Scars or Regeneration?—Dermal Fibroblasts as Drivers of Diverse Skin Wound Responses



Abstract
:1. Introduction
2. Fibroblast Origins and Heterogeneity
2.1. Fibrogenic Fibroblasts Are Defined by Function
2.2. Fibrogenic Fibroblast Functional Subsets Defined by Spatial Location and Surface Markers
2.3. Fibroblastic Cell Plasticity
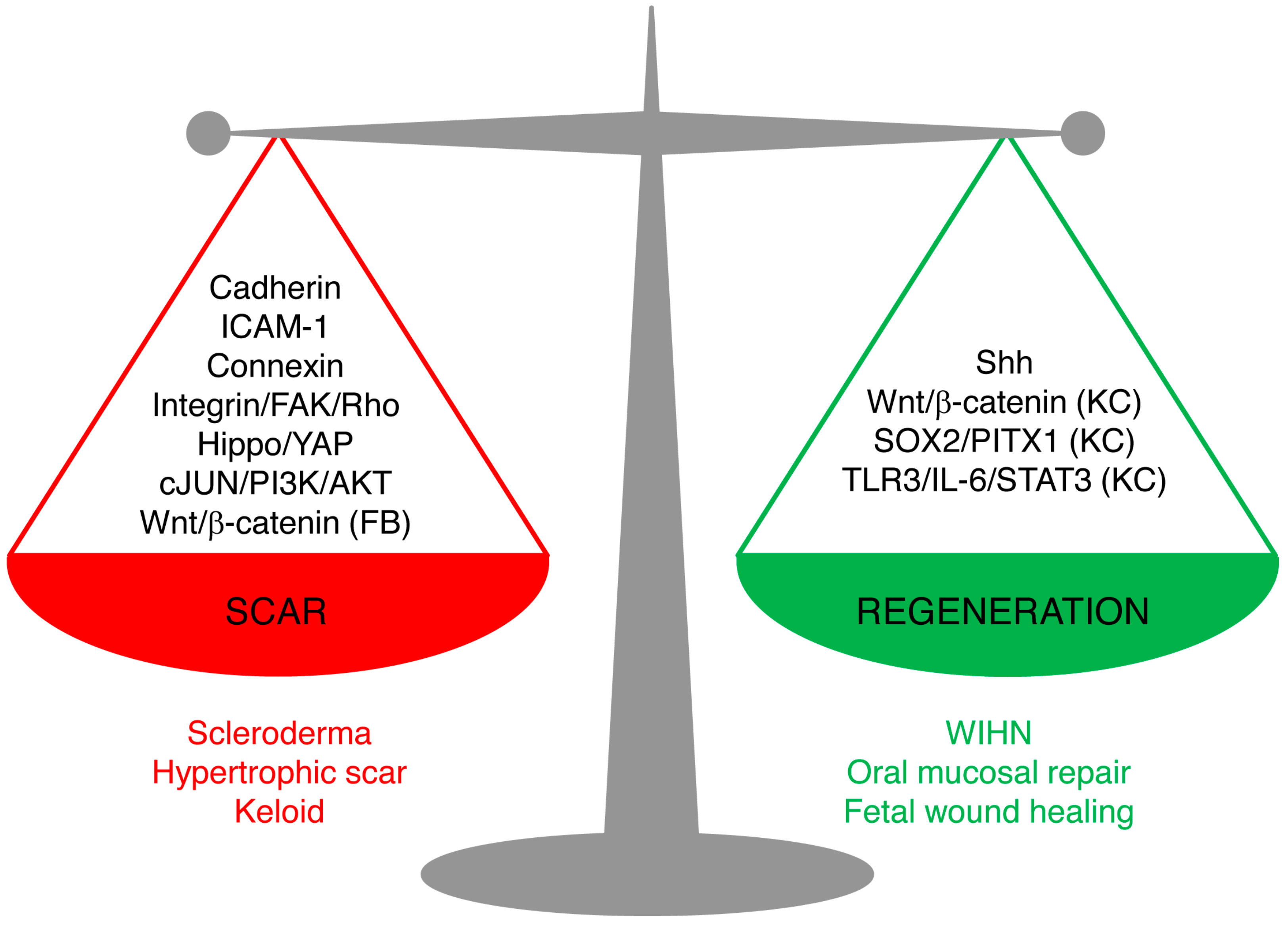
3. Mechanisms of Skin Scarring
3.1. Granulation Tissue and Myofibroblasts
3.2. Cell–ECM Adhesion
3.3. Cell–Cell Adhesion
3.4. Alternative Model
4. Mechanisms of Scarless Healing
4.1. Regeneration across Developmental Age
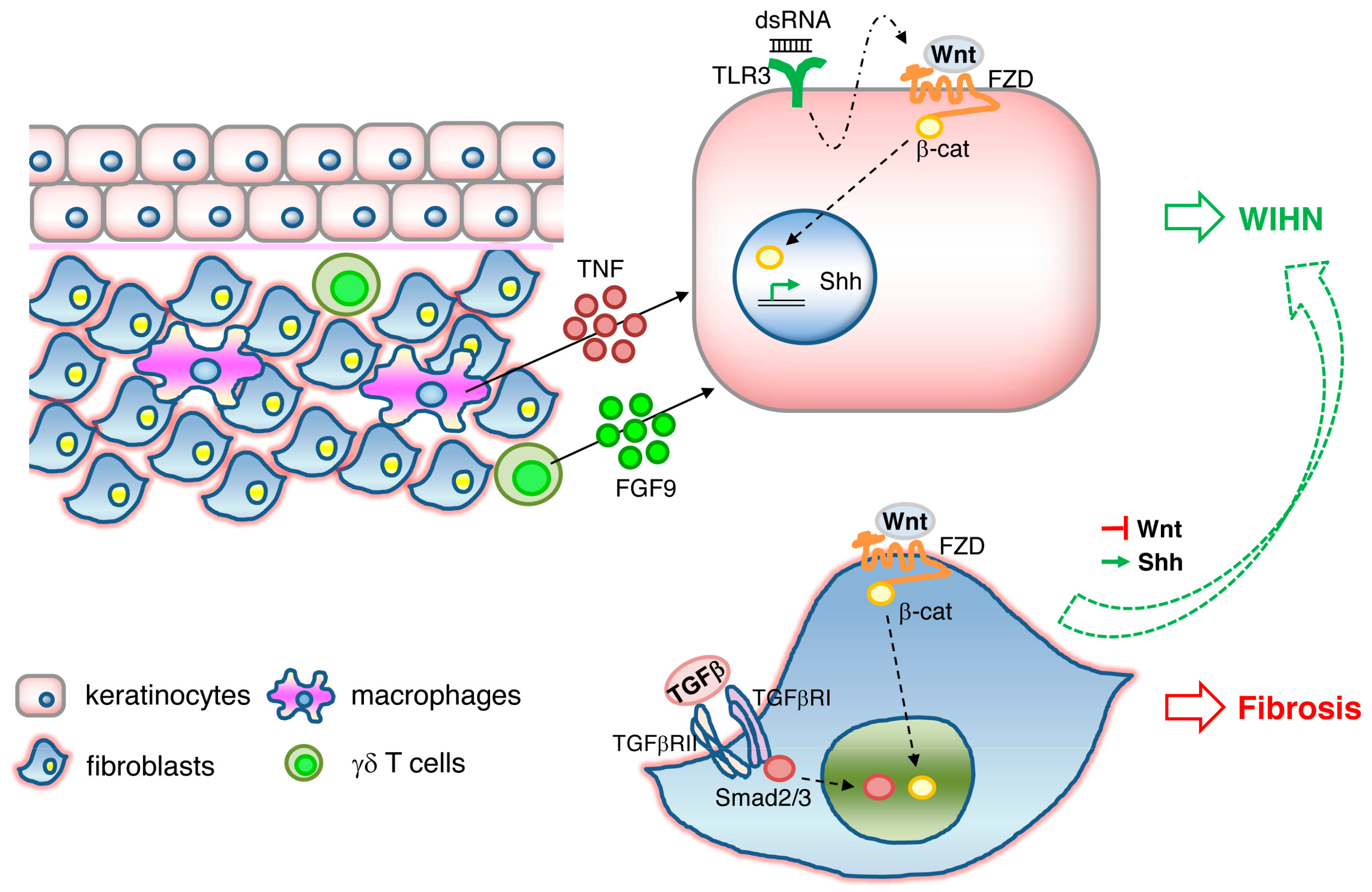
4.2. Wound-Induced Hair Follicle Neogenesis (WIHN)
4.3. Scarless Repair of the Oral Mucosa
4.4. Scarless Skin Regeneration in African Spiny Mice (Genus Acomys)
5. Summary and Perspectives
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| ADAM12 | A disintegrin and metalloprotease domain 12 |
| APs | Adipocyte precursor cells |
| Dlk1 | Delta like non-canonical Notch ligand 1 |
| ENFs | Engrailed-1 lineage negative fibroblasts |
| EPFs | Engrailed-1 lineage positive fibroblasts |
| FAK | Focal adhesion kinase |
| FAP | Fibroblast activation protein |
| GAGs | Glycosaminoglycans |
| Lrig1 | Leucine-rich repeats and immunoglobulin-like domains protein 1 |
| PDGF | Platelet-derived growth factor |
| Prrx1 | Paired-related homeobox 1 |
| Shh | Sonic Hedgehog |
| ST2 | Suppression of tumorigenicity 2 |
| TAZ | Transcriptional coactivator with PDZ-binding motif |
| TGFβ | Transforming growth factor beta |
| TIMP | Tissue inhibitor of metalloproteinases |
| WIHN | Wound-induced hair follicle neogenesis |
| YAP | Yes-associated protein |
References
- Gurtner, G.C.; Werner, S.; Barrandon, Y.; Longaker, M.T. Wound repair and regeneration. Nature 2008, 453, 314–321. [Google Scholar] [CrossRef] [PubMed]
- Bayat, A.; McGrouther, D.A.; Ferguson, M.W. Skin scarring. BMJ 2003, 326, 88–92. [Google Scholar] [CrossRef] [PubMed]
- Longaker, M.T.; Whitby, D.J.; Adzick, N.S.; Crombleholme, T.M.; Langer, J.C.; Duncan, B.W.; Bradley, S.M.; Stern, R.; Ferguson, M.W.; Harrison, M.R. Studies in fetal wound healing, VI. Second and early third trimester fetal wounds demonstrate rapid collagen deposition without scar formation. J. Pediatr. Surg. 1990, 25, 63–68. [Google Scholar] [CrossRef]
- Jones, R.E.; Foster, D.S.; Hu, M.S.; Longaker, M.T. Wound healing and fibrosis: Current stem cell therapies. Transfusion 2019, 59, 884–892. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Padmanabhan, J.; Maan, Z.N.; Kwon, S.H.; Kosaraju, R.; Bonham, C.A.; Gurtner, G.C. In Vivo Models for the Study of Fibrosis. Adv. Wound Care (New Rochelle) 2019. [Google Scholar] [CrossRef] [PubMed]
- Donovan, J.; Shiwen, X.; Norman, J.; Abraham, D. Platelet-derived growth factor alpha and beta receptors have overlapping functional activities towards fibroblasts. Fibrogenes. Tissue Repair 2013, 6, 10. [Google Scholar] [CrossRef] [Green Version]
- Rajkumar, V.S.; Shiwen, X.; Bostrom, M.; Leoni, P.; Muddle, J.; Ivarsson, M.; Gerdin, B.; Denton, C.P.; Bou-Gharios, G.; Black, C.M.; et al. Platelet-derived growth factor-beta receptor activation is essential for fibroblast and pericyte recruitment during cutaneous wound healing. Am. J. Pathol. 2006, 169, 2254–2265. [Google Scholar] [CrossRef] [Green Version]
- Jahoda, C.A.; Whitehouse, J.; Reynolds, A.J.; Hole, N. Hair follicle dermal cells differentiate into adipogenic and osteogenic lineages. Exp. Dermatol. 2003, 12, 849–859. [Google Scholar] [CrossRef]
- Jiang, D.; Rinkevich, Y. Defining Skin Fibroblastic Cell Types Beyond CD90. Front. Cell Dev. Biol. 2018, 6, 133. [Google Scholar] [CrossRef] [Green Version]
- Strutz, F.; Okada, H.; Lo, C.W.; Danoff, T.; Carone, R.L.; Tomaszewski, J.E.; Neilson, E.G. Identification and characterization of a fibroblast marker: FSP1. J. Cell Biol. 1995, 130, 393–405. [Google Scholar] [CrossRef] [Green Version]
