Taming the Sentinels: Microbiome-Derived Metabolites and Polarization of T Cells

Abstract
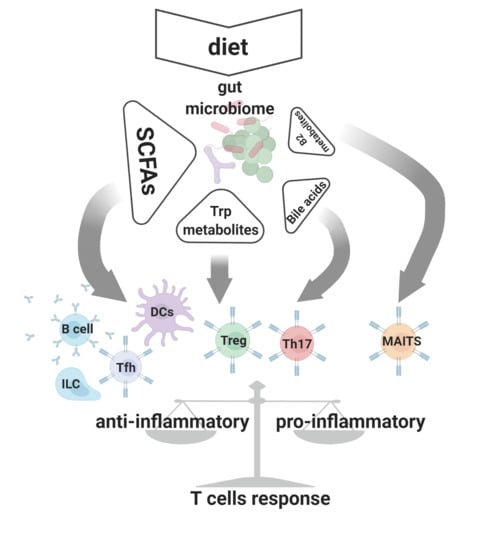
:
1. Introduction
2. Short Chain Fatty Acids (SCFAs)
3. The Role of SCFAs in T Cell Driven Immune Homeostasis
4. Tryptophan Metabolites
4.1. Synthesis and Biological Activity
4.2. Microbial-Derived Indole Derivatives and Control of Adaptive Immunity
4.3. Bile Acid Metabolism
4.4. Vitamin B2 Metabolites
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Josefowicz, S.Z.; Niec, R.E.; Kim, H.Y.; Treuting, P.; Chinen, T.; Zheng, Y.; Umetsu, D.T.; Rudensky, A.Y. Extrathymically generated regulatory T cells control mucosal TH2 inflammation. Nature 2012, 482, 395–399. [Google Scholar] [CrossRef]
- Cebula, A.; Seweryn, M.; Rempala, G.A.; Pabla, S.S.; McIndoe, R.A.; Denning, T.L.; Bry, L.; Kraj, P.; Kisielow, P.; Ignatowicz, L. Thymus-derived regulatory T cells contribute to tolerance to commensal microbiota. Nature 2013, 497, 258–262. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goto, Y.; Panea, C.; Nakato, G.; Cebula, A.; Lee, C.; Diez, M.G.; Laufer, T.M.; Ignatowicz, L.; Ivanov, I.I. Segmented filamentous bacteria antigens presented by intestinal dendritic cells drive mucosal Th17 cell differentiation. Immunity 2014, 40, 594–607. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mayans, S.; Stepniak, D.; Palida, S.F.; Larangé, A.; Dreux, J.; Arlian, B.M.; Shinnakasu, R.; Kronenberg, M.; Cheroutre, H.; Lambolez, F. αβT cell receptors expressed by CD4-CD8αβ- intraepithelial T cells drive their fate into a unique lineage with unusual MHC reactivities. Immunity 2014, 41, 207–218. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McDonald, B.D.; Bunker, J.J.; Ishizuka, I.E.; Jabri, B.; Bendelac, A. Elevated T cell receptor signaling identifies a thymic precursor to the TCRαβ+CD4-CD8β- intraepithelial lymphocyte lineage. Immunity 2014, 41, 219–229. [Google Scholar] [CrossRef] [Green Version]
- Wojciech, L.; Szurek, E.; Kuczma, M.; Cebula, A.; Elhefnawy, W.R.; Pietrzak, M.; Rempala, G.; Ignatowicz, L. Non-canonicaly recruited TCRαβCD8αα IELs recognize microbial antigens. Sci. Rep. 2018, 8, 10848. [Google Scholar] [CrossRef] [Green Version]
- Lee, H.-G.; Lee, J.-U.; Kim, D.-H.; Lim, S.; Kang, I.; Choi, J.-M. Pathogenic function of bystander-activated memory-like CD4+ T cells in autoimmune encephalomyelitis. Nat. Commun. 2019, 10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, J.; Gordon, J.I. Honor thy symbionts. Proc. Natl. Acad. Sci. USA 2003, 100, 10452–10459. [Google Scholar] [CrossRef] [Green Version]
- Almeida, A.; Nayfach, S.; Boland, M.; Strozzi, F.; Beracochea, M.; Shi, Z.J.; Pollard, K.S.; Sakharova, E.; Parks, D.H.; Hugenholtz, P.; et al. A unified catalog of 204,938 reference genomes from the human gut microbiome. Nat. Biotechnol. 2020. [Google Scholar] [CrossRef] [PubMed]
