kopito, R.R. Cotranslational ubiquitination of cystic fibrosis transmembrane conductance regulator in vitro. J. Biol. Chem. 1998, 273, 7189–7192. [Google Scholar] [CrossRef] [Green Version]
- El Khouri, E.; Le Pavec, G.; Toledano, M.B.; Delaunay-Moisan, A. RNF185 is a novel E3 ligase of endoplasmic reticulum-associated degradation (ERAD) that targets cystic fibrosis transmembrane conductance regulator (CFTR). J. Biol. Chem. 2013, 288, 31177–31191. [Google Scholar] [CrossRef] [Green Version]
- Wahlman, J.; DeMartino, G.N.; Skach, W.R.; Bulleid, N.J.; Brodsky, J.L.; Johnson, A.E. Real-time fluorescence detection of ERAD substrate retrotranslocation in a mammalian in vitro system. Cell 2007, 129, 943–955. [Google Scholar] [CrossRef]
- Mehnert, M.; Sommer, T.; Jarosch, E. Der1 promotes movement of misfolded proteins through the endoplasmic reticulum membrane. Nat. Cell Biol. 2014, 16, 77–86. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mehnert, M.; Sommermeyer, F.; Berger, M.; Kumar Lakshmipathy, S.; Gauss, R.; Aebi, M.; Jarosch, E.; Sommer, T. The interplay of Hrd3 and the molecular chaperone system ensures efficient degradation of malfolded secretory proteins. Mol. Biol. Cell 2015, 26, 185–194. [Google Scholar] [CrossRef] [PubMed]
- Claessen, J.H.; Mueller, B.; Spooner, E.; Pivorunas, V.L.; Ploegh, H.L. The transmembrane segment of a tail-anchored protein determines its degradative fate through dislocation from the endoplasmic reticulum. J. Biol. Chem. 2010, 285, 20732–20739. [Google Scholar] [CrossRef] [Green Version]
- Carvalho, P.; Stanley, A.M.; Rapoport, T.A. Retrotranslocation of a misfolded luminal ER protein by the ubiquitin-ligase Hrd1p. Cell 2010, 143, 579–591. [Google Scholar] [CrossRef] [Green Version]
- Tomati, V.; Sondo, E.; Armirotti, A.; Caci, E.; Pesce, E.; Marini, M.; Gianotti, A.; Ju Jeon, Y.; Cilli, M.; Pistorio, A.; et al. Genetic Inhibition of The Ubiquitin Ligase Rnf5 Attenuates Phenotypes Associated to F508del Cystic Fibrosis Mutation. Sci. Rep. 2015, 5, 12138. [Google Scholar] [CrossRef] [PubMed]
- Morito, D.; Hirao, K.; Oda, Y.; Hosokawa, N.; Tokunaga, F.; Cyr, D.M.; Tanaka, K.; Iwai, K.; Nagata, K. Gp78 cooperates with RMA1 in endoplasmic reticulum-associated degradation of CFTR ΔF508. Mol. Biol. Cell 2008, 19, 1328–1336. [Google Scholar] [CrossRef] [Green Version]
- Ballinger, C.A.; Connell, P.; Wu, Y.; Hu, Z.; Thompson, L.J.; Yin, L.Y.; Patterson, C. Identification of CHIP, a novel tetratricopeptide repeat-containing protein that interacts with heat shock proteins and negatively regulates chaperone functions. Mol. Cell Biol. 1999, 19, 4535–4545. [Google Scholar] [CrossRef] [Green Version]
- Meacham, G.C.; Patterson, C.; Zhang, W.; Younger, J.M.; Cyr, D.M. The Hsc70 co-chaperone CHIP targets immature CFTR for proteasomal degradation. Nat. Cell Biol. 2001, 3, 100–105. [Google Scholar] [CrossRef]
- Tannous, A.; Pisoni, G.B.; Hebert, D.N.; Molinari, M. N-linked sugar-regulated protein folding and quality control in the ER. Semin. Cell Dev. Biol. 2015, 41, 79–89. [Google Scholar] [CrossRef] [Green Version]