- Chang, H.Y.; Chi, J.T.; Dudoit, S.; Bondre, C.; van de Rijn, M.; Botstein, D.; Brown, P.O. Diversity, topographic differentiation, and positional memory in human fibroblasts. Proc. Natl. Acad. Sci. USA 2002, 99, 12877–12882. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Driskell, R.R.; Lichtenberger, B.M.; Hoste, E.; Kretzschmar, K.; Simons, B.D.; Charalambous, M.; Ferron, S.R.; Herault, Y.; Pavlovic, G.; Ferguson-Smith, A.C.; et al. Distinct fibroblast lineages determine dermal architecture in skin development and repair. Nature 2013, 504, 277–281. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rinkevich, Y.; Walmsley, G.G.; Hu, M.S.; Maan, Z.N.; Newman, A.M.; Drukker, M.; Januszyk, M.; Krampitz, G.W.; Gurtner, G.C.; Lorenz, H.P.; et al. Skin fibrosis. Identification and isolation of a dermal lineage with intrinsic fibrogenic potential. Science 2015, 348, aaa2151. [Google Scholar] [CrossRef] [Green Version]
- Jiang, D.; Correa-Gallegos, D.; Christ, S.; Stefanska, A.; Liu, J.; Ramesh, P.; Rajendran, V.; De Santis, M.M.; Wagner, D.E.; Rinkevich, Y. Two succeeding fibroblastic lineages drive dermal development and the transition from regeneration to scarring. Nat. Cell Biol. 2018, 20, 422–431. [Google Scholar] [CrossRef] [Green Version]
- Currie, J.D.; Grosser, L.; Murawala, P.; Schuez, M.; Michel, M.; Tanaka, E.M.; Sandoval-Guzman, T. The Prrx1 limb enhancer marks an adult subpopulation of injury-responsive dermal fibroblasts. Biol. Open 2019, 8, bio043711. [Google Scholar] [CrossRef] [Green Version]
- Dulauroy, S.; Di Carlo, S.E.; Langa, F.; Eberl, G.; Peduto, L. Lineage tracing and genetic ablation of ADAM12(+) perivascular cells identify a major source of profibrotic cells during acute tissue injury. Nat. Med. 2012, 18, 1262–1270. [Google Scholar] [CrossRef]
- Wernig, G.; Chen, S.Y.; Cui, L.; Van Neste, C.; Tsai, J.M.; Kambham, N.; Vogel, H.; Natkunam, Y.; Gilliland, D.G.; Nolan, G.; et al. Unifying mechanism for different fibrotic diseases. Proc. Natl. Acad. Sci. USA 2017, 114, 4757–4762. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lichtenberger, B.M.; Mastrogiannaki, M.; Watt, F.M. Epidermal beta-catenin activation remodels the dermis via paracrine signalling to distinct fibroblast lineages. Nat. Commun. 2016, 7, 10537. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Philippeos, C.; Telerman, S.B.; Oules, B.; Pisco, A.O.; Shaw, T.J.; Elgueta, R.; Lombardi, G.; Driskell, R.R.; Soldin, M.; Lynch, M.D.; et al. Spatial and Single-Cell Transcriptional Profiling Identifies Functionally Distinct Human Dermal Fibroblast Subpopulations. J. Investig. Dermatol. 2018, 138, 811–825. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Korosec, A.; Frech, S.; Gesslbauer, B.; Vierhapper, M.; Radtke, C.; Petzelbauer, P.; Lichtenberger, B.M. Lineage Identity and Location within the Dermis Determine the Function of Papillary and Reticular Fibroblasts in Human Skin. J. Investig. Dermatol. 2019, 139, 342–351. [Google Scholar] [CrossRef] [Green Version]
- Shook, B.A.; Wasko, R.R.; Rivera-Gonzalez, G.C.; Salazar-Gatzimas, E.; Lopez-Giraldez, F.; Dash, B.C.; Munoz-Rojas, A.R.; Aultman, K.D.; Zwick, R.K.; Lei, V.; et al. Myofibroblast proliferation and heterogeneity are supported by macrophages during skin repair. Science 2018, 362, eaar2971. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hu, M.S.; Longaker, M.T. Dipeptidyl Peptidase-4, Wound Healing, Scarring, and Fibrosis. Plast. Reconstr. Surg. 2016, 138, 1026–1031. [Google Scholar] [CrossRef] [PubMed]
- Goldstein, L.A.; Ghersi, G.; Pineiro-Sanchez, M.L.; Salamone, M.; Yeh, Y.; Flessate, D.; Chen, W.T. Molecular cloning of seprase: A serine integral membrane protease from human melanoma. Biochim. Biophys. Acta 1997, 1361, 11–19. [Google Scholar] [CrossRef] [Green Version]
- Dienus, K.; Bayat, A.; Gilmore, B.F.; Seifert, O. Increased expression of fibroblast activation protein-alpha in keloid fibroblasts: Implications for development of a novel treatment option. Arch. Dermatol. Res. 2010, 302, 725–731. [Google Scholar] [CrossRef] [PubMed]