- Östman, S.; Rask, C.; Wold, A.; Hultkrantz, S.; Telemo, E. Telemo E. Impaired regulatory T cell function in germ-free mice. Eur. J. Immunol. 2006, 36, 2336–2346. [Google Scholar] [CrossRef]
- Ivanov, I.I.; de Llanos Frutos, R.; Manel, N.; Yoshinaga, K.; Rifkin, D.B.; Sartor, R.B.; Finlay, B.B.; Littman, D.R. Specific microbiota direct the differentiation of Th17 cells in the mucosa of the small intestine. Cell Host Microbe 2008, 4, 337–349. [Google Scholar] [CrossRef] [Green Version]
- Narushima, S.; Sugiura, Y.; Oshima, K.; Atarashi, K.; Hattori, M.; Suematsu, M.; Honda, K. Characterization of the 17 strains of regulatory T cell-inducing human-derived Clostridia. Gut Microbes 2014, 5, 333–339. [Google Scholar] [CrossRef] [PubMed]
- Atarashi, K.; Tanoue, T.; Shima, T.; Imaoka, A.; Kuwahara, T.; Momose, Y.; Cheng, G.; Yamasaki, S.; Saito, T.; Ohba, Y.; et al. Induction of colonic regulatory T cells by indigenous Clostridium species. Science 2010, 331, 337–341. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Luu, M.; Monning, H.; Visekruna, A. Exploring the molecular mechanisms underlying the protective effects of microbial SCFAs on intestinal tolerance and food allergy. Front. Immunol. 2020, 11, 16–21. [Google Scholar] [CrossRef]
- Ahern, P.P.; Faith, J.J.; Gordon, J.I. Mining the human gut microbiota for effector strains that shape the immune system. Immunity 2014, 40, 815–823. [Google Scholar] [CrossRef] [Green Version]
- Ni, J.; Wu, G.D.; Albenberg, L.; Tomov, V.T. Gut microbiota and IBD: Causation or correlation? Nat. Rev. Gastroenterol. Hepatol. 2017, 14, 573–584. [Google Scholar] [CrossRef] [Green Version]
- Britton, G.J.; Contijoch, E.J.; Mogno, I.; Vennaro, O.H.; Llewellyn, S.R.; Ng, R.; Li, Z.; Mortha, A.; Merad, M.; Das, A.; et al. Microbiotas from humans with inflammatory bowel disease alter the balance of gut Th17 and RORγt+ regulatory T cells and exacerbate colitis in mice. Immunity 2019, 50, 212–224. [Google Scholar] [CrossRef] [Green Version]
- Roager, H.M.; Licht, T.R. Microbial tryptophan catabolites in health and disease. Nat. Commun. 2018, 9, 3294. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Besten, G.D.; Van Eunen, K.; Groen, A.K.; Venema, K.; Reijngoud, D.-J.; Bakker, B.M. The role of short-chain fatty acids in the interplay between diet, gut microbiota, and host energy metabolism. J. Lipid Res. 2013, 54, 2325–2340. [Google Scholar] [CrossRef] [Green Version]
- Anderson, J.W.; Baird, P.; Davis, R.H.D.; Ferreri, S.; Knudtson, M.; Koraym, A.; Waters, V.; Williams, C.L. Health benefits of dietary fiber. Nutr. Rev. 2009, 67, 188–205. [Google Scholar] [CrossRef]
- El Kaoutari, A.; Armougom, F.; Gordon, J.I.; Raoult, D.; Henrissat, B. The abundance and variety of carbohydrate-active enzymes in the human gut microbiota. Nat. Rev. Microbiol. 2013, 11, 497–504. [Google Scholar] [CrossRef]
