A Narrative Review on C3 Glomerulopathy: A Rare Renal Disease



Abstract
:1. Introduction
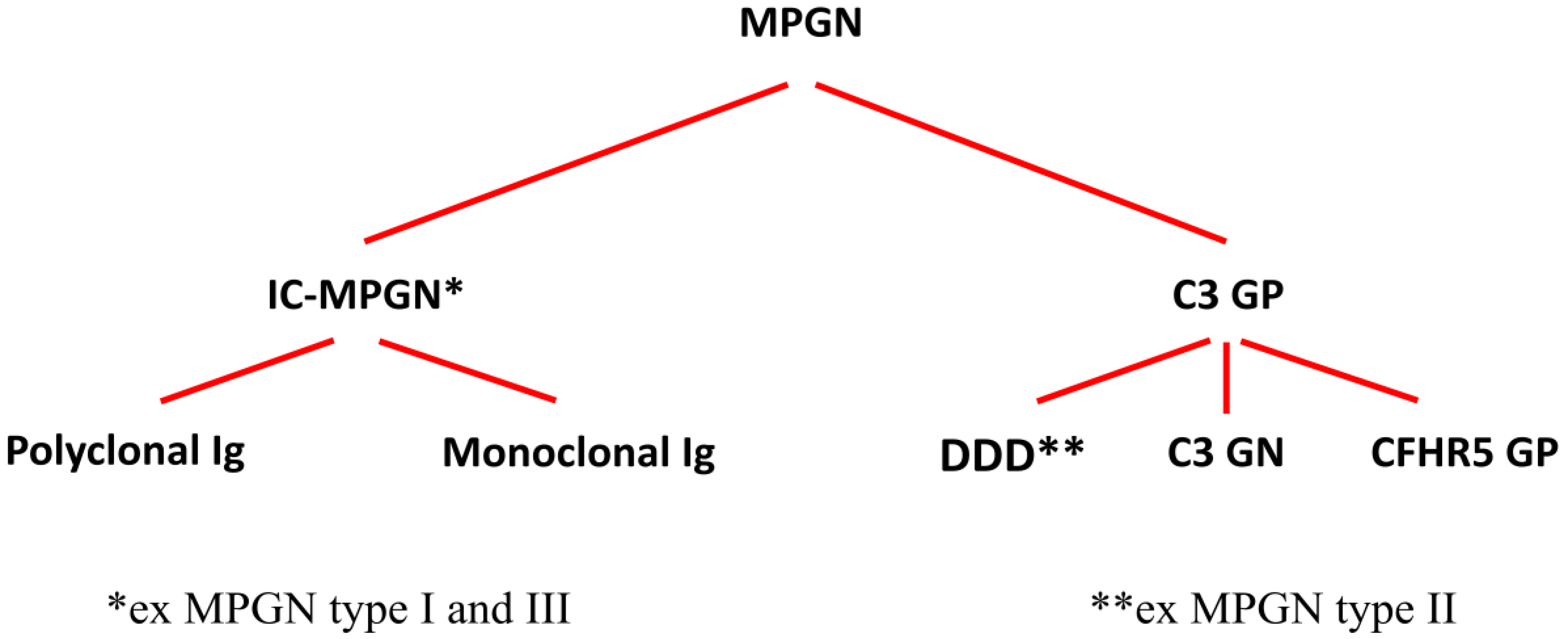
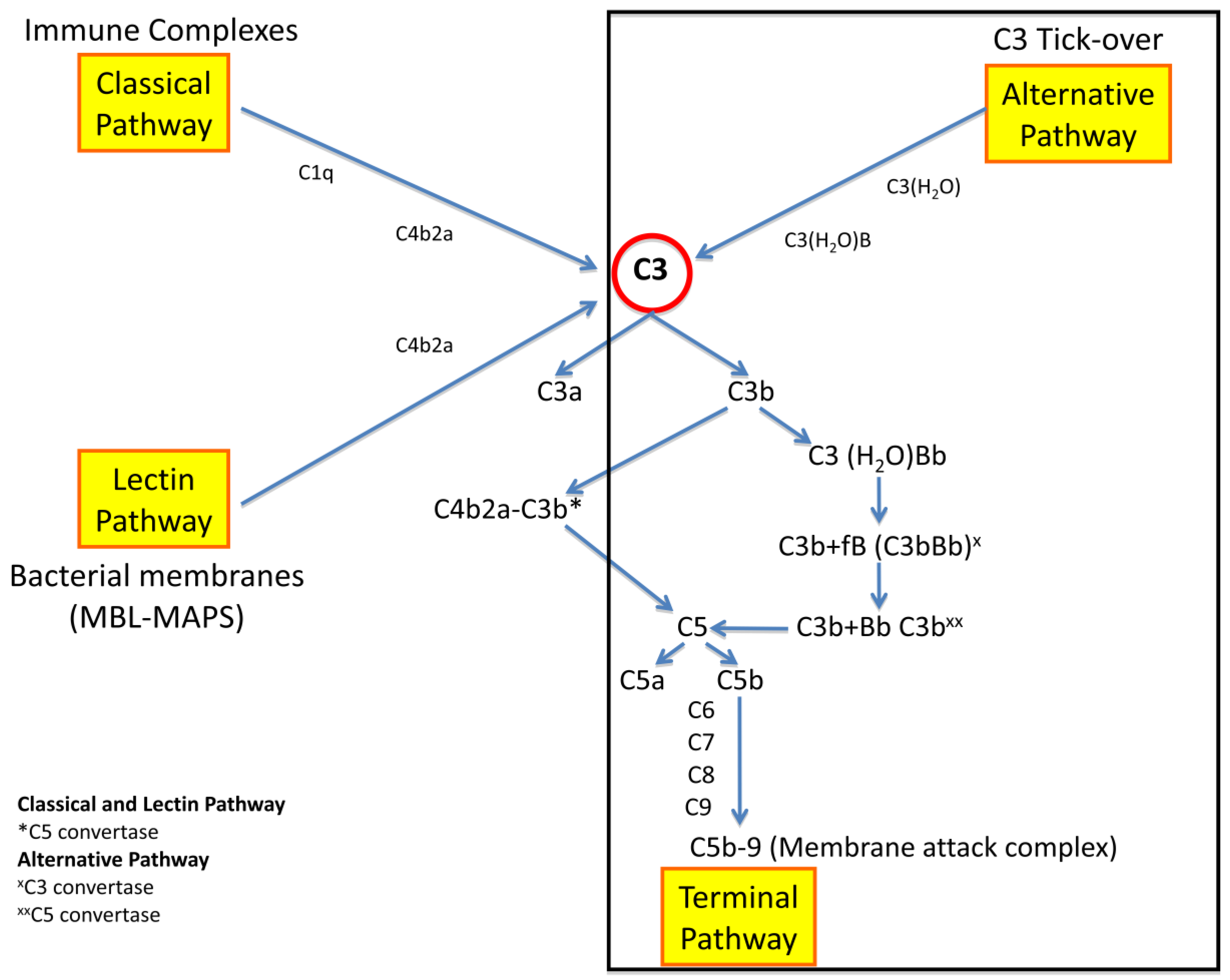
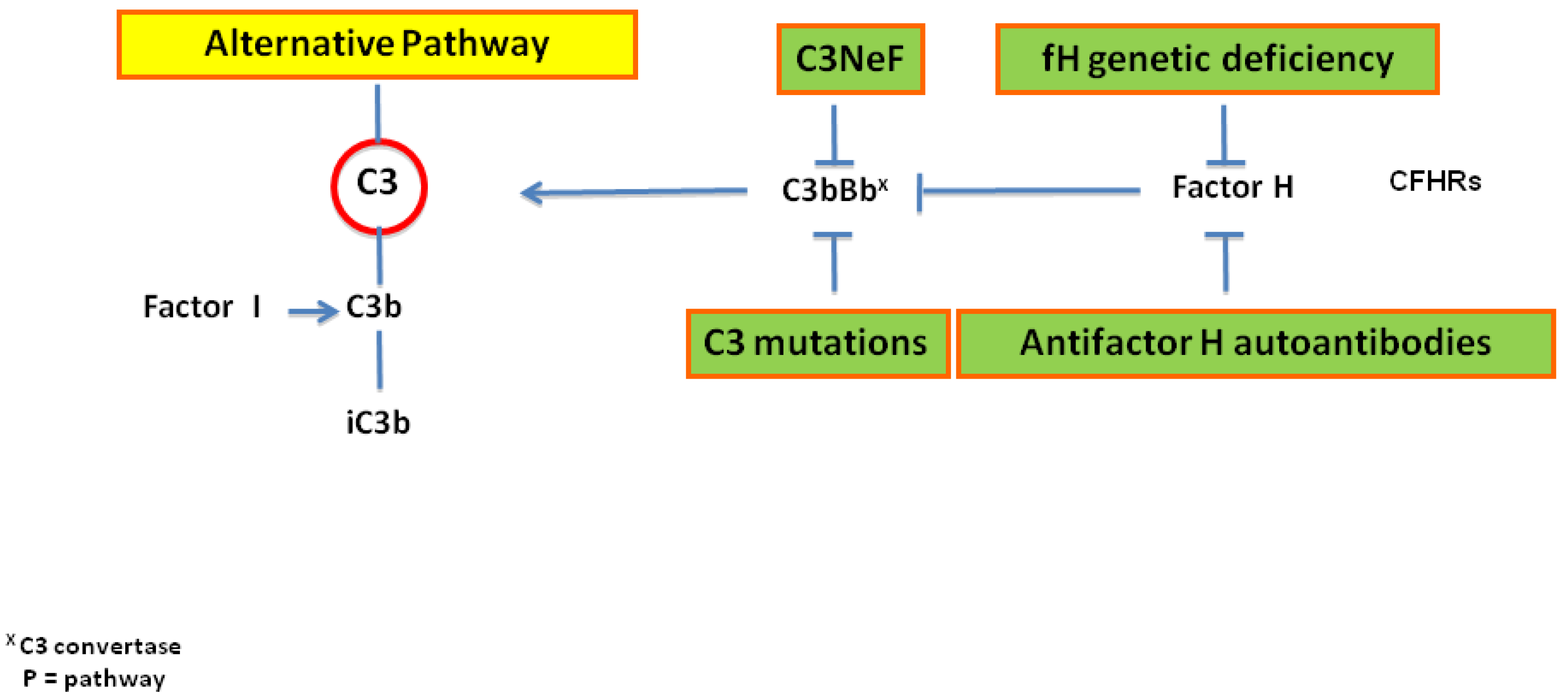
2. Pathogenesis
3. Epidemiology
4. Clinical Manifestations
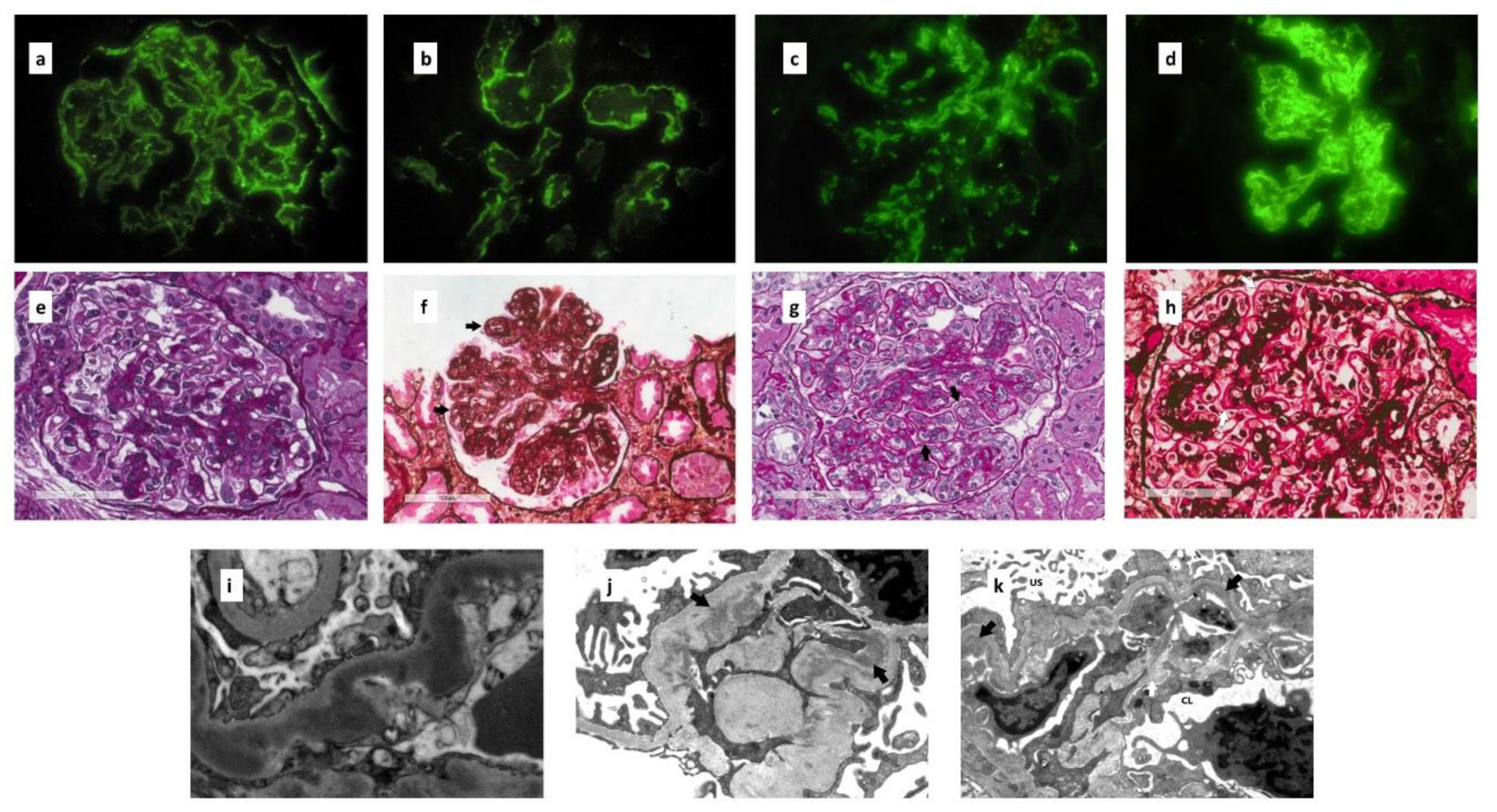
5. Renal Biopsy
5.1. Immunofluorescence Microscopy
5.2. Light Microscopy
5.3. Electron Microscopy
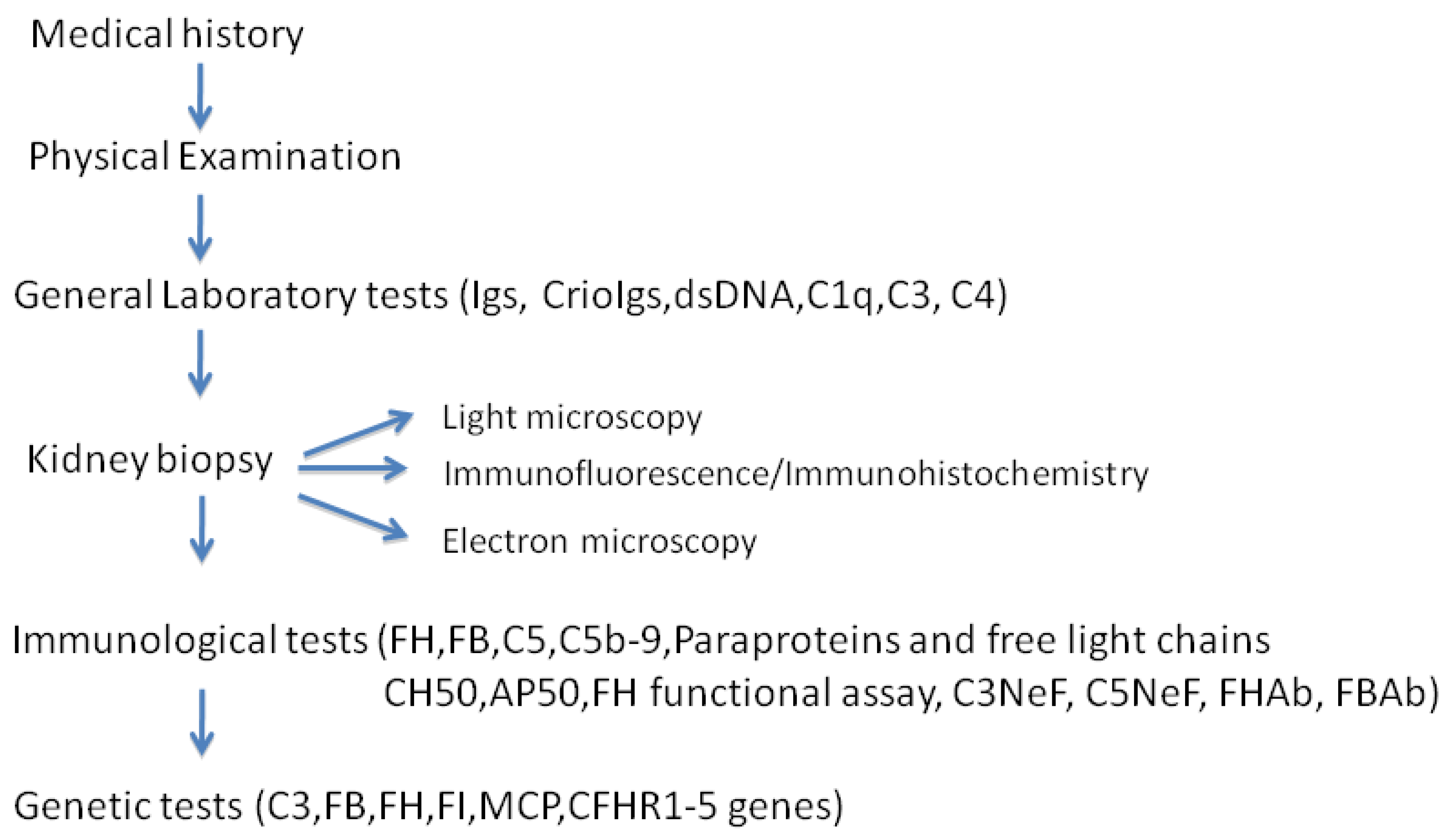
6. Diagnosis
7. Differential Diagnosis
8. Recurrence or De Novo C3 GP after Kidney Transplatation
9. Therapy
10. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Schena, F.P.; Alpers, G.E. Membranoproliferative glomerulonephritis and cryoglobulinemic glomerulonephritis. In Comprehensive Clinical Nephrology, 5th ed.; Elsevier: Sanders, PA, USA, 2015; pp. 253–260. [Google Scholar]
- Pickering, M.C.; D’Agati, V.D.; Nester, C.M.; Smith, R.J.; Haas, M.; Appel, G.B.; Alpers, C.E.; Bajema, I.M.; Bedrosian, C.; Braun, M.; et al. C3 glomerulopathy: Consensus report. Kidney Int. 2013, 84, 1079–1089. [Google Scholar] [CrossRef] [Green Version]
- Fakhouri, F.; Frémeaux-Bacc, V.; Noël, L.H.; Cook, H.T.; Pickering, M.C. C3 glomerulopathy: A new classification. Nat. Rev. Nephrol. 2010, 6, 494–499. [Google Scholar] [CrossRef]
- Cook, H.T.; Pickering, M.C. Clusters Not Classifications: Making Sense of Complement-Mediated Kidney Injury. J. Am. Soc. Nephrol. 2019, 29, 9–12. [Google Scholar] [CrossRef]
- Ricklin, D.; Mastellos, D.C.; Reis, E.S.; Lambris, J.D. The renaissance of complement therapeutics. Nat. Rev. Nephrol. 2018, 14, 26–47. [Google Scholar] [CrossRef] [Green Version]
- Jokiranta, T.S.; Solomon, A.; Pangburn, M.K.; Zipfel, P.F.; Meri, S. Nephritogenic lambda light chain dimer: A unique human miniautoantibody against complement factor H. J Immunol. 1999, 163, 4590–4596. [Google Scholar]
- Goodship, T.H.; Pappworth, I.Y.; Toth, T.; Denton, M.; Houlberg, K.; McCormick, F.; Warland, D.; Moore, I.; Hunze, E.M.; Staniforth, S.J.; et al. Factor H autoantibodies in membranoproliferative glomerulonephritis. Mol. Immunol. 2012, 52, 200–206. [Google Scholar] [CrossRef]
- Nozal, P.; Strobel, S.; Ibernon, M.; López, D.; Sánchez-Corral, P.; Rodríguez de Córdoba, S.; Józsi, M.; López-Trascasa, M. Anti-factor H antibody affecting factor H cofactor activity in a patient with dense deposit disease. Clin. Kidney J. 2012, 5, 133–136. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Blanc, C.; Togarsimalemath, S.K.; Chauvet, S.; Le Quintrec, M.; Moulin, B.; Buchler, M.; Jokiranta, T.S.; Roumenina, L.T.; Fremeaux-Bacchi, V.; Dragon-Durey, M.A. Anti-factor H autoantibodies in C3 glomerulopathies and in atypical hemolytic uremic syndrome: One target, two diseases. J. Immunol. 2015, 194, 5129–5138. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xiao, X.; Pickering, M.C.; Smith, R.J. C3 glomerulopathy: The genetic and clinical findings in dense deposit disease and C3 glomerulonephritis. Semin. Thromb. Hemost. 2014, 40, 465–471. [Google Scholar] [CrossRef] [PubMed]
- Merinero, H.M.; García, S.P.; García-Fernández, J.; Arjona, E.; Tortajada, A.; Rodríguez de Córdoba, S. Complete functional characterization of disease-associated genetic variants in the complement factor H gene. Kidney Int. 2018, 93, 470–481. [Google Scholar] [CrossRef]