- Thielitz, A.; Vetter, R.W.; Schultze, B.; Wrenger, S.; Simeoni, L.; Ansorge, S.; Neubert, K.; Faust, J.; Lindenlaub, P.; Gollnick, H.P.; et al. Inhibitors of dipeptidyl peptidase IV-like activity mediate antifibrotic effects in normal and keloid-derived skin fibroblasts. J. Investig. Dermatol. 2008, 128, 855–866. [Google Scholar] [CrossRef] [Green Version]
- Kollmannsberger, P.; Bidan, C.M.; Dunlop, J.W.C.; Fratzl, P.; Vogel, V. Tensile forces drive a reversible fibroblast-to-myofibroblast transition during tissue growth in engineered clefts. Sci. Adv. 2018, 4, eaao4881. [Google Scholar] [CrossRef] [Green Version]
- Aragona, M.; Panciera, T.; Manfrin, A.; Giulitti, S.; Michielin, F.; Elvassore, N.; Dupont, S.; Piccolo, S. A mechanical checkpoint controls multicellular growth through YAP/TAZ regulation by actin-processing factors. Cell 2013, 154, 1047–1059. [Google Scholar] [CrossRef] [Green Version]
- Dupont, S.; Morsut, L.; Aragona, M.; Enzo, E.; Giulitti, S.; Cordenonsi, M.; Zanconato, F.; Le Digabel, J.; Forcato, M.; Bicciato, S.; et al. Role of YAP/TAZ in mechanotransduction. Nature 2011, 474, 179–183. [Google Scholar] [CrossRef]
- Wada, K.; Itoga, K.; Okano, T.; Yonemura, S.; Sasaki, H. Hippo pathway regulation by cell morphology and stress fibers. Development 2011, 138, 3907–3914. [Google Scholar] [CrossRef] [Green Version]
- Shiraha, H.; Gupta, K.; Drabik, K.; Wells, A. Aging fibroblasts present reduced epidermal growth factor (EGF) responsiveness due to preferential loss of EGF receptors. J. Biol. Chem. 2000, 275, 19343–19351. [Google Scholar] [CrossRef] [Green Version]
- Midgley, A.C.; Rogers, M.; Hallett, M.B.; Clayton, A.; Bowen, T.; Phillips, A.O.; Steadman, R. Transforming growth factor-beta1 (TGF-beta1)-stimulated fibroblast to myofibroblast differentiation is mediated by hyaluronan (HA)-facilitated epidermal growth factor receptor (EGFR) and CD44 co-localization in lipid rafts. J. Biol. Chem. 2013, 288, 14824–14838. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cheng, B.; Fu, X.; Sun, T.; Sun, X.; Sheng, Z. Expression of epidermal growth factor receptor and related phosphorylation proteins in hypertrophic scars and normal skin. Chin. Med. J. (Engl.) 2002, 115, 1525–1528. [Google Scholar] [PubMed]
- Epstein Shochet, G.; Brook, E.; Eyal, O.; Edelstein, E.; Shitrit, D. Epidermal growth factor receptor paracrine upregulation in idiopathic pulmonary fibrosis fibroblasts is blocked by nintedanib. Am. J. Physiol. Lung Cell. Mol. Physiol. 2019, 316, L1025–L1034. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.; Kim, S.Y.; Mun, S.K.; Rhee, S.; Kim, B.J. Epidermal growth factor improves the migration and contractility of aged fibroblasts cultured on 3D collagen matrices. Int. J. Mol. Med. 2015, 35, 1017–1025. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Plikus, M.V.; Guerrero-Juarez, C.F.; Ito, M.; Li, Y.R.; Dedhia, P.H.; Zheng, Y.; Shao, M.; Gay, D.L.; Ramos, R.; Hsi, T.C.; et al. Regeneration of fat cells from myofibroblasts during wound healing. Science 2017, 355, 748–752. [Google Scholar] [CrossRef] [Green Version]
- El Agha, E.; Moiseenko, A.; Kheirollahi, V.; De Langhe, S.; Crnkovic, S.; Kwapiszewska, G.; Szibor, M.; Kosanovic, D.; Schwind, F.; Schermuly, R.T.; et al. Two-Way Conversion between Lipogenic and Myogenic Fibroblastic Phenotypes Marks the Progression and Resolution of Lung Fibrosis. Cell Stem Cell 2017, 20, 261–273.e3. [Google Scholar] [CrossRef] [Green Version]
- Guerrero-Juarez, C.F.; Dedhia, P.H.; Jin, S.; Ruiz-Vega, R.; Ma, D.; Liu, Y.; Yamaga, K.; Shestova, O.; Gay, D.L.; Yang, Z.; et al. Single-cell analysis reveals fibroblast heterogeneity and myeloid-derived adipocyte progenitors in murine skin wounds. Nat. Commun. 2019, 10, 650. [Google Scholar] [CrossRef]