- Wächtershäuser, A.; Stein, J. Rationale for the luminal provision of butyrate in intestinal diseases. Eur. J. Nutr. 2000, 39, 164–171. [Google Scholar] [CrossRef]
- De Groot, P.F.; Belzer, C.; Aydin, Ö.; Levin, E.; Levels, J.H.; Aalvink, S.; Boot, F.; Holleman, F.; Van Raalte, D.H.; Scheithauer, T.P.; et al. Distinct fecal and oral microbiota composition in human type 1 diabetes, an observational study. PLoS ONE 2017, 12, e0188475. [Google Scholar] [CrossRef] [PubMed]
- Bergman, E.N. Energy contributions of volatile fatty acids from the gastrointestinal tract in various species. Physiol. Rev. 1990, 70, 567–590. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sivaprakasam, S.; Bhutia, Y.D.; Yang, S.; Ganapathy, V. Short-Chain Fatty Acid Transporters: Role in Colonic Homeostasis. Compr. Physiol. 2017, 8, 299–314. [Google Scholar] [CrossRef] [PubMed]
- Louis, P.; Duncan, S.H.; McCrae, S.I.; Millar, J.; Jackson, M.S.; Flint, H.J.; Al, L.E.T. Restricted distribution of the butyrate kinase pathway among butyrate-producing bacteria from the human colon. J. Bacteriol. 2004, 186, 2099–2106. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Smith, P.M.; Howitt, M.R.; Panikov, N.; Michaud, M.; Gallini, C.A.; Bohlooly-Y, M.; Glickman, J.N.; Garrett, W.S. The microbial metabolites, short-chain fatty acids, regulate colonic Treg cell homeostasis. Science 2013, 341, 569–573. [Google Scholar] [CrossRef] [Green Version]
- De Smit, S.M.; De Leeuw, K.D.; Buisman, C.J.; Strik, D.P.B.T.B. Continuous n-valerate formation from propionate and methanol in an anaerobic chain elongation open-culture bioreactor. Biotechnol. Biofuels 2019, 12, 132. [Google Scholar] [CrossRef]
- Johnstone, R.W. Histone-deacetylase inhibitors: Novel drugs for the treatment of cancer. Nat. Rev. Drug Discov. 2002, 1, 287–299. [Google Scholar] [CrossRef]
- Bernhard, D.; Ausserlechner, M.J.; Tonko, M.; Löffler, M.; Hartmann, B.L.; Csordas, A.; Kofler, R. Apoptosis induced by the histone deacetylase inhibitor sodium butyrate in human leukemic lymphoblasts. FASEB J. 1999, 13, 1991–2001. [Google Scholar] [CrossRef]
- Martin-Gallausiaux, C.; Larraufie, P.F.; Jarry, A.; Béguet-Crespel, F.; Marinelli, L.; LeDue, F.; Reimann, F.; Blottière, H.M.; Lapaque, N. Butyrate produced by commensal bacteria down-regulates indolamine 2,3-dioxygenase 1 (IDO-1) expression via a dual mechanism in human intestinal epithelial cells. Front. Immunol. 2018, 9, 2838. [Google Scholar] [CrossRef] [PubMed]
- Furusawa, Y.; Obata, Y.; Fukuda, S.; Endo, T.A.; Nakato, G.; Takahashi, D.; Nakanishi, Y.; Uetake, C.; Kato, K.; Kato, T.; et al. Commensal microbe-derived butyrate induces the differentiation of colonic regulatory T cells. Nat. Cell Biol. 2013, 504, 446–450. [Google Scholar] [CrossRef] [PubMed]
- Arpaia, N.; Campbell, C.; Fan, X.; Dikiy, S.; Van Der Veeken, J.; DeRoos, P.; Liu, H.; Cross, J.R.; Pfeffer, K.; Coffer, P.J.; et al. Metabolites produced by commensal bacteria promote peripheral regulatory T-cell generation. Nature 2013, 504, 451–455. [Google Scholar] [CrossRef]