- Chang, X.B.; Cui, L.; Hou, Y.X.; Jensen, T.J.; Aleksandrov, A.A.; Mengos, A.; Riordan, J.R. Removal of multiple arginine-framed trafficking signals overcomes misprocessing of delta F508 CFTR present in most patients with cystic fibrosis. Mol. Cell 1999, 4, 137–142. [Google Scholar] [CrossRef]
- Yoo, J.S.; Moyer, B.D.; Bannykh, S.; Yoo, H.M.; Riordan, J.R.; Balch, W.E. Non-conventional trafficking of the cystic fibrosis transmembrane conductance regulator through the early secretory pathway. J. Biol. Chem. 2002, 277, 11401–11409. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, X.; Matteson, J.; An, Y.; Moyer, B.; Yoo, J.S.; Bannykh, S.; Wilson, I.A.; Riordan, J.R.; Balch, W.E. COPII-dependent export of cystic fibrosis transmembrane conductance regulator from the ER uses a di-acidic exit code. J. Cell Biol. 2004, 167, 65–74. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fromme, J.C.; Orci, L.; Schekman, R. Coordination of COPII vesicle trafficking by Sec23. Trends Cell Biol. 2008, 18, 330–336. [Google Scholar] [CrossRef] [PubMed]
- Chang, X.B.; Hou, Y.X.; Jensen, T.J.; Riordan, J.R. Mapping of cystic fibrosis transmembrane conductance regulator membrane topology by glycosylation site insertion. J. Biol. Chem. 1994, 269, 18572–18575. [Google Scholar] [PubMed]
- Mendoza, J.L.; Schmidt, A.; Li, Q.; Nuvaga, E.; Barrett, T.; Bridges, R.J.; Feranchak, A.P.; Brautigam, C.A.; Thomas, P.J. Requirements for efficient correction of ΔF508 CFTR revealed by analyses of evolved sequences. Cell 2012, 148, 164–174. [Google Scholar] [CrossRef] [Green Version]
- Farhan, H.; Weiss, M.; Tani, K.; Kaufman, R.J.; Hauri, H.P. Adaptation of endoplasmic reticulum exit sites to acute and chronic increases in cargo load. EMBO J. 2008, 27, 2043–2054. [Google Scholar] [CrossRef] [Green Version]
- Piao, H.; Kim, J.; Noh, S.H.; Kweon, H.S.; Kim, J.Y.; Lee, M.G. Sec16A is critical for both conventional and unconventional secretion of CFTR. Sci. Rep. 2017, 7, 39887. [Google Scholar] [CrossRef]
- Kim, J.; Noh, S.H.; Piao, H.; Kim, D.H.; Kim, K.; Cha, J.S.; Chung, W.Y.; Cho, H.S.; Kim, J.Y.; Lee, M.G. Monomerization and ER Relocalization of GRASP Is a Requisite for Unconventional Secretion of CFTR. Traffic 2016, 17, 733–753. [Google Scholar] [CrossRef] [Green Version]
- Gee, H.Y.; Noh, S.H.; Tang, B.L.; Kim, K.H.; Lee, M.G. Rescue of ΔF508-CFTR trafficking via a GRASP-dependent unconventional secretion pathway. Cell 2011, 146, 746–760. [Google Scholar] [CrossRef] [Green Version]
- Collawn, J.F.; Stangel, M.; Kuhn, L.A.; Esekogwu, V.; Jing, S.Q.; Trowbridge, I.S.; Tainer, J.A. Transferrin receptor internalization sequence YXRF implicates a tight turn as the structural recognition motif for endocytosis. Cell 1990, 63, 1061–1072. [Google Scholar] [CrossRef]
- Weixel, K.; Bradbury, N.A. Analysis of CFTR endocytosis by cell surface biotinylation. Methods Mol. Med. 2002, 70, 323–340. [Google Scholar] [PubMed]