- Barbour, T.D.; Ruseva, M.M.; Pickering, M.C. Update on C3 glomerulopathy. Nephrol. Dial. Transplant. 2016, 31, 717–725. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Spitzer, R.E.; Vallota, E.H.; Forristal, J.; Sudora, E.; Stitzel, A.; Davis, N.C.; West, C.D. Serum C’3 lytic system in patients with glomerulonephritis. Science 1969, 164, 436–437. [Google Scholar] [CrossRef] [PubMed]
- Arroyave, C.M.; Vallota, E.H.; Müller-Eberhard, H.J. Lysis of human erythrocytes due to activation of the alternate complement pathway by nephritic factor (C3NeF). J. Immunol. 1974, 113, 764–768. [Google Scholar] [PubMed]
- Daha, M.R.; Fearon, D.T.; Austen, K.F. Formation in the presence of C3 nephritic factor (C3NeF) of an alternative pathway C3 convertase containing uncleaved B. Immunology 1976, 31, 789–796. [Google Scholar] [PubMed]
- Daha, M.R.; Austen, K.F.; Fearon, D.T. The incorporation of C3 nephritic factor (C3NeF) into a stabilized C3 convertase, C3bBb (C3NeF), and its release after decay of convertase function. J. Immunol. 1977, 119, 812–817. [Google Scholar]
- Williams, D.G.; Bartlett, A.; Duffus, P. Identification of nephritic factor as an immunoglobulin. Clin. Exp. Immunol. 1978, 33, 425–429. [Google Scholar]
- Józsi, M.; Reuter, S.; Nozal, P.; López-Trascasa, M.; Sánchez-Corral, P.; Prohászka, Z.; Uzonyi, B. Autoantibodies to complement components in C3 glomerulopathy and atypical hemolytic uremic syndrome. Immunol. Lett. 2014, 160, 163–171. [Google Scholar] [CrossRef] [Green Version]
- Marinozzi, M.C.; Chauvet, S.; Le Quintrec, M.; Mignotet, M.; Petitprez, F.; Legendre, C.; Cailliez, M.; Deschenes, G.; Fischbach, M.; Karras, A.; et al. C5 nephritic factors drive the biological phenotype of C3 glomerulopathies. Kidney Int. 2017, 92, 1232–1241. [Google Scholar] [CrossRef]
- Halbwachs, L.; Leveillé, M.; Lesavre, P.; Wattel, S.; Leibowitch, J. Nephritic factor of the classical pathway of complement: Immunoglobulin G autoantibody directed against the classical pathway C3 convetase enzyme. J. Clin. Investig. 1980, 65, 1249–1256. [Google Scholar] [CrossRef] [Green Version]
- Tanuma, Y.; Ohi, H.; Hatano, M. Two types of C3 nephritic factor: Properdin-dependent C3NeF and properdin-independent C3NeF. Clin. Immunol. Immunopathol. 1990, 56, 226–238. [Google Scholar] [CrossRef]
- Schena, F.P.; Pertosa, G.; Stanziale, P.; Vox, E.; Pecoraro, C.; Andreucci, V.E. Biological significance of the C3 nephritic factor in membranoproliferative glomerulonephritis. Clin. Nephrol. 1982, 18, 240–246. [Google Scholar] [PubMed]
- Ravindran, A.; Fervenza, F.C.; Smith, R.J.H.; De Vriese, A.S.; Sethi, S. C3 Glomerulopathy: Ten Years’ Experience at Mayo Clinic. Mayo Clin. Proc. 2018, 93, 991–1008. [Google Scholar] [CrossRef] [PubMed]
- Donadelli, R.; Pulieri, P.; Piras, R.; Iatropoulos, P.; Valoti, E.; Benigni, A.; Remuzzi, G.; Noris, M. Unraveling the Molecular Mechanisms Underlying Complement Dysregulation by Nephritic Factors in C3G and IC-MPGN. Front. Immunol. 2018, 9, 2329. [Google Scholar] [CrossRef] [PubMed]