- Sinha, M.; Sen, C.K.; Singh, K.; Das, A.; Ghatak, S.; Rhea, B.; Blackstone, B.; Powell, H.M.; Khanna, S.; Roy, S. Direct conversion of injury-site myeloid cells to fibroblast-like cells of granulation tissue. Nat. Commun. 2018, 9, 936. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bucala, R.; Spiegel, L.A.; Chesney, J.; Hogan, M.; Cerami, A. Circulating fibrocytes define a new leukocyte subpopulation that mediates tissue repair. Mol. Med. 1994, 1, 71–81. [Google Scholar] [CrossRef] [Green Version]
- Ling, C.; Nishimoto, K.; Rolfs, Z.; Smith, L.M.; Frey, B.L.; Welham, N.V. Differentiated fibrocytes assume a functional mesenchymal phenotype with regenerative potential. Sci. Adv. 2019, 5, eaav7384. [Google Scholar] [CrossRef] [Green Version]
- Gabbiani, G.; Ryan, G.B.; Majne, G. Presence of modified fibroblasts in granulation tissue and their possible role in wound contraction. Experientia 1971, 27, 549–550. [Google Scholar] [CrossRef] [PubMed]
- Hinz, B. The role of myofibroblasts in wound healing. Curr. Res. Transl. Med. 2016, 64, 171–177. [Google Scholar] [CrossRef]
- Hinz, B.; Gabbiani, G. Mechanisms of force generation and transmission by myofibroblasts. Curr. Opin. Biotechnol. 2003, 14, 538–546. [Google Scholar] [CrossRef]
- Flanagan, M. The characteristics and formation of granulation tissue. J. Wound Care 1998, 7, 508–510. [Google Scholar] [CrossRef] [PubMed]
- Gabbiani, G. Evolution and clinical implications of the myofibroblast concept. Cardiovasc. Res. 1998, 38, 545–548. [Google Scholar] [CrossRef] [Green Version]
- Wong, V.W.; Sorkin, M.; Glotzbach, J.P.; Longaker, M.T.; Gurtner, G.C. Surgical approaches to create murine models of human wound healing. J. BioMed Biotechnol. Int. 2011, 2011, 969618. [Google Scholar] [CrossRef]
- Wong, V.W.; Rustad, K.C.; Akaishi, S.; Sorkin, M.; Glotzbach, J.P.; Januszyk, M.; Nelson, E.R.; Levi, K.; Paterno, J.; Vial, I.N.; et al. Focal adhesion kinase links mechanical force to skin fibrosis via inflammatory signaling. Nat. Med. 2011, 18, 148–152. [Google Scholar] [CrossRef] [Green Version]
- Sakar, M.S.; Eyckmans, J.; Pieters, R.; Eberli, D.; Nelson, B.J.; Chen, C.S. Cellular forces and matrix assembly coordinate fibrous tissue repair. Nat. Commun. 2016, 7, 11036. [Google Scholar] [CrossRef] [Green Version]
- Henderson, N.C.; Arnold, T.D.; Katamura, Y.; Giacomini, M.M.; Rodriguez, J.D.; McCarty, J.H.; Pellicoro, A.; Raschperger, E.; Betsholtz, C.; Ruminski, P.G.; et al. Targeting of alphav integrin identifies a core molecular pathway that regulates fibrosis in several organs. Nat. Med. 2013, 19, 1617–1624. [Google Scholar] [CrossRef] [Green Version]
- Reed, N.I.; Jo, H.; Chen, C.; Tsujino, K.; Arnold, T.D.; DeGrado, W.F.; Sheppard, D. The alphavbeta1 integrin plays a critical in vivo role in tissue fibrosis. Sci. Transl. Med. 2015, 7, 288ra79. [Google Scholar] [CrossRef] [Green Version]
- Kim, N.G.; Gumbiner, B.M. Adhesion to fibronectin regulates Hippo signaling via the FAK-Src-PI3K pathway. J. Cell Biol. 2015, 210, 503–515. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, B.; Li, L.; Wang, L.; Wang, C.Y.; Yu, J.; Guan, K.L. Cell detachment activates the Hippo pathway via cytoskeleton reorganization to induce anoikis. Genes Dev. 2012, 26, 54–68. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Elosegui-Artola, A.; Andreu, I.; Beedle, A.E.M.; Lezamiz, A.; Uroz, M.; Kosmalska, A.J.; Oria, R.; Kechagia, J.Z.; Rico-Lastres, P.; Le Roux, A.L.; et al. Force Triggers YAP Nuclear Entry by Regulating Transport across Nuclear Pores. Cell 2017, 171, 1397–1410. [Google Scholar] [CrossRef] [PubMed]
- Shiu, J.Y.; Aires, L.; Lin, Z.; Vogel, V. Nanopillar force measurements reveal actin-cap-mediated YAP mechanotransduction. Nat. Cell Biol. 2018, 20, 262–271. [Google Scholar] [CrossRef]
- Liu, F.; Lagares, D.; Choi, K.M.; Stopfer, L.; Marinkovic, A.; Vrbanac, V.; Probst, C.K.; Hiemer, S.E.; Sisson, T.H.; Horowitz, J.C.; et al. Mechanosignaling through YAP and TAZ drives fibroblast activation and fibrosis. Am. J. Physiol. Lung Cell. Mol. Physiol. 2015, 308, L344–L357. [Google Scholar] [CrossRef] [Green Version]