- Peter, K.; Varga, K.; Bebök, Z.; McNicholas-Bevensee, C.M.; Schwiebert, L.; Sorscher, E.J.; Schwiebert, E.M.; Collawn, J.F. Ablation of internalization signals in the carboxyl-terminal tail of the cystic fibrosis transmembrane conductance regulator enhances cell surface expression. J. Biol. Chem. 2002, 277, 49952–49957. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bradbury, N.A.; Cohn, J.A.; Venglarik, C.J.; Bridges, R.J. Biochemical and biophysical identification of cystic fibrosis transmembrane conductance regulator chloride channels as components of endocytic clathrin-coated vesicles. J. Biol. Chem. 1994, 269, 8296–8302. [Google Scholar] [PubMed]
- Weixel, K.M.; Bradbury, N.A. The carboxyl terminus of the cystic fibrosis transmembrane conductance regulator binds to AP-2 clathrin adaptors. J. Biol. Chem. 2000, 275, 3655–3660. [Google Scholar] [CrossRef] [Green Version]
- Kumari, V.; Desai, S.; Ameen, N.A. AP2 alpha modulates cystic fibrosis transmembrane conductance regulator function in the human intestine. J. Cyst. Fibros. 2017, 16, 327–334. [Google Scholar] [CrossRef]
- Swiatecka-Urban, A.; Boyd, C.; Coutermarsh, B.; Karlson, K.H.; Barnaby, R.; Aschenbrenner, L.; Langford, G.M.; Hasson, T.; Stanton, B.A. Myosin VI regulates endocytosis of the cystic fibrosis transmembrane conductance regulator. J. Biol. Chem. 2004, 279, 38025–38031. [Google Scholar] [CrossRef] [Green Version]
- Ameen, N.; Apodaca, G. Defective CFTR apical endocytosis and enterocyte brush border in myosin VI-deficient mice. Traffic 2007, 8, 998–1006. [Google Scholar] [CrossRef]
- Fu, L.; Rab, A.; Tang, L.P.; Rowe, S.M.; Bebök, Z.; Collawn, J.F. Dab2 is a key regulator of endocytosis and post-endocytic trafficking of the cystic fibrosis transmembrane conductance regulator. Biochem. J. 2012, 441, 633–643. [Google Scholar] [CrossRef] [Green Version]
- Fu, L.; Rab, A.; Tang, L.; Bebök, Z.; Rowe, S.M.; Bartoszewski, R.; Collawn, J.F. ΔF508 CFTR surface stability is regulated by DAB2 and CHIP-mediated ubiquitination in post-endocytic compartments. PLoS ONE 2015, 10, e0123131. [Google Scholar] [CrossRef] [Green Version]
- Gentzsch, M.; Chang, X.B.; Cui, L.; Wu, Y.; Ozols, V.V.; Choudhury, A.; Pagano, R.E.; Riordan, J.R. Endocytic trafficking routes of wild type and ΔF508 cystic fibrosis transmembrane conductance regulator. Mol. Biol. Cell 2004, 15, 2684–2696. [Google Scholar] [CrossRef]
- Okiyoneda, T.; Barriere, H.; Bagdany, M.; Rabeh, W.M.; Du, K.; Hohfeld, J.; Young, J.C.; Lukacs, G.L. Peripheral protein quality control removes unfolded CFTR from the plasma membrane. Science 2010, 329, 805–810. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eckford, P.D.; Ramjeesingh, M.; Molinski, S.; Pasyk, S.; Dekkers, J.F.; Li, C.; Ahmadi, S.; Ip, W.; Chung, T.E.; Du, K.; et al. VX-809 and related corrector compounds exhibit secondary activity stabilizing active F508del-CFTR after its partial rescue to the cell surface. Chem. Biol. 2014, 21, 666–678. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Okiyoneda, T.; Veit, G.; Sakai, R.; Aki, M.; Fujihara, T.; Higashi, M.; Susuki-Miyata, S.; Miyata, M.; Fukuda, N.; Yoshida, A.; et al. Chaperone-Independent Peripheral Quality Control of CFTR by RFFL E3 Ligase. Dev. Cell 2018, 44, 694–708. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Loureiro, C.A.; Matos, A.M.; Dias-Alves, A.; Pereira, J.F.; Uliyakina, I.; Barros, P.; Amaral, M.D.; Matos, P. A molecular switch in the scaffold NHERF1 enables misfolded CFTR to evade the peripheral quality control checkpoint. Sci. Signal 2015, 8, ra48. [Google Scholar] [CrossRef] [PubMed]
