Moderate Aerobic Exercise Training Prevents the Augmented Hepatic Glucocorticoid Response Induced by High-Fat Diet in Mice


 , , ,
, , ,
Abstract
:1. Introduction
2. Results
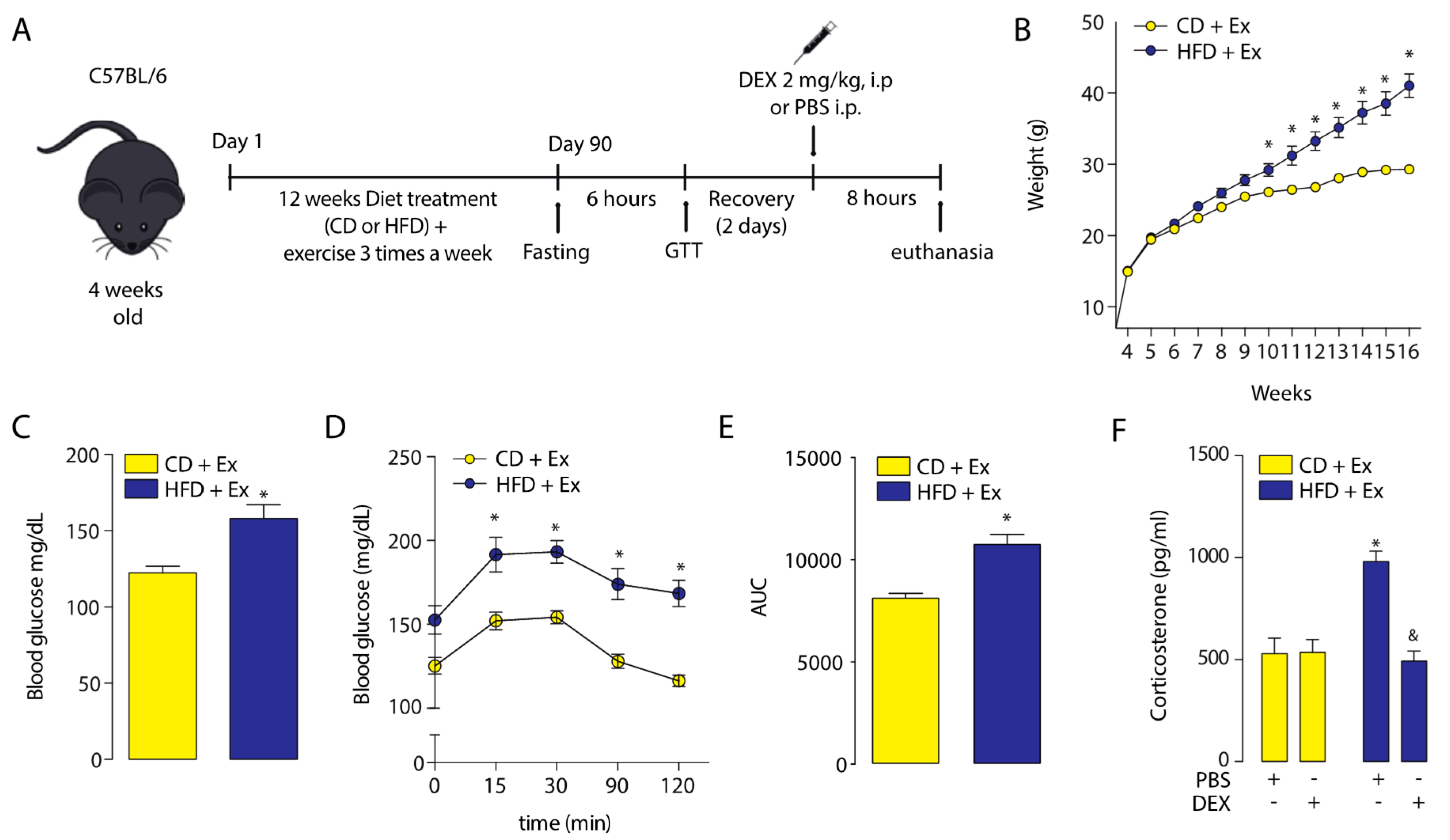
2.1. Corticosterone Plasmatic Levels Were Altered in High-Fat Diet Mice
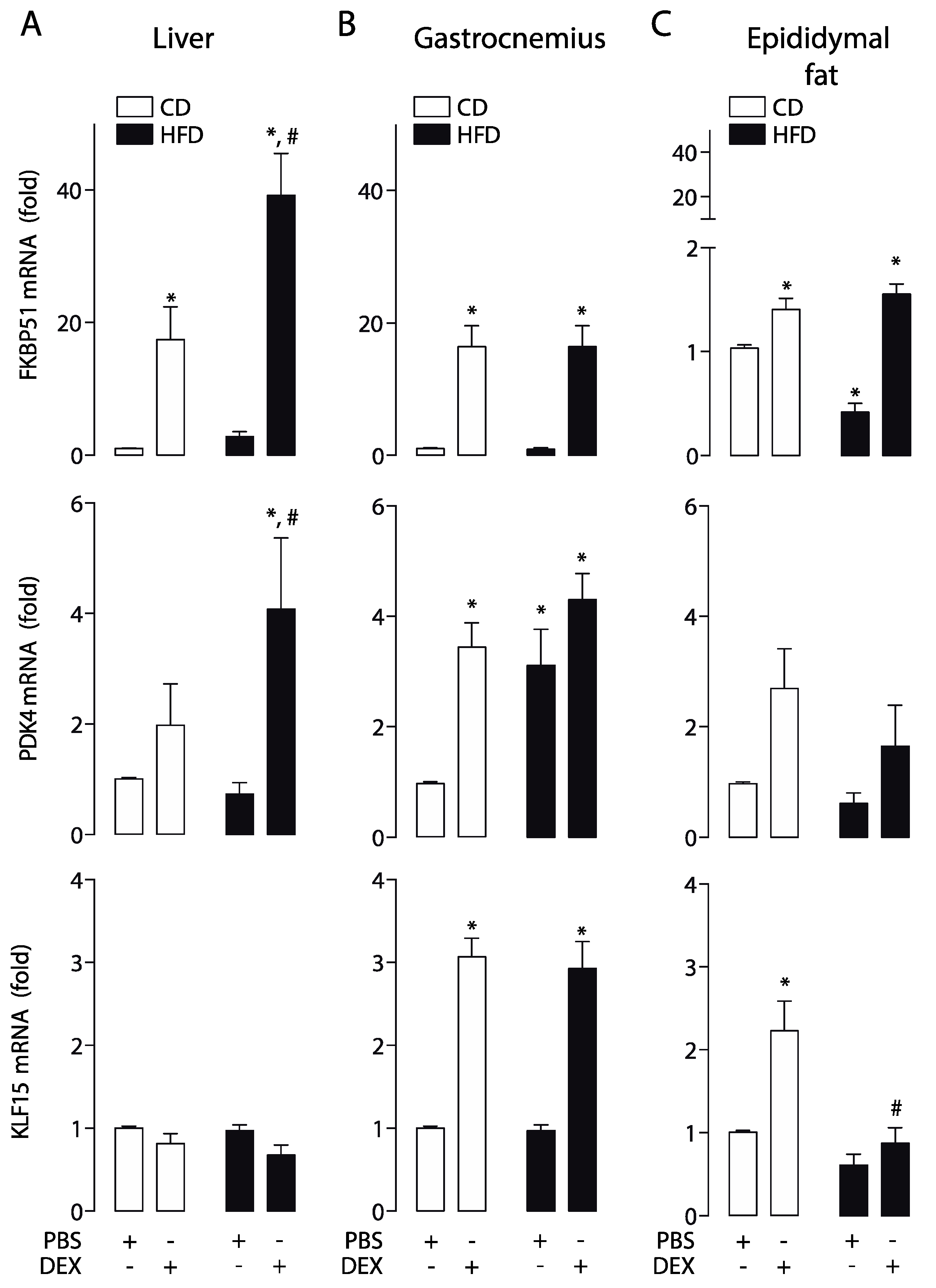
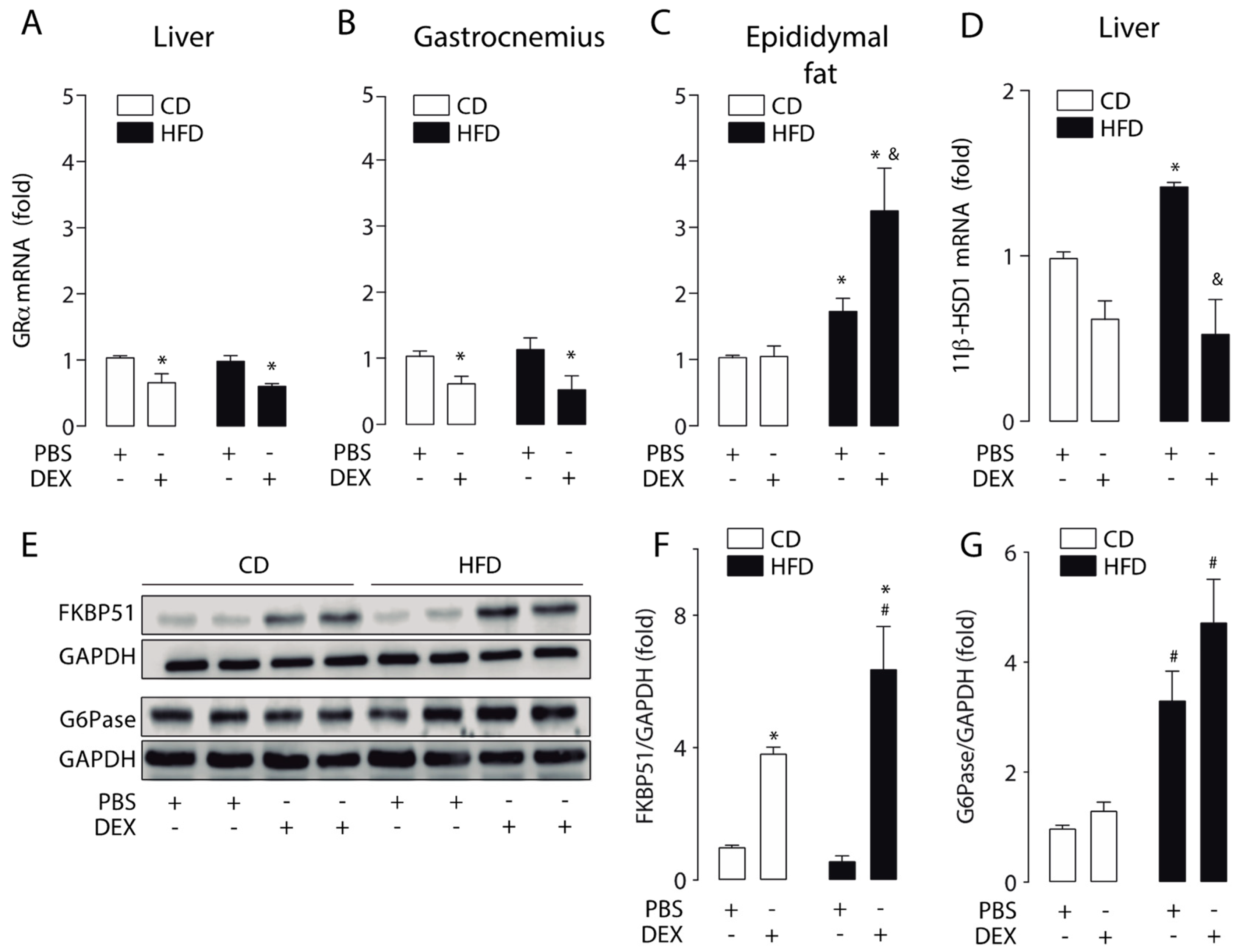
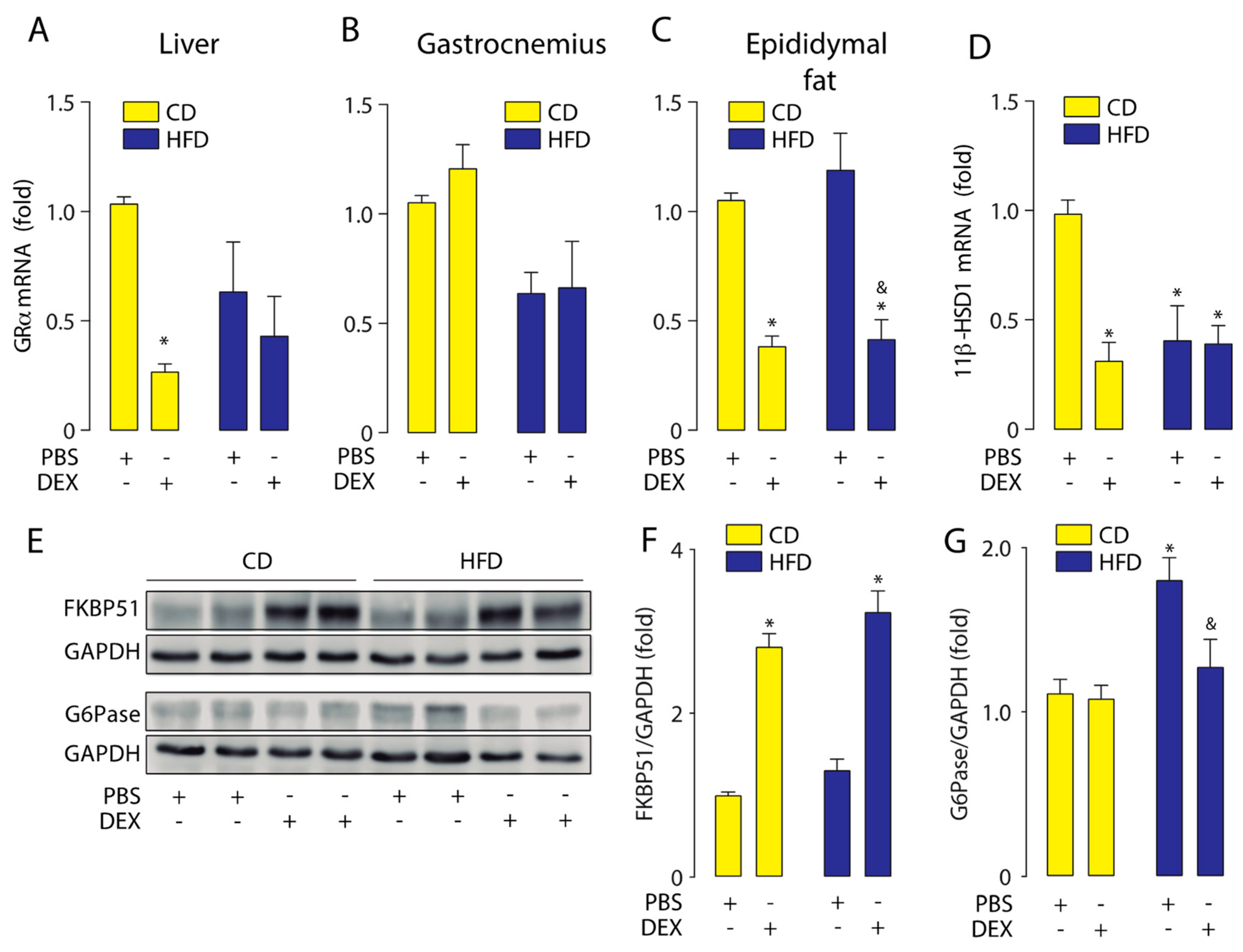
2.2. High-Fat Diet Altered GC Response in the Liver, Skeletal Muscle, and Epididymal White Adipose Tissue
2.3. Physical Training Increased the Corticosterone Plasmatic Levels
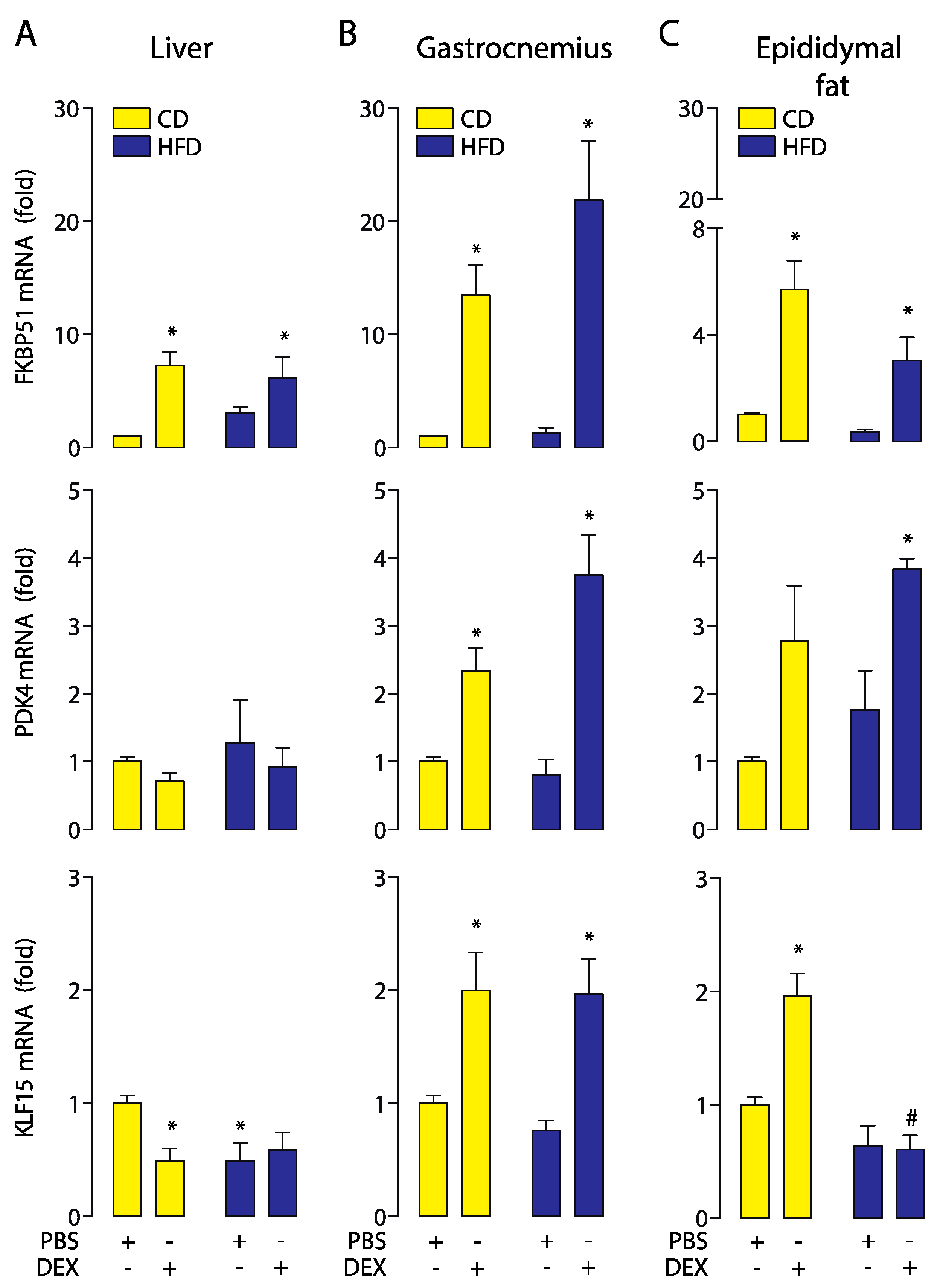
2.4. Moderate Aerobic Exercise Prevented the Hypersensitivity of Hepatic GC Response
3. Discussion
4. Materials and Methods
4.1. Animals and Study Design
4.2. Glucose Tolerance Test (GTT)
4.3. Corticosterone
4.4. RNA Extraction, cDNA Synthesis, and Real-Time PCR
4.5. Western Blot
4.6. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| 11β-HSD1 | 11β-hydroxysteroid dehydrogenase type 1 |
| CD | Control diet |
| DEX | Dexamethasone |
| FKBP51 | FK506-binding protein 51 |
| GAPDH | Glyceraldehyde-3-phosphate dehydrogenase |
| G6Pase | Glucose-6-phosphatase |
| GTT | Glucose tolerance test |
| HFD | High-fat diet |
| HPA | Hypothalamic–pituitary–adrenal |
| i.p. | Intraperitoneally |
| GCs | Glucocorticoids |
| GR | Glucocorticoid receptor |
| GRα | Glucocorticoid receptor alpha |
| KLF15 | Krüppel-like factor 15 |
| PDK4 | Pyruvate dehydrogenase kinase 4 |
References
- Schetz, M.; De Jong, A.; Deane, A.M.; Druml, W.; Hemelaar, P.; Pelosi, P.; Pickkers, P.; Reintam-Blaser, A.; Roberts, J.; Sakr, Y.; et al. Obesity in the critically ill: A narrative review. Intensiv. Care Med. 2019, 45, 757–769. [Google Scholar] [CrossRef] [PubMed]
- Galgani, J.E.; Moro, C.; Ravussin, E. Metabolic flexibility and insulin resistance. Am. J. Physiol. Endocrinol. Metab. 2008, 295, E1009–E1017. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- van Jaarsveld, C.H.M.; Fidler, J.A.; Steptoe, A.; Boniface, D.; Wardle, J. Perceived stress and weight gain in adolescence: A longitudinal analysis. Obes. Silver Spring Md. 2009, 17, 2155–2161. [Google Scholar] [CrossRef]
- Daousi, C.; Dunn, A.J.; Foy, P.M.; MacFarlane, I.A.; Pinkney, J.H. Endocrine and neuroanatomic features associated with weight gain and obesity in adult patients with hypothalamic damage. Am. J. Med. 2005, 118, 45–50. [Google Scholar] [CrossRef] [PubMed]
- Patel, S.R.; Malhotra, A.; White, D.P.; Gottlieb, D.J.; Hu, F.B. Association between reduced sleep and weight gain in women. Am. J. Epidemiol. 2006, 164, 947–954. [Google Scholar] [CrossRef] [Green Version]
- Raskind, M.A.; Burke, B.L.; Crites, N.J.; Tapp, A.M.; Rasmussen, D.D. Olanzapine-induced weight gain and increased visceral adiposity is blocked by melatonin replacement therapy in rats. Neuropsychopharmacol. Off. Publ. Am. Coll. Neuropsychopharmacol. 2007, 32, 284–288. [Google Scholar] [CrossRef]