- Lu, Y.; Fan, C.; Li, P.; Lu, Y.; Chang, X.; Qi, K. Short chain fatty acids prevent high-fat-diet-induced obesity in mice by regulating G protein-coupled receptors and gut Microbiota. Sci. Rep. 2016, 6, 37589. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sivaprakasam, S.; Prasad, P.D.; Singh, N. Benefits of short-chain fatty acids and their receptors in inflammation and carcinogenesis. Pharmacol. Ther. 2016, 164, 144–151. [Google Scholar] [CrossRef] [Green Version]
- Chen, H.; Wettschureck, N.; Schwaninger, M.; Chen, H.; Assmann, J.C.; Krenz, A.; Rahman, M.; Grimm, M.; Karsten, C.M.; Köhl, J.; et al. Hydroxycarboxylic acid receptor 2 mediates dimethyl fumarate’s protective effect in EAE. J. Clin. Investig. 2014, 124, 2188–2192. [Google Scholar] [CrossRef]
- Brown, K.; Godovannyi, A.; Ma, C.; Zhang, Y.; Ahmadi-Vand, Z.; Dai, C.; Gorzelak, M.A.; Chan, Y.; Chan, J.M.; Lochner, A.; et al. The orphan G protein-coupled receptors GPR41 and GPR43 are activated by propionate and other short chain carboxylic acids. ISME J. 2015, 10, 321–332. [Google Scholar] [CrossRef] [PubMed]
- De Oliveira, P.G.; Ramos, M.L.S.; Amaro, A.J.; Dias, R.A.; Vieira, S.I. Gi/O-protein coupled receptors in the aging brain. Front. Aging Neurosci. 2019, 11, 89. [Google Scholar] [CrossRef] [Green Version]
- Zhang, L.; Shi, G. Gq-coupled receptors in autoimmunity. J. Immunol. Res. 2016, 2016, 3969023. [Google Scholar] [CrossRef] [Green Version]
- Ji, L.-S.; Sun, X.-H.; Zhang, X.; Zhou, Z.-H.; Yu, Z.; Zhu, X.-J.; Huang, L.-Y.; Fang, M.; Gao, Y.-T.; Li, M.; et al. Mechanism of follicular helper T cell differentiation regulated by transcription factors. J. Immunol. Res. 2020, 2020, 1826587. [Google Scholar] [CrossRef]
- Baidya, M.; Kumari, P.; Dwivedi, H.; Pandey, S.; Chaturvedi, M.; Stepniewski, T.M.; Kawakami, K.; Cao, Y.; Laporte, S.A.; Selent, J.; et al. Key phosphorylation sites in GPCRs orchestrate the contribution of β-Arrestin 1 in ERK1/2 activation. EMBO Rep. 2020, 21, e49886. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Kokrashvili, Z.; Mosinger, B.; Margolskee, R.F. Gustducin couples fatty acid receptors to GLP-1 release in colon. Am. J. Physiol. Metab. 2013, 304, E651–E660. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tolhurst, G.; Heffron, H.; Lam, Y.S.; Parker, H.E.; Habib, A.M.; Diakogiannaki, E.; Cameron, J.; Grosse, J.; Reimann, F.; Gribble, F.M. Short-chain fatty acids stimulate glucagon-like peptide-1 secretion via the G-protein-coupled receptor FFAR2. Diabetes 2011, 61, 364–371. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Le Bras, S.; Geha, R.S. IPEX and the role of Foxp3 in the development and function of human Tregs. J. Clin. Investig. 2006, 116, 1473–1475. [Google Scholar] [CrossRef]