- Haggie, P.M.; Kim, J.K.; Lukacs, G.L.; Verkman, A.S. Tracking of quantum dot-labeled CFTR shows near immobilization by C-terminal PDZ interactions. Mol. Biol. Cell 2006, 17, 4937–4945. [Google Scholar] [CrossRef] [Green Version]
- Valentine, C.D.; Lukacs, G.L.; Verkman, A.S.; Haggie, P.M. Reduced PDZ interactions of rescued ΔF508CFTR increases its cell surface mobility. J. Biol. Chem. 2012, 287, 43630–43638. [Google Scholar] [CrossRef] [Green Version]
- Cheng, J.; Wang, H.; Guggino, W.B. Modulation of mature cystic fibrosis transmembrane regulator protein by the PDZ domain protein CAL. J. Biol. Chem. 2004, 279, 1892–1898. [Google Scholar] [CrossRef] [Green Version]
- Cheng, J.; Cebotaru, V.; Cebotaru, L.; Guggino, W.B. Syntaxin 6 and CAL mediate the degradation of the cystic fibrosis transmembrane conductance regulator. Mol. Biol. Cell 2010, 21, 1178–1187. [Google Scholar] [CrossRef]
- Cheng, J.; Guggino, W. Ubiquitination and degradation of CFTR by the E3 ubiquitin ligase MARCH2 through its association with adaptor proteins CAL and STX6. PLoS ONE 2013, 8, e68001. [Google Scholar] [CrossRef] [Green Version]
- Bergbower, E.; Boinot, C.; Sabirzhanova, I.; Guggino, W.; Cebotaru, L. The CFTR-Associated Ligand Arrests the Trafficking of the Mutant ΔF508 CFTR Channel in the ER Contributing to Cystic Fibrosis. Cell Physiol. Biochem. 2018, 45, 639–655. [Google Scholar] [CrossRef]
- Wolde, M.; Fellows, A.; Cheng, J.; Kivenson, A.; Coutermarsh, B.; Talebian, L.; Karlson, K.; Piserchio, A.; Mierke, D.F.; Stanton, B.A.; et al. Targeting CAL as a negative regulator of ΔF508-CFTR cell-surface expression: An RNA interference and structure-based mutagenetic approach. J. Biol. Chem. 2007, 282, 8099–8109. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, Y.; Cushing, P.R.; Smithson, D.C.; Pellegrini, M.; Pletnev, A.A.; Al-Ayyoubi, S.; Grassetti, A.V.; Gerber, S.A.; Guy, R.K.; Madden, D.R. Cysteine modifiers suggest an allosteric inhibitory site on the CAL PDZ domain. Biosci. Rep. 2018, 38, BSR20180231. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lopes-Pacheco, M.; Boinot, C.; Sabirzhanova, I.; Rapino, D.; Cebotaru, L. Combination of Correctors Rescues CFTR Transmembrane-Domain Mutants by Mitigating their Interactions with Proteostasis. Cell Physiol. Biochem. 2017, 41, 2194–2210. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brown, C.R.; Hong-Brown, L.Q.; Biwersi, J.; Verkman, A.S.; Welch, W.J. Chemical chaperones correct the mutant phenotype of the delta F508 cystic fibrosis transmembrane conductance regulator protein. Cell Stress Chaperones 1996, 1, 117–125. [Google Scholar] [CrossRef]