- Ghanemi, A.; Yoshioka, M.; St-Amand, J. Broken Energy Homeostasis and Obesity Pathogenesis: The Surrounding Concepts. J. Clin. Med. 2018, 7, 453. [Google Scholar] [CrossRef] [Green Version]
- Lucassen, E.A.; Cizza, G. The Hypothalamic-Pituitary-Adrenal Axis, Obesity, and Chronic Stress Exposure: Sleep and the HPA Axis in Obesity. Curr. Obes. Rep. 2012, 1, 208–215. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ramamoorthy, S.; Cidlowski, J.A. Corticosteroids: Mechanisms of Action in Health and Disease. Rheum. Dis. Clin. N. Am. 2016, 42, 15–31, vii. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Magomedova, L.; Cummins, C.L. Glucocorticoids and Metabolic Control. Handb. Exp. Pharmacol. 2016, 233, 73–93. [Google Scholar] [CrossRef] [PubMed]
- Vermeer, H.; Hendriks-Stegeman, B.I.; van der Burg, B.; van Buul-Offers, S.C.; Jansen, M. Glucocorticoid-Induced Increase in Lymphocytic FKBP51 Messenger Ribonucleic Acid Expression: A Potential Marker for Glucocorticoid Sensitivity, Potency, and Bioavailability. J. Clin. Endocrinol. Metab. 2003, 88, 277–284. [Google Scholar] [CrossRef]
- Bali, U.; Phillips, T.; Hunt, H.; Unitt, J. FKBP5 mRNA Expression Is a Biomarker for GR Antagonism. J. Clin. Endocrinol. Metab. 2016, 101, 4305–4312. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Häusl, A.S.; Balsevich, G.; Gassen, N.C.; Schmidt, M.V. Focus on FKBP51: A molecular link between stress and metabolic disorders. Mol. Metab. 2019, 29, 170–181. [Google Scholar] [CrossRef] [PubMed]
- Connaughton, S.; Chowdhury, F.; Attia, R.R.; Song, S.; Zhang, Y.; Elam, M.B.; Cook, G.A.; Park, E.A. Regulation of pyruvate dehydrogenase kinase isoform 4 (PDK4) gene expression by glucocorticoids and insulin. Mol. Cell. Endocrinol. 2010, 315, 159–167. [Google Scholar] [CrossRef] [Green Version]
- Lee, I.-K. The Role of Pyruvate Dehydrogenase Kinase in Diabetes and Obesity. Diabetes Metab. J. 2014, 38, 181. [Google Scholar] [CrossRef] [Green Version]
- Morrison-Nozik, A.; Anand, P.; Zhu, H.; Duan, Q.; Sabeh, M.; Prosdocimo, D.A.; Lemieux, M.E.; Nordsborg, N.; Russell, A.P.; MacRae, C.A.; et al. Glucocorticoids enhance muscle endurance and ameliorate Duchenne muscular dystrophy through a defined metabolic program. Proc. Natl. Acad. Sci. USA 2015, 112, E6780–E6789. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sefton, C.; Harno, E.; Davies, A.; Small, H.; Allen, T.-J.; Wray, J.R.; Lawrence, C.B.; Coll, A.P.; White, A. Elevated Hypothalamic Glucocorticoid Levels Are Associated With Obesity and Hyperphagia in Male Mice. Endocrinology 2016, 157, 4257–4265. [Google Scholar] [CrossRef] [Green Version]
- Ramamoorthy, S.; Cidlowski, J.A. Exploring the Molecular Mechanisms of Glucocorticoid Receptor Action from Sensitivity to Resistance. In Endocrine Development; Maghnie, M., Loche, S., Cappa, M., Ghizzoni, L., Lorini, R., Eds.; S. KARGER AG: Basel, Switzerland, 2013; Volume 24, pp. 41–56. ISBN 978-3-318-02267-4. [Google Scholar]