- Brunkow, M.E.; Jeffery, E.W.; Hjerrild, K.A.; Paeper, B.; Clark, L.B.; Yasayko, S.-A.; Wilkinson, J.E.; Galas, D.; Ziegler, S.F.; Ramsdell, F. Disruption of a new forkhead/winged-helix protein, scurfin, results in the fatal lymphoproliferative disorder of the scurfy mouse. Nat. Genet. 2001, 27, 68–73. [Google Scholar] [CrossRef]
- Josefowicz, S.Z.; Lu, L.-F.; Rudensky, A.Y. Regulatory T cells: Mechanisms of differentiation and function. Annu. Rev. Immunol. 2012, 30, 531–564. [Google Scholar] [CrossRef]
- Coombes, J.L.; Powrie, F. Dendritic cells in intestinal immune regulation. Nat. Rev. Immunol. 2008, 8, 435–446. [Google Scholar] [CrossRef]
- Tan, J.; McKenzie, C.; Vuillermin, P.J.; Goverse, G.; Vinuesa, C.G.; Mebius, R.E.; Macia, L.; Mackay, C.R. Dietary fiber and Bbacterial SCFA enhance oral tolerance and protect against food allergy through diverse cellular pathways. Cell Rep. 2016, 15, 2809–2824. [Google Scholar] [CrossRef] [Green Version]
- Pabst, O. New concepts in the generation and functions of IgA. Nat. Rev. Immunol. 2012, 12, 821–832. [Google Scholar] [CrossRef]
- Bunker, J.J.; Flynn, T.M.; Koval, J.C.; Shaw, D.G.; Meisel, M.; McDonald, B.D.; Ishizuka, I.E.; Dent, A.L.; Wilson, P.C.; Jabri, B.; et al. Innate and adaptive humoral responses coat distinct commensal bacteria with immunoglobulin A. Immunity 2015, 43, 541–553. [Google Scholar] [CrossRef] [Green Version]
- Melo-Gonzalez, F.; Kammoun, H.; Evren, E.; Dutton, E.E.; Papadopoulou, M.; Bradford, B.M.; Tanes, C.; Fardus-Reid, F.; Swann, J.; Bittinger, K.; et al. Antigen-presenting ILC3 regulate T cell-dependent IgA responses to colonic mucosal bacteria. J. Exp. Med. 2019, 216, 728–742. [Google Scholar] [CrossRef]
- Sepahi, A.; Liu, Q.; Friesen, L.; Kim, C.H. Dietary fiber metabolites regulate innate lymphoid cell responses. Mucosal Immunol. 2020. [Google Scholar] [CrossRef] [PubMed]
- Luu, M.; Pautz, S.; Kohl, V.; Singh, R.; Romero, R.; Lucas, S.; Hofmann, J.; Raifer, H.; Vachharajani, N.; Carrascosa, L.C.; et al. The short-chain fatty acid pentanoate suppresses autoimmunity by modulating the metabolic-epigenetic crosstalk in lymphocytes. Nat. Commun. 2019, 10, 760. [Google Scholar] [CrossRef] [Green Version]
- Bluestone, J.A.; Tang, Q.; Sedwick, C.E. T regulatory cells in autoimmune diabetes: Past challenges, future prospects. J. Clin. Immunol. 2008, 28, 677–684. [Google Scholar] [CrossRef]
- Gavin, P.G.; Hamilton-Williams, E.E. The gut microbiota in type 1 diabetes: Friend or foe? Curr. Opin. Endocrinol. Diabetes Obes. 2019, 26, 207–212. [Google Scholar] [CrossRef] [PubMed]
- Endesfelder, D.; Zu Castell, W.; Ardissone, A.; Davis-Richardson, A.G.; Achenbach, P.; Hagen, M.; Pflueger, M.; Gano, K.A.; Fagen, J.R.; Drew, J.C.; et al. Compromised gut microbiota networks in children with anti-islet cell autoimmunity. Diabetes 2014, 63, 2006–2014. [Google Scholar] [CrossRef] [Green Version]