- Medjeral-Thomas, N.R.; O’Shaughnessy, M.M.; O’Regan, J.A.; Traynor, C.; Flanagan, M.; Wong, L.; Teoh, C.W.; Awan, A.; Waldron, M.; Cairns, T.; et al. C3 glomerulopathy: Clinicopathologic features and predictors of outcome. Clin. J. Am. Soc. Nephrol. 2014, 9, 46–53. [Google Scholar] [CrossRef]
- Strobel, S.; Zimmering, M.; Papp, K.; Prechl, J.; Józsi, M. Anti-factor B autoantibody in dense deposit disease. Mol. Immunol. 2010, 47, 1476–1483. [Google Scholar] [CrossRef] [Green Version]
- Chen, Q.; Müller, D.; Rudolph, B.; Hartmann, A.; Kuwertz-Bröking, E.; Wu, K.; Kirschfink, M.; Skerka, C.; Zipfel, P.F. Combined C3b and factor B autoantibodies and MPGN type II. N Engl. J. Med. 2011, 365, 2340–2342. [Google Scholar] [CrossRef]
- Smith, R.J.H.; Alexander, J.; Barlow, P.N.; Botto, M.; Cassavant, T.L.; Cook, H.T.; deCórdoba, S.R.; Hageman, G.S.; Jokiranta, T.S.; Kimberling, W.J.; et al. Dense Deposit Disease Focus Group. New approaches to the treatment of dense deposit disease. J. Am. Soc. Nephrol. 2007, 18, 2447–2456. [Google Scholar] [CrossRef] [Green Version]
- Bomback, A.S.; Santoriello, D.; Avasare, R.S.; Regunathan-Shenk, R.; Canetta, P.A.; Ahn, W.; Radhakrishnan, J.; Marasa, M.; Rosenstiel, P.E.; Herlitz, L.C.; et al. C3 glomerulonephritis and dense deposit disease share a similar disease course in a large United States cohort of patients with C3 glomerulopathy. Kidney Int. 2018, 93, 977–985. [Google Scholar] [CrossRef]
- Smith, R.J.H.; Appel, G.B.; Blom, A.M.; Cook, H.T.; D’Agati, V.D.; Fakhouri, F.; Fremeaux-Bacchi, V.; Józsi, M.; Kavanagh, D.; Lambris, J.D.; et al. C3 glomerulopathy—Understanding a rare complement-driven renal disease. Nat. Rev. Nephrol. 2019, 15, 129–143. [Google Scholar] [CrossRef]
- Athanasiou, Y.; Voskarides, K.; Gale, D.P.; Damianou, L.; Patsias, C.; Zavros, M.; Maxwell, P.H.; Cook, H.T.; Demosthenous, P.; Hadjisavvas, A.; et al. Familial C3 glomerulopathy associated with CFHR5 mutations: Clinical characteristics of 91 patients in 16 pedigrees. Clin. J. Am. Soc. Nephrol. 2011, 6, 1436–1446. [Google Scholar] [CrossRef] [Green Version]
- Servais, A.; Noël, L.H.; Roumenina, L.T.; Le Quintrec, M.; Ngo, S.; Dragon-Durey, M.A.; Macher, M.A.; Zuber, J.; Karras, A.; Provot, F.; et al. Acquired and genetic complement abnormalities play a critical role in dense deposit disease and other C3 glomerulopathies. Kidney Int. 2012, 82, 454–464. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rabasco, C.; Cavero, T.; Román, E.; Rojas-Rivera, J.; Olea, T.; Espinosa, M.; Cabello, V.; Fernández-Juarez, G.; González, F.; Ávila, A.; et al. Spanish Group for the Study of Glomerular Diseases (GLOSEN). Effectiveness of mycophenolate mofetil in C3 glomerulonephritis. Kidney Int. 2015, 88, 1153–1160. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Iatropoulos, P.; Noris, M.; Mele, C.; Piras, R.; Valoti, E.; Bresin, E.; Curreri, M.; Mondo, E.; Zito, A.; Gamba, S.; et al. Complement gene variants determine the risk of immunoglobulin-associated MPGN and C3 glomerulopathy and predict long-term renal outcome. Mol. Immunol. 2016, 71, 131–142. [Google Scholar] [CrossRef] [PubMed]
- Caliskan, Y.; Torun, E.S.; Tiryaki, T.O.; Oruc, A.; Ozluk, Y.; Akgul, S.U.; Temurhan, S.; Oztop, N.; Kilicaslan, I.; Sever, M.S. Immunosuppressive Treatment in C3 Glomerulopathy: Is it Really Effective? Am. J. Nephrol. 2017, 46, 96–107. [Google Scholar] [CrossRef] [PubMed]