- Shpilberg, Y.; Beaudry, J.L.; D’Souza, A.; Campbell, J.E.; Peckett, A.; Riddell, M.C. A rodent model of rapid-onset diabetes induced by glucocorticoids and high-fat feeding. Dis. Model. Mech. 2012, 5, 671–680. [Google Scholar] [CrossRef] [Green Version]
- Liu, L.; Wang, X.; Jiao, H.; Zhao, J.; Lin, H. Glucocorticoids inhibited hypothalamic target of rapamycin in high fat diet-fed chicks. Poult. Sci. 2015, 94, 2221–2227. [Google Scholar] [CrossRef]
- Church, T. Exercise in Obesity, Metabolic Syndrome, and Diabetes. Prog. Cardiovasc. Dis. 2011, 53, 412–418. [Google Scholar] [CrossRef] [PubMed]
- Campbell, J.E.; Király, M.A.; Atkinson, D.J.; D’souza, A.M.; Vranic, M.; Riddell, M.C. Regular exercise prevents the development of hyperglucocorticoidemia via adaptations in the brain and adrenal glands in male Zucker diabetic fatty rats. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2010, 299, R168–R176. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Coutinho, A.E.; Campbell, J.E.; Fediuc, S.; Riddell, M.C. Effect of voluntary exercise on peripheral tissue glucocorticoid receptor content and the expression and activity of 11β-HSD1 in the Syrian hamster. J. Appl. Physiol. 2006, 100, 1483–1488. [Google Scholar] [CrossRef] [PubMed]
- Barel, M.; Perez, O.A.B.; Giozzet, V.A.; Rafacho, A.; Bosqueiro, J.R.; do Amaral, S.L. Exercise training prevents hyperinsulinemia, muscular glycogen loss and muscle atrophy induced by dexamethasone treatment. Eur. J. Appl. Physiol. 2010, 108, 999–1007. [Google Scholar] [CrossRef] [PubMed]
- Dionísio, T.; Louzada, J.; Viscelli, B.; Dionísio, E.; Martuscelli, A.; Barel, M.; Perez, O.; Bosqueiro, J.; Brozoski, D.; Santos, C.; et al. Aerobic Training Prevents Dexamethasone-Induced Peripheral Insulin Resistance. Horm. Metab. Res. 2014, 46, 484–489. [Google Scholar] [CrossRef] [PubMed]
- Krug, A.L.O.; Macedo, A.G.; Zago, A.S.; Rush, J.W.E.; Santos, C.F.; Amaral, S.L. High-intensity resistance training attenuates dexamethasone-induced muscle atrophy: Attenuation of Muscle Atrophy. Muscle Nerve 2016, 53, 779–788. [Google Scholar] [CrossRef] [PubMed]
- Nicastro, H.; Zanchi, N.E.; da Luz, C.R.; de Moraes, W.M.A.M.; Ramona, P.; de Siqueira Filho, M.A.; Chaves, D.F.S.; Medeiros, A.; Brum, P.C.; Dardevet, D.; et al. Effects of leucine supplementation and resistance exercise on dexamethasone-induced muscle atrophy and insulin resistance in rats. Nutrition 2012, 28, 465–471. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vander Kooi, B.T.; Onuma, H.; Oeser, J.K.; Svitek, C.A.; Allen, S.R.; Vander Kooi, C.W.; Chazin, W.J.; O’Brien, R.M. The Glucose-6-Phosphatase Catalytic Subunit Gene Promoter Contains Both Positive and Negative Glucocorticoid Response Elements. Mol. Endocrinol. 2005, 19, 3001–3022. [Google Scholar] [CrossRef] [PubMed]