- Alimperti, S.; Andreadis, S.T. CDH2 and CDH11 act as regulators of stem cell fate decisions. Stem Cell Res. 2015, 14, 270–282. [Google Scholar] [CrossRef] [Green Version]
- Hazan, R.B.; Kang, L.; Whooley, B.P.; Borgen, P.I. N-cadherin promotes adhesion between invasive breast cancer cells and the stroma. Cell Adhes. Commun. 1997, 4, 399–411. [Google Scholar] [CrossRef] [Green Version]
- Whitfield, M.L.; Finlay, D.R.; Murray, J.I.; Troyanskaya, O.G.; Chi, J.T.; Pergamenschikov, A.; McCalmont, T.H.; Brown, P.O.; Botstein, D.; Connolly, M.K. Systemic and cell type-specific gene expression patterns in scleroderma skin. Proc. Natl. Acad. Sci. USA 2003, 100, 12319–12324. [Google Scholar] [CrossRef] [Green Version]
- Wu, M.; Pedroza, M.; Lafyatis, R.; George, A.T.; Mayes, M.D.; Assassi, S.; Tan, F.K.; Brenner, M.B.; Agarwal, S.K. Identification of cadherin 11 as a mediator of dermal fibrosis and possible role in systemic sclerosis. Arthritis Rheumatol. 2014, 66, 1010–1021. [Google Scholar] [CrossRef] [Green Version]
- Pedroza, M.; Welschhans, R.L.; Agarwal, S.K. Targeting of cadherin-11 decreases skin fibrosis in the tight skin-1 mouse model. PLoS ONE 2017, 12, e0187109. [Google Scholar] [CrossRef]
- Lodyga, M.; Cambridge, E.; Karvonen, H.M.; Pakshir, P.; Wu, B.; Boo, S.; Kiebalo, M.; Kaarteenaho, R.; Glogauer, M.; Kapoor, M.; et al. Cadherin-11-mediated adhesion of macrophages to myofibroblasts establishes a profibrotic niche of active TGF-beta. Sci. Signal. 2019, 12, eaao3469. [Google Scholar] [CrossRef] [PubMed]
- Heuberger, J.; Birchmeier, W. Interplay of cadherin-mediated cell adhesion and canonical Wnt signaling. Cold Spring Harb. Perspect. Biol. 2010, 2, a002915. [Google Scholar] [CrossRef] [PubMed]
- Howard, S.; Deroo, T.; Fujita, Y.; Itasaki, N. A positive role of cadherin in Wnt/beta-catenin signalling during epithelial-mesenchymal transition. PLoS ONE 2011, 6, e23899. [Google Scholar] [CrossRef] [Green Version]
- Hu, X.; Zhang, H.; Li, X.; Li, Y.; Chen, Z. Activation of mTORC1 in fibroblasts accelerates wound healing and induces fibrosis in mice. Wound Repair Regen. 2020, 28, 6–15. [Google Scholar] [CrossRef] [PubMed]
- Matsushita, Y.; Hasegawa, M.; Matsushita, T.; Fujimoto, M.; Horikawa, M.; Fujita, T.; Kawasuji, A.; Ogawa, F.; Steeber, D.A.; Tedder, T.F.; et al. Intercellular adhesion molecule-1 deficiency attenuates the development of skin fibrosis in tight-skin mice. J. Immunol. 2007, 179, 698–707. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pincha, N.; Hajam, E.Y.; Badarinath, K.; Batta, S.P.R.; Masudi, T.; Dey, R.; Andreasen, P.; Kawakami, T.; Samuel, R.; George, R.; et al. PAI1 mediates fibroblast-mast cell interactions in skin fibrosis. J. Clin. Investig. 2018, 128, 1807–1819. [Google Scholar] [CrossRef]
- Wong, P.; Tan, T.; Chan, C.; Laxton, V.; Chan, Y.W.; Liu, T.; Wong, W.T.; Tse, G. The Role of Connexins in Wound Healing and Repair: Novel Therapeutic Approaches. Front. Physiol. 2016, 7, 596. [Google Scholar] [CrossRef] [Green Version]
- Chen, Q.; Boire, A.; Jin, X.; Valiente, M.; Er, E.E.; Lopez-Soto, A.; Jacob, L.; Patwa, R.; Shah, H.; Xu, K.; et al. Carcinoma-astrocyte gap junctions promote brain metastasis by cGAMP transfer. Nature 2016, 533, 493–498. [Google Scholar] [CrossRef]
- Li, X.; Guo, L.; Yang, X.; Wang, J.; Hou, Y.; Zhu, S.; Du, J.; Feng, J.; Xie, Y.; Zhuang, L.; et al. TGF-beta1-Induced Connexin43 Promotes Scar Formation via the Erk/MMP-1/Collagen III Pathway. J. Oral Rehabil. 2019. [Google Scholar] [CrossRef]
- Qiu, C.; Coutinho, P.; Frank, S.; Franke, S.; Law, L.Y.; Martin, P.; Green, C.R.; Becker, D.L. Targeting connexin43 expression accelerates the rate of wound repair. Curr. Biol. 2003, 13, 1697–1703. [Google Scholar] [CrossRef] [Green Version]