- Brown, C.R.; Hong-Brown, L.Q.; Welch, W.J. Correcting temperature-sensitive protein folding defects. J. Clin. Investig. 1997, 99, 1432–1444. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sato, S.; Ward, C.L.; Krouse, M.E.; Wine, J.J.; Kopito, R.R. Glycerol reverses the misfolding phenotype of the most common cystic fibrosis mutation. J. Biol. Chem. 1996, 271, 635–638. [Google Scholar] [CrossRef] [Green Version]
- Hanrahan, J.W.; Sato, Y.; Carlile, G.W.; Jansen, G.; Young, J.C.; Thomas, D.Y. Cystic Fibrosis: Proteostatic correctors of CFTR trafficking and alternative therapeutic targets. Expert Opin. Targets 2019, 23, 711–724. [Google Scholar] [CrossRef]
- Rubenstein, R.C.; Egan, M.E.; Zeitlin, P.L. In vitro pharmacologic restoration of CFTR-mediated chloride transport with sodium 4-phenylbutyrate in cystic fibrosis epithelial cells containing delta F508-CFTR. J. Clin. Investig. 1997, 100, 2457–2465. [Google Scholar] [CrossRef] [Green Version]
- Rubenstein, R.C.; Zeitlin, P.L. Sodium 4-phenylbutyrate downregulates Hsc70: Implications for intracellular trafficking of ΔF508-CFTR. Am. J. Physiol. Cell Physiol. 2000, 278, C259–C267. [Google Scholar] [CrossRef]
- Rubenstein, R.C.; Zeitlin, P.L. A pilot clinical trial of oral sodium 4-phenylbutyrate (Buphenyl) in deltaF508-homozygous cystic fibrosis patients: Partial restoration of nasal epithelial CFTR function. Am. J. Respir. Crit. Care Med. 1998, 157, 484–490. [Google Scholar] [CrossRef]
- Zeitlin, P.L.; Diener-West, M.; Rubenstein, R.C.; Boyle, M.P.; Lee, C.K.; Brass-Ernst, L. Evidence of CFTR function in cystic fibrosis after systemic administration of 4-phenylbutyrate. Mol. Ther. 2002, 6, 119–126. [Google Scholar] [CrossRef] [PubMed]
- Ma, T.; Vetrivel, L.; Yang, H.; Pedemonte, N.; Zegarra-Moran, O.; Galietta, L.J.; Verkman, A.S. High-affinity activators of cystic fibrosis transmembrane conductance regulator (CFTR) chloride conductance identified by high-throughput screening. J. Biol. Chem. 2002, 277, 37235–37241. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, H.; Shelat, A.A.; Guy, R.K.; Gopinath, V.S.; Ma, T.; Du, K.; Lukacs, G.L.; Taddei, A.; Folli, C.; Pedemonte, N.; et al. Nanomolar affinity small molecule correctors of defective ΔF508-CFTR chloride channel gating. J. Biol. Chem. 2003, 278, 35079–35085. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pedemonte, N.; Lukacs, G.L.; Du, K.; Caci, E.; Zegarra-Moran, O.; Galietta, L.J.; Verkman, A.S. Small-molecule correctors of defective ΔF508-CFTR cellular processing identified by high-throughput screening. J. Clin. Investig. 2005, 115, 2564–2571. [Google Scholar] [CrossRef]
- Van Goor, F.; Straley, K.S.; Cao, D.; Gonzalez, J.; Hadida, S.; Hazlewood, A.; Joubran, J.; Knapp, T.; Makings, L.R.; Miller, M.; et al. Rescue of ΔF508-CFTR trafficking and gating in human cystic fibrosis airway primary cultures by small molecules. Am. J. Physiol. Lung Cell Mol. Physiol. 2006, 290, L1117–L1130. [Google Scholar] [CrossRef]
- Van Goor, F.; Hadida, S.; Grootenhuis, P.D.; Burton, B.; Stack, J.H.; Straley, K.S.; Decker, C.J.; Miller, M.; McCartney, J.; Olson, E.R.; et al. Correction of the F508del-CFTR protein processing defect in vitro by the investigational drug VX-809. Proc. Natl. Acad. Sci. USA 2011, 108, 18843–18848. [Google Scholar] [CrossRef] [Green Version]