- Nemati, M.; Zardooz, H.; Rostamkhani, F.; Abadi, A.; Foroughi, F. High-fat diet effects on metabolic responses to chronic stress. Arch. Physiol. Biochem. 2017, 123, 182–191. [Google Scholar] [CrossRef]
- Brown, J.L.; Spicer, M.T.; Spicer, L.J. Effect of High-Fat Diet on Body Composition and Hormone Responses to Glucose Tolerance Tests. Endocrine 2002, 19, 327–332. [Google Scholar] [CrossRef]
- Kamara, K.; Eskay, R.; Castonguay, T. High-Fat Diets and Stress Responsivity. Physiol. Behav. 1998, 64, 1–6. [Google Scholar] [CrossRef]
- Paredes, S.; Ribeiro, L. Cortisol: The villain in metabolic syndrome? Rev. Assoc. Medica Bras. 1992 2014, 60, 84–92. [Google Scholar] [CrossRef]
- Díaz-Castro, F.; Monsalves-Álvarez, M.; Rojo, L.E.; del Campo, A.; Troncoso, R. Mifepristone for Treatment of Metabolic Syndrome: Beyond Cushing’s Syndrome. Front. Pharmacol. 2020, 11, 429. [Google Scholar] [CrossRef] [PubMed]
- Koorneef, L.L.; van den Heuvel, J.K.; Kroon, J.; Boon, M.R.; Hoen, P.A.C.; Hettne, K.M.; van de Velde, N.M.; Kolenbrander, K.B.; Streefland, T.C.M.; Mol, I.M.; et al. Selective Glucocorticoid Receptor Modulation Prevents and Reverses Nonalcoholic Fatty Liver Disease in Male Mice. Endocrinology 2018, 159, 3925–3936. [Google Scholar] [CrossRef] [Green Version]
- Moraes, R.C.; Blondet, A.; Birkenkamp-Demtroeder, K.; Tirard, J.; Orntoft, T.F.; Gertler, A.; Durand, P.; Naville, D.; Bégeot, M. Study of the Alteration of Gene Expression in Adipose Tissue of Diet-Induced Obese Mice by Microarray and Reverse Transcription-Polymerase Chain Reaction Analyses. Endocrinology 2003, 144, 4773–4782. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morton, N.M.; Ramage, L.; Seckl, J.R. Down-Regulation of Adipose 11β-Hydroxysteroid Dehydrogenase Type 1 by High-Fat Feeding in Mice: A Potential Adaptive Mechanism Counteracting Metabolic Disease. Endocrinology 2004, 145, 2707–2712. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McClean, P.L.; Irwin, N.; Cassidy, R.S.; Holst, J.J.; Gault, V.A.; Flatt, P.R. GIP receptor antagonism reverses obesity, insulin resistance, and associated metabolic disturbances induced in mice by prolonged consumption of high-fat diet. Am. J. Physiol. Endocrinol. Metab. 2007, 293, E1746–E1755. [Google Scholar] [CrossRef] [PubMed]
- Michailidou, Z.; Carter, R.N.; Marshall, E.; Sutherland, H.G.; Brownstein, D.G.; Owen, E.; Cockett, K.; Kelly, V.; Ramage, L.; Al-Dujaili, E.A.S.; et al. Glucocorticoid receptor haploinsufficiency causes hypertension and attenuates hypothalamic-pituitary-adrenal axis and blood pressure adaptions to high-fat diet. FASEB J. 2008, 22, 3896–3907. [Google Scholar] [CrossRef] [Green Version]
- Liu, Y.; Nakagawa, Y.; Wang, Y.; Liu, L.; Du, H.; Wang, W.; Ren, X.; Lutfy, K.; Friedman, T.C. Reduction of hepatic glucocorticoid receptor and hexose-6-phosphate dehydrogenase expression ameliorates diet-induced obesity and insulin resistance in mice. J. Mol. Endocrinol. 2008, 41, 53–64. [Google Scholar] [CrossRef]