- Cogliati, B.; Vinken, M.; Silva, T.C.; Araujo, C.M.M.; Aloia, T.P.A.; Chaible, L.M.; Mori, C.M.C.; Dagli, M.L.Z. Connexin 43 deficiency accelerates skin wound healing and extracellular matrix remodeling in mice. J. Dermatol. Sci. 2015, 79, 50–56. [Google Scholar] [CrossRef] [Green Version]
- Becker, D.L.; Thrasivoulou, C.; Phillips, A.R. Connexins in wound healing; perspectives in diabetic patients. Biochim. Biophys. Acta 2012, 1818, 2068–2075. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kotini, M.; Barriga, E.H.; Leslie, J.; Gentzel, M.; Rauschenberger, V.; Schambony, A.; Mayor, R. Gap junction protein Connexin-43 is a direct transcriptional regulator of N-cadherin in vivo. Nat. Commun. 2018, 9, 3846. [Google Scholar] [CrossRef] [PubMed]
- Gross, J.; Farinelli, W.; Sadow, P.; Anderson, R.; Bruns, R. On the mechanism of skin wound “contraction”: A granulation tissue “knockout” with a normal phenotype. Proc. Natl. Acad. Sci. USA 1995, 92, 5982–5986. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grillo, H.C.; Watts, G.T.; Gross, J. Studies in wound healing: I. Contraction and the wound contents. Ann. Surg. 1958, 148, 145–160. [Google Scholar] [CrossRef] [PubMed]
- Watts, G.T.; Grillo, H.C.; Gross, J. Studies in Wound Healing: II. The Role of Granulation Tissue in Contraction. Ann. Surg. 1958, 148, 153–160. [Google Scholar] [CrossRef] [PubMed]
- Armstrong, J.R.; Ferguson, M.W. Ontogeny of the skin and the transition from scar-free to scarring phenotype during wound healing in the pouch young of a marsupial, Monodelphis domestica. Dev. Biol. 1995, 169, 242–260. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lorenz, H.P.; Whitby, D.J.; Longaker, M.T.; Adzick, N.S. Fetal wound healing. The ontogeny of scar formation in the non-human primate. Ann. Surg. 1993, 217, 391–396. [Google Scholar] [CrossRef]
- Eming, S.A.; Wynn, T.A.; Martin, P. Inflammation and metabolism in tissue repair and regeneration. Science 2017, 356, 1026–1030. [Google Scholar] [CrossRef] [Green Version]
- Hesketh, M.; Sahin, K.B.; West, Z.E.; Murray, R.Z. Macrophage Phenotypes Regulate Scar Formation and Chronic Wound Healing. Int. J. Mol. Sci. 2017, 18, 1545. [Google Scholar] [CrossRef] [Green Version]
- Julier, Z.; Park, A.J.; Briquez, P.S.; Martino, M.M. Promoting tissue regeneration by modulating the immune system. Acta Biomater. 2017, 53, 13–28. [Google Scholar] [CrossRef] [PubMed]
- Keightley, M.C.; Wang, C.H.; Pazhakh, V.; Lieschke, G.J. Delineating the roles of neutrophils and macrophages in zebrafish regeneration models. Int. J. Biochem. Cell Biol. 2014, 56, 92–106. [Google Scholar] [CrossRef] [PubMed]
- Smigiel, K.S.; Parks, W.C. Macrophages, Wound Healing, and Fibrosis: Recent Insights. Curr. Rheumatol. Rep. 2018, 20, 17. [Google Scholar] [CrossRef] [PubMed]
- Sorg, H.; Tilkorn, D.J.; Hager, S.; Hauser, J.; Mirastschijski, U. Skin Wound Healing: An Update on the Current Knowledge and Concepts. Eur. Surg. Res. 2017, 58, 81–94. [Google Scholar] [CrossRef] [PubMed]
- Gawronska-Kozak, B.; Bogacki, M.; Rim, J.S.; Monroe, W.T.; Manuel, J.A. Scarless skin repair in immunodeficient mice. Wound Repair Regen. 2006, 14, 265–276. [Google Scholar] [CrossRef] [PubMed]
- Larson, B.J.; Longaker, M.T.; Lorenz, H.P. Scarless fetal wound healing: A basic science review. Plast. Reconstr. Surg. 2010, 126, 1172–1180. [Google Scholar] [CrossRef] [Green Version]
- Xue, M.; Jackson, C.J. Extracellular Matrix Reorganization During Wound Healing and Its Impact on Abnormal Scarring. Adv. Wound Care (New Rochelle) 2015, 4, 119–136. [Google Scholar] [CrossRef] [Green Version]
- Bond, J.S.; Duncan, J.A.; Sattar, A.; Boanas, A.; Mason, T.; O’Kane, S.; Ferguson, M.W. Maturation of the human scar: An observational study. Plast. Reconstr. Surg. 2008, 121, 1650–1658. [Google Scholar] [CrossRef]