- Le Quintrec, M.; Lapeyraque, A.L.; Lionet, A.; Sellier-Leclerc, A.L.; Delmas, Y.; Baudouin, V.; Daugas, E.; Decramer, S.; Tricot, L.; Cailliez, M.; et al. Patterns of Clinical Response to Eculizumab in Patients with C3 Glomerulopathy. Am. J. Kidney Dis. 2018, 72, 84–92. [Google Scholar] [CrossRef] [PubMed]
- Gale, D.P.; Goicoechea de Jorge, E.G.; Cook, H.T.; Martinez-Barricate, R.; Hadjisavvas, A.; McLean, A.G. Identification of a mutation in complement factor H-related protein 5 in patients of Cypriot origin with glomerulonephritis. Lancet 2010, 376, 794–801. [Google Scholar] [CrossRef] [Green Version]
- Martínez-Barricarte, R.; Heurich, M.; Valdes-Cañedo, F.; Vazquez-Martul, E.; Torreira, E.; Montes, T.; Tortajada, A.; Pinto, S.; Lopez-Trascasa, M.; Morgan, B.P.; et al. Human C3 mutation reveals a mechanism of dense deposit disease pathogenesis and provides insights into complement activation and regulation. J. Clin. Investig. 2010, 120, 3702–3712. [Google Scholar] [CrossRef] [Green Version]
- Malik, T.H.; Lavin, P.J.; Goicoechea de Jorge, E.; Vernon, K.A.; Rose, K.L.; Patel, M.P.A. A hybrid CFHR3-1 gene causes familial C3 glomerulopathy. J. Am. Soc. Nephrol. 2012, 23, 1155–1160. [Google Scholar] [CrossRef] [Green Version]
- Tortajada, A.; Ye’benes, H.; Abarrategui-Garrido, C.; Anter, J.; García-Fernández, J.M.; Martínez-Barricarte, R.; Alba-Domínguez, M.; Malik, T.H.; Bedoya, R.; Cabrera Pérez, R.; et al. C3 glomerulopathy associated CFHR1 mutation alters FHR oligomerization and complement regulation. J. Clin. Investig. 2013, 123, 2434–2446. [Google Scholar] [CrossRef] [Green Version]
- Medjeral-Thomas, N.R.; Troldborg, A.; Constantinou, N.; Lomax-Browne, H.J.; Hansen, A.G.; Willicombe, M.; Pusey, C.D.; Cook, H.T.; Thiel, S.; Pickering, M.C. Progressive IgA Nephropathy is Associated with low circulating mannan-binding lectin-associated serine protease-3 (MASP-3) and increased glomerular factor H-related protein-5 (FHR5) Deposition. Kidney Int. Rep. 2017, 3, 426–438. [Google Scholar] [CrossRef] [Green Version]
- Hou, J.; Markowitz, G.S.; Bomback, A.S.; Appel, G.B.; Herlitz, L.C.; Barry Stokes, M.; D’Agati, V.D. Toward a working definition of C3 glomerulopathy by immunofluorescence. Kidney Int. 2014, 85, 450–456. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sethi, S.; Nasr, S.H.; De Vriese, A.S.; Fervenza, F.C. C4d as a diagnostic tool in proliferative GN. J. Am. Soc. Nephrol. 2015, 26, 2852–2859. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Singh, G.; Singh, S.K.; Nalwa, A.; Lavleen, S.; Pradeep, I.; Barwad, A.; Sinha, A.; Hari, P.; Bagga, A.; Bagchi, S.; et al. Glomerular C4d staining does not exclude a C3 Glomerulopathy. Kidney Int. Rep. 2019, 4, 698–709. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leung, N.; Bridoux, F.; Batuman, V.; Chaidos, A.; Cockwell, P.; D’Agati, V.D.; Dispenzieri, A.; Fervenza, F.C.; Fermand, J.P.; Gibbs, S.; et al. The evaluation of monoclonal gammopathy of renal significance: A consensus report of the International Kidney and Monoclonal Gammopathy Research Group. Nat. Rev. Nephrol. 2019, 15, 121–135. [Google Scholar] [CrossRef] [Green Version]