- Bamberger, C.M.; Schulte, H.M.; Chrousos, G.P. Molecular Determinants of Glucocorticoid Receptor Function and Tissue Sensitivity to Glucocorticoids. Endocr. Rev. 1996, 17, 245–261. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez, J.M.; Monsalves-Alvarez, M.; Henriquez, S.; Llanos, M.N.; Troncoso, R. Glucocorticoid resistance in chronic diseases. Steroids 2016, 115, 182–192. [Google Scholar] [CrossRef] [PubMed]
- Iida, S.; Nakamura, Y.; Fujii, H.; Nishimura, J.-I.; Tsugawa, M.; Gomi, M.; Fukata, J.; Tarui, S.; Moriwaki, K.; Kitani, T. A Patient with Hypocortisolism and Cushing’s Syndrome-Like Manifestations: Cortisol Hyperreactive Syndrome. J. Clin. Endocrinol. Metab. 1990, 70, 729–737. [Google Scholar] [CrossRef]
- Santen, R.J.; Jewell, C.M.; Yue, W.; Heitjan, D.F.; Raff, H.; Katen, K.S.; Cidlowski, J.A. Glucocorticoid Receptor Mutations and Hypersensitivity to Endogenous and Exogenous Glucocorticoids. J. Clin. Endocrinol. Metab. 2018, 103, 3630–3639. [Google Scholar] [CrossRef] [PubMed]
- Quax, R.A.; Manenschijn, L.; Koper, J.W.; Hazes, J.M.; Lamberts, S.W.J.; van Rossum, E.F.C.; Feelders, R.A. Glucocorticoid sensitivity in health and disease. Nat. Rev. Endocrinol. 2013, 9, 670–686. [Google Scholar] [CrossRef] [PubMed]
- Quinn, M.A.; Xu, X.; Ronfani, M.; Cidlowski, J.A. Estrogen Deficiency Promotes Hepatic Steatosis via a Glucocorticoid Receptor-Dependent Mechanism in Mice. Cell Rep. 2018, 22, 2690–2701. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shimizu, N.; Yoshikawa, N.; Ito, N.; Maruyama, T.; Suzuki, Y.; Takeda, S.; Nakae, J.; Tagata, Y.; Nishitani, S.; Takehana, K.; et al. Crosstalk between Glucocorticoid Receptor and Nutritional Sensor mTOR in Skeletal Muscle. Cell Metab. 2011, 13, 170–182. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Takashima, M.; Ogawa, W.; Hayashi, K.; Inoue, H.; Kinoshita, S.; Okamoto, Y.; Sakaue, H.; Wataoka, Y.; Emi, A.; Senga, Y.; et al. Role of KLF15 in Regulation of Hepatic Gluconeogenesis and Metformin Action. Diabetes 2010, 59, 1608–1615. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thoudam, T.; Ha, C.-M.; Leem, J.; Chanda, D.; Park, J.-S.; Kim, H.-J.; Jeon, J.-H.; Choi, Y.-K.; Liangpunsakul, S.; Huh, Y.H.; et al. PDK4 Augments ER–Mitochondria Contact to Dampen Skeletal Muscle Insulin Signaling During Obesity. Diabetes 2019, 68, 571–586. [Google Scholar] [CrossRef]
- Naowaboot, J.; Wannasiri, S.; Pannangpetch, P. Morin attenuates hepatic insulin resistance in high-fat-diet-induced obese mice. J. Physiol. Biochem. 2016, 72, 269–280. [Google Scholar] [CrossRef] [PubMed]