- Salzer, M.C.; Lafzi, A.; Berenguer-Llergo, A.; Youssif, C.; Castellanos, A.; Solanas, G.; Peixoto, F.O.; Stephan-Otto Attolini, C.; Prats, N.; Aguilera, M.; et al. Identity Noise and Adipogenic Traits Characterize Dermal Fibroblast Aging. Cell 2018, 175, 1575–1590.e22. [Google Scholar] [CrossRef] [Green Version]
- Sole-Boldo, L.; Raddatz, G.; Schütz, S.; Mallm, J.-P.; Rippe, K.; Lonsdorf, A.S.; Rodriguez-Paredes, M.; Lyko, F. Single-cell transcriptomes of the aging human skin reveal loss of fibroblast priming. bioRxiv 2019. [Google Scholar] [CrossRef] [Green Version]
- Nishiguchi, M.A.; Spencer, C.A.; Leung, D.H.; Leung, T.H. Aging Suppresses Skin-Derived Circulating SDF1 to Promote Full-Thickness Tissue Regeneration. Cell Rep. 2018, 24, 3383–3392. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ito, M.; Yang, Z.; Andl, T.; Cui, C.; Kim, N.; Millar, S.E.; Cotsarelis, G. Wnt-dependent de novo hair follicle regeneration in adult mouse skin after wounding. Nature 2007, 447, 316–320. [Google Scholar] [CrossRef]
- Myung, P.S.; Takeo, M.; Ito, M.; Atit, R.P. Epithelial Wnt ligand secretion is required for adult hair follicle growth and regeneration. J. Investig. Dermatol. 2013, 133, 31–41. [Google Scholar] [CrossRef] [Green Version]
- Lee, S.H.; Seo, S.H.; Lee, D.H.; Pi, L.Q.; Lee, W.S.; Choi, K.Y. Targeting of CXXC5 by a Competing Peptide Stimulates Hair Regrowth and Wound-Induced Hair Neogenesis. J. Investig. Dermatol. 2017, 137, 2260–2269. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gay, D.; Kwon, O.; Zhang, Z.; Spata, M.; Plikus, M.V.; Holler, P.D.; Ito, M.; Yang, Z.; Treffeisen, E.; Kim, C.D.; et al. Fgf9 from dermal gammadelta T cells induces hair follicle neogenesis after wounding. Nat. Med. 2013, 19, 916–923. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, X.; Chen, H.; Tian, R.; Zhang, Y.; Drutskaya, M.S.; Wang, C.; Ge, J.; Fan, Z.; Kong, D.; Cai, T.; et al. Macrophages induce AKT/beta-catenin-dependent Lgr5(+) stem cell activation and hair follicle regeneration through TNF. Nat. Commun. 2017, 8, 14091. [Google Scholar] [CrossRef]
- Hughes, M.W.; Jiang, T.X.; Plikus, M.V.; Guerrero-Juarez, C.F.; Lin, C.H.; Schafer, C.; Maxson, R.; Widelitz, R.B.; Chuong, C.M. Msx2 Supports Epidermal Competency during Wound-Induced Hair Follicle Neogenesis. J. Investig. Dermatol. 2018, 138, 2041–2050. [Google Scholar] [CrossRef] [Green Version]
- Nelson, A.M.; Loy, D.E.; Lawson, J.A.; Katseff, A.S.; Fitzgerald, G.A.; Garza, L.A. Prostaglandin D2 inhibits wound-induced hair follicle neogenesis through the receptor, Gpr44. J. Investig. Dermatol. 2013, 133, 881–889. [Google Scholar] [CrossRef] [Green Version]
- Bastakoty, D.; Saraswati, S.; Cates, J.; Lee, E.; Nanney, L.B.; Young, P.P. Inhibition of Wnt/beta-catenin pathway promotes regenerative repair of cutaneous and cartilage injury. FASEB J. 2015, 29, 4881–4892. [Google Scholar] [CrossRef] [Green Version]
- Lam, A.P.; Gottardi, C.J. beta-catenin signaling: A novel mediator of fibrosis and potential therapeutic target. Curr. Opin. Rheumatol. 2011, 23, 562–567. [Google Scholar] [CrossRef]
- Rognoni, E.; Gomez, C.; Pisco, A.O.; Rawlins, E.L.; Simons, B.D.; Watt, F.M.; Driskell, R.R. Inhibition of beta-catenin signalling in dermal fibroblasts enhances hair follicle regeneration during wound healing. Development 2016, 143, 2522–2535. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sato, M. Upregulation of the Wnt/beta-catenin pathway induced by transforming growth factor-beta in hypertrophic scars and keloids. Acta Derm. Venereol. 2006, 86, 300–307. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nelson, A.M.; Reddy, S.K.; Ratliff, T.S.; Hossain, M.Z.; Katseff, A.S.; Zhu, A.S.; Chang, E.; Resnik, S.R.; Page, C.; Kim, D.; et al. dsRNA Released by Tissue Damage Activates TLR3 to Drive Skin Regeneration. Cell Stem Cell 2015, 17, 139–151. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Madaro, L.; Passafaro, M.; Sala, D.; Etxaniz, U.; Lugarini, F.; Proietti, D.; Alfonsi, M.V.; Nicoletti, C.; Gatto, S.; De Bardi, M.; et al. Denervation-activated STAT3-IL-6 signalling in fibro-adipogenic progenitors promotes myofibres atrophy and fibrosis. Nat. Cell Biol. 2018, 20, 917–927. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.S.; Liang, Y.C.; Wu, P.; Kulber, D.A.; Tanabe, K.; Chuong, C.M.; Widelitz, R.; Tuan, T.L. STAT3 signalling pathway is implicated in keloid pathogenesis by preliminary transcriptome and open chromatin analyses. Exp. Dermatol. 2019, 28, 480–484. [Google Scholar] [CrossRef] [PubMed]