- Ren, H.Y.; Grove, D.E.; De La Rosa, O.; Houck, S.A.; Sopha, P.; Van Goor, F.; Hoffman, B.J.; Cyr, D.M. VX-809 corrects folding defects in cystic fibrosis transmembrane conductance regulator protein through action on membrane-spanning domain 1. Mol. Biol. Cell 2013, 24, 3016–3024. [Google Scholar] [CrossRef]
- Loo, T.W.; Bartlett, M.C.; Clarke, D.M. Corrector VX-809 stabilizes the first transmembrane domain of CFTR. Biochem. Pharm. 2013, 86, 612–619. [Google Scholar] [CrossRef]
- Farinha, C.M.; King-Underwood, J.; Sousa, M.; Correia, A.R.; Henriques, B.J.; Roxo-Rosa, M.; Da Paula, A.C.; Williams, J.; Hirst, S.; Gomes, C.M.; et al. Revertants, low temperature, and correctors reveal the mechanism of F508del-CFTR rescue by VX-809 and suggest multiple agents for full correction. Chem. Biol. 2013, 20, 943–955. [Google Scholar] [CrossRef] [Green Version]
- Illek, B.; Fischer, H.; Santos, G.F.; Widdicombe, J.H.; Machen, T.E.; Reenstra, W.W. cAMP-independent activation of CFTR Cl channels by the tyrosine kinase inhibitor genistein. Am. J. Physiol. 1995, 268, C886–C893. [Google Scholar] [CrossRef]
- Illek, B.; Fischer, H.; Machen, T.E. Alternate stimulation of apical CFTR by genistein in epithelia. Am. J. Physiol. 1996, 270, C265–C275. [Google Scholar] [CrossRef] [PubMed]
- Wellhauser, L.; Kim Chiaw, P.; Pasyk, S.; Li, C.; Ramjeesingh, M.; Bear, C.E. A small-molecule modulator interacts directly with deltaPhe508-CFTR to modify its ATPase activity and conformational stability. Mol. Pharm. 2009, 75, 1430–1438. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, F.; Zhang, Z.; Levit, A.; Levring, J.; Touhara, K.K.; Shoichet, B.K.; Chen, J. Structural identification of a hotspot on CFTR for potentiation. Science 2019, 364, 1184–1188. [Google Scholar] [CrossRef] [PubMed]
- Kim Chiaw, P.; Eckford, P.D.; Bear, C.E. Insights into the mechanisms underlying CFTR channel activity, the molecular basis for cystic fibrosis and strategies for therapy. Essays Biochem. 2011, 50, 233–248. [Google Scholar]
- Ramsey, B.W.; Davies, J.; McElvaney, N.G.; Tullis, E.; Bell, S.C.; Drevinek, P.; Griese, M.; McKone, E.F.; Wainwright, C.E.; Konstan, M.W.; et al. A CFTR potentiator in patients with cystic fibrosis and the G551D mutation. N. Engl. J. Med. 2011, 365, 1663–1672. [Google Scholar] [CrossRef] [Green Version]
- Van Goor, F.; Yu, H.; Burton, B.; Hoffman, B.J. Effect of ivacaftor on CFTR forms with missense mutations associated with defects in protein processing or function. J. Cyst. Fibros. 2014, 13, 29–36. [Google Scholar] [CrossRef] [Green Version]
- Flume, P.A.; Liou, T.G.; Borowitz, D.S.; Li, H.; Yen, K.; Ordonez, C.L.; Geller, D.E. VX 08-770-104 Study Group, Ivacaftor in subjects with cystic fibrosis who are homozygous for the F508del-CFTR mutation. Chest 2012, 142, 718–724. [Google Scholar] [CrossRef] [Green Version]
- Wainwright, C.E.; Elborn, J.S.; Ramsey, B.W.; Marigowda, G.; Huang, X.; Cipolli, M.; Colombo, C.; Davies, J.C.; De Boeck, K.; Flume, P.A.; et al. Lumacaftor-Ivacaftor in Patients with Cystic Fibrosis Homozygous for Phe508del CFTR. N. Engl. J. Med. 2015, 373, 220–231. [Google Scholar] [CrossRef] [Green Version]