- Kabir, M.; Catalano, K.J.; Ananthnarayan, S.; Kim, S.P.; Van Citters, G.W.; Dea, M.K.; Bergman, R.N. Molecular evidence supporting the portal theory: A causative link between visceral adiposity and hepatic insulin resistance. Am. J. Physiol. Endocrinol. Metab. 2005, 288, E454–E461. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, J.; Wang, J.; Shao, J.; Gao, Y.; Xu, J.; Yu, S.; Liu, Z.; Jia, L. The unique pharmacological characteristics of mifepristone (RU486): From terminating pregnancy to preventing cancer metastasis. Med. Res. Rev. 2014, 34, 979–1000. [Google Scholar] [CrossRef] [PubMed]
- Grad, I.; Picard, D. The glucocorticoid responses are shaped by molecular chaperones. Mol. Cell. Endocrinol. 2007, 275, 2–12. [Google Scholar] [CrossRef]
- Denny, W.B.; Valentine, D.L.; Reynolds, P.D.; Smith, D.F.; Scammell, J.G. Squirrel Monkey Immunophilin FKBP51 Is a Potent Inhibitor of Glucocorticoid Receptor Binding 1. Endocrinology 2000, 141, 4107–4113. [Google Scholar] [CrossRef] [PubMed]
- Wochnik, G.M.; Rüegg, J.; Abel, G.A.; Schmidt, U.; Holsboer, F.; Rein, T. FK506-binding Proteins 51 and 52 Differentially Regulate Dynein Interaction and Nuclear Translocation of the Glucocorticoid Receptor in Mammalian Cells. J. Biol. Chem. 2005, 280, 4609–4616. [Google Scholar] [CrossRef] [Green Version]
- Fries, G.; Gassen, N.; Rein, T. The FKBP51 Glucocorticoid Receptor Co-Chaperone: Regulation, Function, and Implications in Health and Disease. Int. J. Mol. Sci. 2017, 18, 2614. [Google Scholar] [CrossRef] [Green Version]
- Fries, G.R.; Gassen, N.C.; Schmidt, U.; Rein, T. The FKBP51-Glucocorticoid Receptor Balance in Stress-Related Mental Disorders. Curr. Mol. Pharmacol. 2015, 9, 126–140. [Google Scholar] [CrossRef]
- Pereira, M.J.; Palming, J.; Svensson, M.K.; Rizell, M.; Dalenbäck, J.; Hammar, M.; Fall, T.; Sidibeh, C.O.; Svensson, P.-A.; Eriksson, J.W. FKBP5 expression in human adipose tissue increases following dexamethasone exposure and is associated with insulin resistance. Metabolism 2014, 63, 1198–1208. [Google Scholar] [CrossRef] [PubMed]
- Stechschulte, L.A.; Qiu, B.; Warrier, M.; Hinds, T.D.; Zhang, M.; Gu, H.; Xu, Y.; Khuder, S.S.; Russo, L.; Najjar, S.M.; et al. FKBP51 Null Mice Are Resistant to Diet-Induced Obesity and the PPARγ Agonist Rosiglitazone. Endocrinology 2016, 157, 3888–3900. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Balsevich, G.; Häusl, A.S.; Meyer, C.W.; Karamihalev, S.; Feng, X.; Pöhlmann, M.L.; Dournes, C.; Uribe-Marino, A.; Santarelli, S.; Labermaier, C.; et al. Stress-responsive FKBP51 regulates AKT2-AS160 signaling and metabolic function. Nat. Commun. 2017, 8, 1725. [Google Scholar] [CrossRef] [Green Version]
- Gleeson, M.; Bishop, N.C.; Stensel, D.J.; Lindley, M.R.; Mastana, S.S.; Nimmo, M.A. The anti-inflammatory effects of exercise: Mechanisms and implications for the prevention and treatment of disease. Nat. Rev. Immunol. 2011, 11, 607–615. [Google Scholar] [CrossRef] [PubMed]
- Viru, A.; Viru, M. Cortisol—Essential Adaptation Hormone in Exercise. Int. J. Sports Med. 2004, 25, 461–464. [Google Scholar] [CrossRef]