- Zand, L.; Kattah, A.; Fervenza, F.C.; Smith, R.J.; Nasr, S.H.; Zhang, Y.; Vrana, J.A.; Leung, N.; Cornell, L.D.; Sethi, S. C3 glomerulonephritis associated with monoclonal gammopathy: A case series. Am. J. Kidney Dis. 2013, 62, 506–514. [Google Scholar] [CrossRef] [Green Version]
- Angioi, A.; Fervenza, F.C.; Sethi, S.; Zhang, Y.; Smith, R.J.; Murray, D.; Van Praet, J.; Pani, A.; De Vriese, A.S. Diagnosis of complement alternative pathway disorders. Kidney Int. 2016, 89, 278–288. [Google Scholar] [CrossRef] [Green Version]
- Grumach, A.S.; Kirschfink, M. Are complement deficiencies really rare? Overview on prevalence, clinical importance and modern diagnostic approach. Mol. Immunol. 2014, 61, 110–117. [Google Scholar] [CrossRef]
- Zhang, Y.; Nester, C.M.; Martin, B.; Skjoedt, M.O.; Meyer, N.C.; Shao, D.; Borsa, N.; Palarasah, Y.; Smith, R.J. Defining the complement biomarker profile of C3 glomerulopathy. Clin. J. Am. Soc. Nephrol. 2014, 9, 1876–1882. [Google Scholar] [CrossRef] [Green Version]
- Fervenza, F.C.; Sethi, S. Circulating complement levels and C3 glomerulopathy. Clin. J. Am. Soc. Nephrol. 2014, 9, 1829–1831. [Google Scholar] [CrossRef] [Green Version]
- Corvillo, F.; Okrój, M.; Nozal, P.; Melgosa, M.; Sánchez-Corral, P.; López-Trascasa, M. Nephritic Factors: An Overview of Classification, Diagnostic Tools and Clinical Associations. Front. Immunol. 2019, 10, 886. [Google Scholar] [CrossRef]
- Zhang, Y.; Meyer, N.C.; Wang, K.; Nishimura, C.; Frees, K.; Jones, M.; Katz, L.M.; Sethi, S.; Smith, R.J. Causes of alternative pathway dysregulation in dense deposit disease. Clin. J. Am. Soc. Nephrol. 2012, 7, 265–274. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ohi, H.; Yasugi, T. Occurrence of C3 nephritic factor and C4 nephritic factor in membranoproliferative glomerulonephritis (MPGN). Clin. Exp. Immunol. 1994, 95, 316–321. [Google Scholar] [CrossRef] [PubMed]
- Watson, R.; Lindner, S.; Bordereau, P.; Hunze, E.M.; Tak, F.; Ngo, S.; Zipfel, P.F.; Skerka, C.; Dragon-Durey, M.A.; Marchbank, K.J. Standardisation of the factor H autoantibody assay. Immunobiology 2014, 219, 9–16. [Google Scholar] [CrossRef] [PubMed]
- Osborne, A.J.; Breno, M.; Borsa, N.G.; Bu, F.; Frémeaux-Bacchi, V.; Gale, D.P.; van den Heuvel, L.P.; Kavanagh, D.; Noris, M.; Pinto, S.; et al. Statistical Validation of Rare Complement Variants Provides Insights into the Molecular Basis of Atypical Hemolytic Uremic Syndrome and C3 Glomerulopathy. J. Immunol. 2018, 200, 2464–2478. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sánchez-Corral, P.; Pouw, R.B.; López-Trascasa, M.; Józsi, M. Self-Damage Caused by Dysregulation of the Complement Alternative Pathway: Relevance of the Factor H Protein Family. Front. Immunol. 2018, 12, 1607. [Google Scholar] [CrossRef]
- Vernon, K.A.; Gale, D.P.; de Jorge, E.G.; McLean, A.G.; Galliford, J.; Pierides, A.; Maxwell, P.H.; Taube, D.; Pickering, M.C.; Cook, H.T. Recurrence of complement factor H-related protein 5 nephropathy in a renal transplant. Am. J. Transplant. 2011, 11, 152–155. [Google Scholar] [CrossRef]
- Cochat, P.; Fargue, S.; Mestrallet, G.; Jungraithmayr, T.; Koch-Nogueira, P.; Ranchin, B.; Zimmerhackl, L.B. Disease recurrence in paediatric renal transplantation. Pediatr. Nephrol. 2009, 11, 2097–2108. [Google Scholar] [CrossRef] [Green Version]