- Veit, G.; Avramescu, R.G.; Perdomo, D.; Phuan, P.W.; Bagdany, M.; Apaja, P.M.; Borot, F.; Szollosi, D.; Wu, Y.S.; Finkbeiner, W.E.; et al. Some gating potentiators, including VX-770, diminish ΔF508-CFTR functional expression. Sci. Transl. Med. 2014, 6, 246ra97. [Google Scholar] [CrossRef] [Green Version]
- Cholon, D.M.; Quinney, N.L.; Fulcher, M.L.; Esther, C.R., Jr.; Das, J.; Dokholyan, N.V.; Randell, S.H.; Boucher, R.C.; Gentzsch, M. Potentiator ivacaftor abrogates pharmacological correction of ΔF508 CFTR in cystic fibrosis. Sci. Transl. Med. 2014, 6, 246ra96. [Google Scholar] [CrossRef] [Green Version]
- Keating, D.; Marigowda, G.; Burr, L.; Daines, C.; Mall, M.A.; McKone, E.F.; Ramsey, B.W.; Rowe, S.M.; Sass, L.A.; Tullis, E.; et al. VX-445-Tezacaftor-Ivacaftor in Patients with Cystic Fibrosis and One or Two Phe508del Alleles. N. Engl. J. Med. 2018, 379, 1612–1620. [Google Scholar] [CrossRef] [PubMed]
- Heijerman, H.G.M.; McKone, E.F.; Downey, D.G.; Van Braeckel, E.; Rowe, S.M.; Tullis, E.; Mall, M.A.; Welter, J.J.; Ramsey, B.W.; McKee, C.M.; et al. Efficacy and safety of the elexacaftor plus tezacaftor plus ivacaftor combination regimen in people with cystic fibrosis homozygous for the F508del mutation: A double-blind, randomised, phase 3 trial. Lancet 2019, 394, 1940–1948. [Google Scholar] [CrossRef]
- Middleton, P.G.; Mall, M.A.; Drevinek, P.; Lands, L.C.; McKone, E.F.; Polineni, D.; Ramsey, B.W.; Taylor-Cousar, J.L.; Tullis, E.; Vermeulen, F.; et al. Elexacaftor-Tezacaftor-Ivacaftor for Cystic Fibrosis with a Single Phe508del Allele. N. Engl. J. Med. 2019, 381, 1809–1819. [Google Scholar] [CrossRef] [PubMed]
- Davies, J.C.; Wainwright, C.E.; Canny, G.J.; Chilvers, M.A.; Howenstine, M.S.; Munck, A.; Mainz, J.G.; Rodriguez, S.; Li, H.; Yen, K.; et al. Efficacy and safety of ivacaftor in patients aged 6 to 11 years with cystic fibrosis with a G551D mutation. Am. J. Respir. Crit. Care Med. 2013, 187, 1219–1225. [Google Scholar] [CrossRef] [Green Version]
- Ratjen, F.; Hug, C.; Marigowda, G.; Tian, S.; Huang, X.; Stanojevic, S.; Milla, C.E.; Robinson, P.D.; Waltz, D.; Davies, J.C.; et al. Efficacy and safety of lumacaftor and ivacaftor in patients aged 6-11 years with cystic fibrosis homozygous for F508del-CFTR: A randomised, placebo-controlled phase 3 trial. Lancet Respir. Med. 2017, 5, 557–567. [Google Scholar] [CrossRef]
- Clancy, J.P.; Cotton, C.U.; Donaldson, S.H.; Solomon, G.M.; VanDevanter, D.R.; Boyle, M.P.; Gentzsch, M.; Nick, J.A.; Illek, B.; Wallenburg, J.C.; et al. CFTR modulator theratyping: Current status, gaps and future directions. J. Cyst. Fibros. 2019, 18, 22–34. [Google Scholar] [CrossRef] [Green Version]