- Zorgati, H.; Prieur, F.; Vergniaud, T.; Cottin, F.; Do, M.-C.; Labsy, Z.; Amarantini, D.; Gagey, O.; Lasne, F.; Collomp, K. Ergogenic and metabolic effects of oral glucocorticoid intake during repeated bouts of high-intensity exercise. Steroids 2014, 86, 10–15. [Google Scholar] [CrossRef]
- la Fuente, F.P.; Quezada, L.; Sepúlveda, C.; Monsalves-Alvarez, M.; Rodríguez, J.M.; Sacristán, C.; Chiong, M.; Llanos, M.; Espinosa, A.; Troncoso, R. Exercise regulates lipid droplet dynamics in normal and fatty liver. Biochim. Biophys. Acta BBA Mol. Cell Biol. Lipids 2019, 1864, 158519. [Google Scholar] [CrossRef] [PubMed]
- Ok, D.-P.; Ko, K.; Bae, J.Y. Exercise without dietary changes alleviates nonalcoholic fatty liver disease without weight loss benefits. Lipids Health Dis. 2018, 17, 207. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brownlee, K.K.; Moore, A.W.; Hackney, A.C. Relationship between circulating cortisol and testosterone: Influence of physical exercise. J. Sports Sci. Med. 2005, 4, 76–83. [Google Scholar]
- Malisch, J.L.; Saltzman, W.; Gomes, F.R.; Rezende, E.L.; Jeske, D.R.; Garland, T., Jr. Baseline and Stress-Induced Plasma Corticosterone Concentrations of Mice Selectively Bred for High Voluntary Wheel Running. Physiol. Biochem. Zool. 2007, 80, 146–156. [Google Scholar] [CrossRef] [Green Version]
- Stranahan, A.M.; Lee, K.; Mattson, M.P. Central Mechanisms of HPA Axis Regulation by Voluntary Exercise. NeuroMol. Med. 2008, 10, 118–127. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, W.; Zhai, X.; Li, H.; Ji, L. Depression-like behaviors in mice subjected to co-treatment of high-fat diet and corticosterone are ameliorated by AICAR and exercise. J. Affect. Disord. 2014, 156, 171–177. [Google Scholar] [CrossRef] [PubMed]
- Beaudry, J.L.; Dunford, E.C.; Leclair, E.; Mandel, E.R.; Peckett, A.J.; Haas, T.L.; Riddell, M.C. Voluntary exercise improves metabolic profile in high-fat fed glucocorticoid-treated rats. J. Appl. Physiol. 2015, 118, 1331–1343. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Duclos, M.; Gouarne, C.; Bonnemaison, D. Acute and chronic effects of exercise on tissue sensitivity to glucocorticoids. J. Appl. Physiol. 2003, 94, 869–875. [Google Scholar] [CrossRef]
- DeRijk, R.H.; Petrides, J.; Deuster, P.; Gold, P.W.; Sternberg, E.M. Changes in corticosteroid sensitivity of peripheral blood lymphocytes after strenuous exercise in humans. J. Clin. Endocrinol. Metab. 1996, 81, 228–235. [Google Scholar] [CrossRef]
- Disanzo, B.L.; You, T. Effects of exercise training on indicators of adipose tissue angiogenesis and hypoxia in obese rats. Metabolism 2014, 63, 452–455. [Google Scholar] [CrossRef] [PubMed]
- Sidibeh, C.O.; Pereira, M.J.; Abalo, X.M.; Boersma, G.J.; Skrtic, S.; Lundkvist, P.; Katsogiannos, P.; Hausch, F.; Castillejo-López, C.; Eriksson, J.W. FKBP5 expression in human adipose tissue: Potential role in glucose and lipid metabolism, adipogenesis and type 2 diabetes. Endocrine 2018, 62, 116–128. [Google Scholar] [CrossRef] [PubMed] [Green Version]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Exercise | ||||