- Wong, L.; Moran, S.; Lavin, P.J.; Dorman, A.M.; Conlon, P.J. Kidney transplant outcomes in familial C3 glomerulopathy. Clin. Kidney J. 2016, 3, 403–407. [Google Scholar] [CrossRef] [Green Version]
- Bomback, A.S.; Smith, R.J.; Barile, G.R.; Zhang, Y.; Heher, E.C.; Herlitz, L.; Stokes, M.B.; Markowitz, G.S.; D’Agati, V.D.; Canetta, P.A.; et al. Eculizumab for dense deposit disease and C3 glomerulonephritis. Clin. J. Am. Soc. Nephrol. 2012, 7, 748–756. [Google Scholar] [CrossRef]
- Abbas, F.; El Kossi, M.; Kim, J.J.; Shaheen, I.S.; Sharma, A.; Halawa, A. Complement-mediated renal diseases after kidney transplantation—Current diagnostic and therapeutic options in de novo and recurrent diseases. World J. Transplant. 2018, 8, 203–219. [Google Scholar] [CrossRef]
- Sánchez-Moreno, A.; De la Cerda, F.; Cabrera, R.; Fijo, J.; López-Trascasa, M.; Bedoya, R.; Rodríguez de Córdoba, S.; Ybot-González, P. Eculizumab in dense-deposit disease after renal transplantation. Pediatr. Nephrol. 2014, 29, 2055–2059. [Google Scholar] [CrossRef] [PubMed]
- Lim, W.H.; Shingde, M.; Wong, G. Recurrent and de novo Glomerulonephritis after Kidney Transplantation. Front. Immunol. 2019, 10, 1944. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Uffing, A.; Pérez-Sáez, M.J.; La Manna, G.; Comai, G.; Fischman, C.; Farouk, S.; Manfro, R.C.; Bauer, A.C.; Lichtenfels, B.; Mansur, J.B.; et al. A large, international study on post-transplant glomerular diseases: The TANGO project. BMC Nephrol. 2018, 19, 229. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Legendre, C.M.; Licht, C.; Muus, P.; Greenbaum, L.A.; Babu, S.; Bedrosian, C.; Bingham, C.; Cohen, D.J.; Delmas, Y.; Douglas, K.; et al. Terminal complement inhibitor eculizumab in atypical hemolytic-uremic syndrome. N. Engl. J. Med. 2013, 368, 2169–2181. [Google Scholar] [CrossRef] [Green Version]
- Welte, T.; Arnold, F.; Kappes, J.; Seidl, M.; Häffner, K.; Bergmann, C.; Walz, G.; Neumann-Haefelin, E. Treating C3 glomerulopathy with eculizumab. BMC Nephrol. 2018, 19, 7. [Google Scholar] [CrossRef]
- Von Visger, J.; Cassol, C.; Nori, U.; Franco-Ahumada, G.; Nadasdy, T.; Satoskar, A.A. Complete biopsy-proven resolution of deposits in recurrent proliferative glomerulonephritis with monoclonal IgG deposits (PGNMIGD) following rituximab treatment in renal allograft. BMC Nephrol. 2019, 20, 53. [Google Scholar] [CrossRef] [Green Version]
- McCaughan, J.A.; O’Rourke, D.M.; Courtney, A.E. Recurrent dense deposit disease after renal transplantation: An emerging role for complementary therapies. Am. J. Transplant. 2012, 12, 1046–1051. [Google Scholar] [CrossRef]
- Zand, L.; Lorenz, E.C.; Cosio, F.G.; Fervenza, F.C.; Nasr, S.H.; Gandhi, M.J.; Smith, R.J.; Sethi, S. Clinical findings, pathology, and outcomes of C3GN after kidney transplantation. J. Am. Soc. Nephrol. 2014, 25, 1110–1117. [Google Scholar] [CrossRef]
- Rudnicki, M. Rituximab for Treatment of Membranoproliferative Glomerulonephritis and C3 Glomerulopathies. BioMed Res. Int. 2017, 2017, 2180508. [Google Scholar] [CrossRef]
- Koopman, J.J.E.; Teng, Y.K.O.; Boon, C.J.F.; van den Heuvel, L.P.; Rabelink, T.J.; van Kooten, C.; de Vries, A.P.J. Diagnosis and treatment of C3 glomerulopathy in a center of expertise. Neth. J. Med. 2019, 77, 10–18. [Google Scholar]