- Lim, C.H.; Sun, Q.; Ratti, K.; Lee, S.H.; Zheng, Y.; Takeo, M.; Lee, W.; Rabbani, P.; Plikus, M.V.; Cain, J.E.; et al. Hedgehog stimulates hair follicle neogenesis by creating inductive dermis during murine skin wound healing. Nat. Commun. 2018, 9, 4903. [Google Scholar] [CrossRef]
- Wang, X.; Hsi, T.C.; Guerrero-Juarez, C.F.; Pham, K.; Cho, K.; McCusker, C.D.; Monuki, E.S.; Cho, K.W.; Gay, D.L.; Plikus, M.V. Principles and mechanisms of regeneration in the mouse model for wound-induced hair follicle neogenesis. Regeneration (Oxf.) 2015, 2, 169–181. [Google Scholar] [CrossRef]
- Guerrero-Juarez, C.F.; Astrowski, A.A.; Murad, R.; Dang, C.T.; Shatrova, V.O.; Astrowskaja, A.; Lim, C.H.; Ramos, R.; Wang, X.; Liu, Y.; et al. Wound Regeneration Deficit in Rats Correlates with Low Morphogenetic Potential and Distinct Transcriptome Profile of Epidermis. J. Investig. Dermatol. 2018, 138, 1409–1419. [Google Scholar] [CrossRef] [Green Version]
- Iglesias-Bartolome, R.; Uchiyama, A.; Molinolo, A.A.; Abusleme, L.; Brooks, S.R.; Callejas-Valera, J.L.; Edwards, D.; Doci, C.; Asselin-Labat, M.L.; Onaitis, M.W.; et al. Transcriptional signature primes human oral mucosa for rapid wound healing. Sci. Transl. Med. 2018, 10, eaap8798. [Google Scholar] [CrossRef] [Green Version]
- Seifert, A.W.; Kiama, S.G.; Seifert, M.G.; Goheen, J.R.; Palmer, T.M.; Maden, M. Skin shedding and tissue regeneration in African spiny mice (Acomys). Nature 2012, 489, 561–565. [Google Scholar] [CrossRef] [Green Version]
- Maden, M.; Brant, J.O. Insights into the regeneration of skin from Acomys, the spiny mouse. Exp. Dermatol. 2019, 28, 436–441. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jiang, T.X.; Harn, H.I.; Ou, K.L.; Lei, M.; Chuong, C.M. Comparative regenerative biology of spiny (Acomys cahirinus) and laboratory (Mus musculus) mouse skin. Exp. Dermatol. 2019, 28, 442–449. [Google Scholar] [CrossRef] [PubMed]
- Simkin, J.; Gawriluk, T.R.; Gensel, J.C.; Seifert, A.W. Macrophages are necessary for epimorphic regeneration in African spiny mice. Elife 2017, 6, e24623. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
| Fibrotic Subset | Nonfibrotic Subset | Species | Reference |
|---|---|---|---|
| En1 lineage+ | En1 lineage− | mouse | [13,14] |
| Prrx1 enhancer+ | Prrx1 enhancer− | mouse | [15] |
| ADAM12+ | mouse | [16] | |
| nuclear c-JUN+ | human | [17] | |
| Dlk1+ | Lrig1+ | mouse (E16.5) | [12,18] |
| CD36+CD90+ | CD39+CD90+ | human | [19] |
| FAP-CD90+ | FAP+CD90− | human | [20] |
| Sca1+CD34+CD29+ | mouse | [21] | |
| CD26+ | mouse | [13,22] |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jiang, D.; Rinkevich, Y. Scars or Regeneration?—Dermal Fibroblasts as Drivers of Diverse Skin Wound Responses. Int. J. Mol. Sci. 2020, 21, 617. https://doi.org/10.3390/ijms21020617
Jiang D, Rinkevich Y. Scars or Regeneration?—Dermal Fibroblasts as Drivers of Diverse Skin Wound Responses. International Journal of Molecular Sciences. 2020; 21(2):617. https://doi.org/10.3390/ijms21020617
Chicago/Turabian StyleJiang, Dongsheng, and Yuval Rinkevich. 2020. "Scars or Regeneration?—Dermal Fibroblasts as Drivers of Diverse Skin Wound Responses" International Journal of Molecular Sciences 21, no. 2: 617. https://doi.org/10.3390/ijms21020617
APA StyleJiang, D., & Rinkevich, Y. (2020). Scars or Regeneration?—Dermal Fibroblasts as Drivers of Diverse Skin Wound Responses. International Journal of Molecular Sciences, 21(2), 617. https://doi.org/10.3390/ijms21020617