|---|---|---|---|---|
| CD | HFD | CD | HFD | |
| Weight | ||||
| Body (g) | 30 ± 0.7 | 44.6 ± 1.1 * | 29.3 ± 0.4 | 41 ± 1.6 * |
| Liver (g) | 1.46 ± 0.2 | 1.54 ± 0.2 | 1.46 + 0.2 | 1.63 ± 0.2 |
| eWAT (g) | 0.72 ± 0.2 | 2.51 ± 0.4 * | 0.71 ± 0.2 | 2.77 + 0.2 * |
| Glucose | ||||
| Fasting blood (mg/dL) | 131 ± 3.7 | 178 ± 5.7 * | 122 ± 4.4 | 158 ± 9.2 # |
| GTT AUC | 9912 ± 295 | 13,424 ± 315 * | 9200 ± 228 | 12,053 ± 484 *,# |
| Exercise | ||||
|---|---|---|---|---|
| CD + DEX | HFD + DEX | CD + DEX | HFD + DEX | |
| Liver | ||||
| FKBP51 | 17.46 ± 4.9 | 39.21 ± 6.3 * | 7.23 ± 1.2# | 6.17 ± 1.6 # |
| PDK4 | 1.98 ± 0.7 | 4.08 ± 1.3 * | 0.71 + 0.1# | 0.92 ± 0.3 # |
| KLF15 | 0.81 ± 0.1 | 0.68 ± 0.1 | 0.49 ± 0.1 | 0.59 + 0.1 |
| Gastrocnemius | ||||
| FKBP51 | 14.07 ± 3.7 | 13.61 ± 3.5 | 13.51 ± 4.3 | 21.9 ± 5.2 |
| PDK4 | 3.44 ± 0.4 | 4.3 ± 0.47 | 2.53 ± 0.4 | 3.75 ± 0.6 |
| KFLF15 | 3.07 ± 0.2 | 2.93 ± 0.3 | 1.99 ± 0.3 | 1.97 ± 0.3 |
| Epididymal fat | ||||
| FKBP51 | 1.40 ± 0.1 | 1.55 ± 0.1 | 5.70 ± 1.1 * | 3.05 ± 0.9 |
| PDK4 | 2.70 ± 0.7 | 1.05 ± 0.5 | 2.78 ± 0.8 | 3.84 ± 0.2 # |
| KFLF15 | 2.23 ± 0.4 | 0.87 ± 0.2 * | 1.96 ± 0.2 | 0.60 ± 0.1 * |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dassonvalle, J.; Díaz-Castro, F.; Donoso-Barraza, C.; Sepúlveda, C.; Pino-de la Fuente, F.; Pino, P.; Espinosa, A.; Chiong, M.; Llanos, M.; Troncoso, R. Moderate Aerobic Exercise Training Prevents the Augmented Hepatic Glucocorticoid Response Induced by High-Fat Diet in Mice. Int. J. Mol. Sci. 2020, 21, 7582. https://doi.org/10.3390/ijms21207582
Dassonvalle J, Díaz-Castro F, Donoso-Barraza C, Sepúlveda C, Pino-de la Fuente F, Pino P, Espinosa A, Chiong M, Llanos M, Troncoso R. Moderate Aerobic Exercise Training Prevents the Augmented Hepatic Glucocorticoid Response Induced by High-Fat Diet in Mice. International Journal of Molecular Sciences. 2020; 21(20):7582. https://doi.org/10.3390/ijms21207582
Chicago/Turabian StyleDassonvalle, Jonatan, Francisco Díaz-Castro, Camila Donoso-Barraza, Carlos Sepúlveda, Francisco Pino-de la Fuente, Pamela Pino, Alejandra Espinosa, Mario Chiong, Miguel Llanos, and Rodrigo Troncoso. 2020. "Moderate Aerobic Exercise Training Prevents the Augmented Hepatic Glucocorticoid Response Induced by High-Fat Diet in Mice" International Journal of Molecular Sciences 21, no. 20: 7582. https://doi.org/10.3390/ijms21207582
APA StyleDassonvalle, J., Díaz-Castro, F., Donoso-Barraza, C., Sepúlveda, C., Pino-de la Fuente, F., Pino, P., Espinosa, A., Chiong, M., Llanos, M., & Troncoso, R. (2020). Moderate Aerobic Exercise Training Prevents the Augmented Hepatic Glucocorticoid Response Induced by High-Fat Diet in Mice. International Journal of Molecular Sciences, 21(20), 7582. https://doi.org/10.3390/ijms21207582