- Chauvet, S.; Roumenina, L.T.; Aucouturier, P.; Marinozzi, M.C.; Dragon-Durey, M.A.; Karras, A.; Delmas, Y.; Le Quintrec, M.; Guerrot, D.; Jourde-Chiche, N.; et al. Both Monoclonal and Polyclonal Immunoglobulin Contingents Mediate Complement Activation in Monoclonal Gammopathy Associated-C3 Glomerulopathy. Front. Immunol. 2018, 9, 2260. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chauvet, S.; Frémeaux-Bacchi, V.; Petitprez, F.; Karras, A.; Daniel, L.; Burtey, S.; Choukroun, G.; Delmas, Y.; Guerrot, D.; François, A.; et al. Treatment of B-cell disorder improves renal outcome of patients with monoclonal gammopathy-associated C3 glomerulopathy. Blood 2017, 129, 1437–1447. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sethi, S.; Sukov, W.R.; Zhang, Y.; Fervenza, F.C.; Lager, D.J.; Miller, D.V.; Cornell, L.D.; Krishnan, S.G.; Smith, R.J. Dense deposit disease associated with monoclonal gammopathy of undetermined significance. Am. J. Kidney Dis. 2010, 56, 977–982. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Y.; Shao, D.; Ricklin, D.; Hilkin, B.M.; Nester, C.M.; Lambris, J.D.; Smith, R.J. Compstatin analog Cp40 inhibits complement dysregulation in vitro in C3 glomerulopathy. Immunobiology 2015, 220, 993–998. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Paixão-Cavalcante, D.; Torreira, E.; Lindorfer, M.A.; Rodriguez de Cordoba, S.; Morgan, B.P.; Taylor, R.P.; Llorca, O.; Harris, C.L. A humanized antibody that regulates the alternative pathway convertase: Potential for therapy of renal disease associated with nephritic factors. J. Immunol. 2014, 192, 4844–4851. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Subías, M.; Tortajada, A.; Gastoldi, S.; Galbusera, M.; López-Perrote, A.; Lopez, L.J.; González-Fernández, F.A.; Villegas-Martínez, A.; Dominguez, M.; Llorca, O.; et al. A novel antibody against human factor B that blocks formation of the C3bB proconvertase and inhibits complement activation in disease models. J. Immunol. 2014, 193, 5567–5575. [Google Scholar] [CrossRef] [Green Version]
- Pauly, D.; Nagel, B.M.; Reinders, J.; Killian, T.; Wulf, M.; Ackerman, S.; Ehrenstein, B.; Zipfel, P.F.; Skerka, C.; Weber, B.H. A novel antibody against human properdin inhibits the alternative complement system and specifically detects properdin from blood samples. PLoS ONE 2014, 9, e96371. [Google Scholar] [CrossRef] [Green Version]
- Krmar, R.T.; Holtbäck, U.; Linné, T.; Berg, U.B.; Celsi, G.; Söderberg, M.P.; Wernerson, A.; Szakos, A.; Larsson, S.; Skattum, L.; et al. Acute renal failure in dense deposit disease: Complete recovery after combination therapy with immunosuppressant and plasma exchange. Clin. Nephrol. 2011, 75, 4–10. [Google Scholar]
- Häffner, K.; Michelfelder, S.; Pohl, M. Successful therapy of C3Nef-positive C3 glomerulopathy with plasma therapy and immunosuppression. Pediatr. Nephrol. 2015, 30, 1951–1959. [Google Scholar] [CrossRef]