- Haggie, P.M.; Phuan, P.W.; Tan, J.A.; Xu, H.; Avramescu, R.G.; Perdomo, D.; Zlock, L.; Nielson, D.W.; Finkbeiner, W.E.; Lukacs, G.L.; et al. Correctors and Potentiators Rescue Function of the Truncated W1282X-CFTR Translation Product. J. Biol. Chem. 2016, 292, 771–785. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Joshi, D.; Ehrhardt, A.; Hong, J.S.; Sorscher, E.J. Cystic fibrosis precision therapeutics: Emerging considerations. Pediatr. Pulmonol. 2019, 54, S13–S17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sawicki, G.S.; McKone, E.F.; Pasta, D.J.; Millar, S.J.; Wagener, J.S.; Johnson, C.A.; Konstan, M.W. Sustained Benefit from ivacaftor demonstrated by combining clinical trial and cystic fibrosis patient registry data. Am. J. Respir. Crit. Care Med. 2015, 192, 836–842. [Google Scholar] [CrossRef] [PubMed]
- Fu, L.; Sztul, E. ER-associated complexes (ERACs) containing aggregated cystic fibrosis transmembrane conductance regulator (CFTR) are degraded by autophagy. Eur. J. Cell Biol. 2009, 88, 215–226. [Google Scholar] [CrossRef] [Green Version]
- Liu, Q.; Sabirzhanova, I.; Yanda, M.K.; Bergbower, E.A.S.; Boinot, C.; Guggino, W.B.; Cebotaru, L. Rescue of CFTR NBD2 mutants N1303K and S1235R is influenced by the functioning of the autophagosome. J. Cyst. Fibros. 2018, 17, 582–594. [Google Scholar] [CrossRef] [PubMed]
- Luciani, A.; Villella, V.R.; Esposito, S.; Brunetti-Pierri, N.; Medina, D.; Settembre, C.; Gavina, M.; Pulze, L.; Giardino, I.; Pettoello-Mantovani, M.; et al. Defective CFTR induces aggresome formation and lung inflammation in cystic fibrosis through ROS-mediated autophagy inhibition. Nat. Cell Biol. 2010, 12, 863–875. [Google Scholar] [CrossRef] [PubMed]
- Chung, W.J.; Goeckeler-Fried, J.L.; Havasi, V.; Chiang, A.; Rowe, S.M.; Plyler, Z.E.; Hong, J.S.; Mazur, M.; Piazza, G.A.; Keeton, A.B.; et al. Increasing the Endoplasmic Reticulum Pool of the F508del Allele of the Cystic Fibrosis Transmembrane Conductance Regulator Leads to Greater Folding Correction by Small Molecule Therapeutics. PLoS ONE 2016, 11, e0163615. [Google Scholar] [CrossRef] [PubMed]
- Labbadia, J.; Morimoto, R.I. The biology of proteostasis in aging and disease. Annu. Rev. Biochem. 2015, 84, 435–464. [Google Scholar] [CrossRef] [Green Version]
- Kelly, J.W. Pharmacologic Approaches for Adapting Proteostasis in the Secretory Pathway to Ameliorate Protein Conformational Diseases. Cold Spring Harb. Perspect. Biol. 2019, 2019, a034108. [Google Scholar] [CrossRef]



© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Estabrooks, S.; Brodsky, J.L. Regulation of CFTR Biogenesis by the Proteostatic Network and Pharmacological Modulators. Int. J. Mol. Sci. 2020, 21, 452. https://doi.org/10.3390/ijms21020452
Estabrooks S, Brodsky JL. Regulation of CFTR Biogenesis by the Proteostatic Network and Pharmacological Modulators. International Journal of Molecular Sciences. 2020; 21(2):452. https://doi.org/10.3390/ijms21020452
Chicago/Turabian StyleEstabrooks, Samuel, and Jeffrey L. Brodsky. 2020. "Regulation of CFTR Biogenesis by the Proteostatic Network and Pharmacological Modulators" International Journal of Molecular Sciences 21, no. 2: 452. https://doi.org/10.3390/ijms21020452
APA StyleEstabrooks, S., & Brodsky, J. L. (2020). Regulation of CFTR Biogenesis by the Proteostatic Network and Pharmacological Modulators. International Journal of Molecular Sciences, 21(2), 452. https://doi.org/10.3390/ijms21020452