- Kurtz, K.A.; Schlueter, A.J. Management of membranoproliferative glomerulonephritis type II with plasmapheresis. J. Clin. Apher. 2002, 17, 135–137. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Study Population | Clinical Findings | Histology | Complement Findings § | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Author-Year (Ref. No.) | Country | C3 GP Class | N | Age at Diagnosis (Years) | Male Sex (%) | Median SCr at Diagnosis (mg/dL) | Median UP at Diagnosis (g/24 h) | Main Clinical Presentation (%) | Main Histological Patterns (%) | Low C3 (%) | Low C4 (%) | C3Nef No. (%) | Complement Gene Variant Identified No. (%) | Other Complement Antibodies No. (%) |
| Athanasiou, 2011 [31] | Cyprus | CFHR5 GP | 91 | Range 12–88 | 47 | N/A | N/A | H 90 Pu 38 | MES | 0 | 0 | N/A | 91/91 (100) | N/A |
| Servais, 2012 [32] | France | C3 GN | 56 | 30 | 58.6 | 1.3 | 3.6 | H 64 NS 27 | MPGN (71), MES (29) | 40 | 0 | 25 (45) | 10 (18) | N/A |
| DDD | 29 | 12 | 43 | 1.6 | 5.6 | H 76 NS 30 | 59 | 4 | 25 (86) | 5 (17) | N/A | |||
| Mejeral-Thomas, 2014 [25] | UK/ Ireland | C3 GN | 59 | 26 | 54 | 1.4 | 3 | H 87 NS 44 | MPGN (52), MES (24), Crescentic (5) | 48 | 36 | N/A | N/A | N/A |
| DDD | 21 | 12 | 43 | 0.9 | 3 | H 86 NS 43 | MPGN (62), MES (14), Crescentic (19) | 79 | 15 | N/A | N/A | N/A | ||
| Rabasco, 2015 [33] | Spain | C3 GN | 60 | 27 | 57 | 1.4 | 3.8 | NS 52 H 49 | MPGN (75), MES (15) | 63 | N/A | 11/23 (48) | 3/23 (13) | 0 |
| Iatropoulos, 2016 [34] | Italy | C3 GN | 52 | 14.6 | 42 | N/A | N/A | H 86 NS 29 | N/A | 69 | N/A | 23/52 (44) | 10/52 (20) | N/A |
| DDD | 21 | 15.9 | 38 | N/A | N/A | H 90 NS29 | 86 | N/A | 16/21 (78) | 3/21 (14) | N/A | |||
| Caliskan, 2017 [35] | Turkey | C3 GN (no subclass) | 66 | 36 | 54 | 1.6 | 3.8 | H 89 NS 48 | MPGN (75), MES (24) | 41 | 4 | N/A | N/A | N/A |
| Bomback, 2018 [29] | USA | C3 GN | 87 | 28.3 | 63.2 | 2 | 3.7 | H 59 NS 33 | MPGN (68.8), MES (17.5) | 65 | 14 | 13/42 (31) | 12/42 (28) | 3/42 (7) |
| DDD | 24 | 40 | 67 | 2.1 | 4.4 | H 82 NS 17 | MPGN (45.8), MES (29.3) | 64 | 14 | 1/9 (11) | 3/9 (33) | 1/9 (11) | ||
| Le Quintec, 2018 [36] | France | C3 GN (no subclass) | 26 | 17.5 | 48 | 1 | 4.1 | NS 73 | MPGN (62) | 88 | N/A | 16/24 (67) | 5/22 (23) | 2/22 (9) |
| Ravindran, 2018 [23] | USA * | C3 GN | 102 | 41.5 | 59 | 1.6 | 2.5 | H 87 Pu 38 | MPGN (52), MES (34), Crescentic (7) | 42 | 12 | 27/60 (46) | 21/61 (39) | 8/60 (13) |
| DDD | 12 | 31.5 | 25 | 1.4 | 6.5 | H 92 Pu 75 | MPGN (45), MES (27), Crescentic (16) | 58 | 8 | 3/10 (30) | 2/9 (22) | 1/8 (12) | ||
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Schena, F.P.; Esposito, P.; Rossini, M. A Narrative Review on C3 Glomerulopathy: A Rare Renal Disease. Int. J. Mol. Sci. 2020, 21, 525. https://doi.org/10.3390/ijms21020525
Schena FP, Esposito P, Rossini M. A Narrative Review on C3 Glomerulopathy: A Rare Renal Disease. International Journal of Molecular Sciences. 2020; 21(2):525. https://doi.org/10.3390/ijms21020525
Chicago/Turabian StyleSchena, Francesco Paolo, Pasquale Esposito, and Michele Rossini. 2020. "A Narrative Review on C3 Glomerulopathy: A Rare Renal Disease" International Journal of Molecular Sciences 21, no. 2: 525. https://doi.org/10.3390/ijms21020525
APA StyleSchena, F. P., Esposito, P., & Rossini, M. (2020). A Narrative Review on C3 Glomerulopathy: A Rare Renal Disease. International Journal of Molecular Sciences, 21(2), 525. https://doi.org/10.3390/ijms21020525