- Mariño, E.; Richards, J.L.; McLeod, K.H.; Stanley, D.; Yap, Y.A.; Knight, J.; McKenzie, C.; Kranich, J.; Oliveira, A.C.; Rossello, F.J.; et al. Gut microbial metabolites limit the frequency of autoimmune T cells and protect against type 1 diabetes. Nat. Immunol. 2017, 18, 552–562. [Google Scholar] [CrossRef]
- Fehlbaum, S.; Prudence, K.; Kieboom, J.; Heerikhuisen, M.; Broek, T.J.V.D.; Schuren, F.H.J.; Steinert, R.E.; Raederstorff, D. In vitro fermentation of selected prebiotics and their effects on the composition and activity of the adult gut microbiota. Int. J. Mol. Sci. 2018, 19, 3097. [Google Scholar] [CrossRef] [Green Version]
- Brown, K.; Godovannyi, A.; Ma, C.; Zhang, Y.; Ahmadi-Vand, Z.; Dai, C.; Gorzelak, M.A.; Chan, Y.; Chan, J.M.; Lochner, A.; et al. Prolonged antibiotic treatment induces a diabetogenic intestinal microbiome that accelerates diabetes in NOD mice. ISME J. 2016, 10, 321–332. [Google Scholar] [CrossRef]
- Hu, M.; Eviston, D.; Hsu, P.; Mariño, E.; Chidgey, A.; Santner-Nanan, B.; Wong, K.; Richards, J.L.; Yap, Y.A.; Collier, F.; et al. Decreased maternal serum acetate and impaired fetal thymic and regulatory T cell development in preeclampsia. Nat. Commun. 2019, 10, 3031. [Google Scholar] [CrossRef] [PubMed]
- El-Chennawi, F.; Rageh, I.M.; Mansour, A.; Darwish, M.I.; Elghzaly, A.A.; Sakr, B.E.S.; Elbaz, K.M. Comparison of the percentages of CD4+ CD25high FOXP3+, CD4+ CD25low FOXP3+, and CD4+ FOXP3+ Tregs, in the umbilical cord blood of babies born to mothers with and without preeclampsia. Am. J. Reprod. Immunol. 2017, 78, 1–7. [Google Scholar] [CrossRef]
- Loewendorf, A.I.; Nguyen, T.A.; Yesayan, M.N.; Kahn, D.A. Preeclampsia is Characterized by Fetal NK Cell Activation and a Reduction in Regulatory T Cells. Am. J. Reprod. Immunol. 2015, 74, 258–267. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Santner-Nanan, B.; Straubinger, K.; Hsu, P.; Parnell, G.; Tang, B.; Xu, B.; Makris, A.; Hennessy, A.; Peek, M.J.; Busch, D.H.; et al. Fetal–maternal alignment of regulatory T cells correlates with IL-10 and Bcl-2 upregulation in pregnancy. J. Immunol. 2013, 191, 145–153. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Byberg, K.K.; Ogland, B.; Eide, G.E.; Øymar, K. Birth after preeclamptic pregnancies: Association with allergic sensitization and allergic rhinoconjunctivitis in late childhood; a historically matched cohort study. BMC Pediatr. 2014, 14, 101. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Birukov, A.; Herse, F.; Nielsen, J.H.; Kyhl, H.B.; Golic, M.; Kräker, K.; Haase, N.; Busjahn, A.; Bruun, S.; Jensen, B.L.; et al. Blood pressure and angiogenic markers in pregnancy. Hypertension 2020, 76, 901–909. [Google Scholar] [CrossRef] [PubMed]
- Aschenbrenner, K.; D’Cruz, L.M.; Vollmann, E.H.; Hinterberger, M.; Emmerich, J.; Swee, L.K.; Rolink, A.; Klein, L. Selection of Foxp3+ regulatory T cells specific for self antigen expressed and presented by Aire+ medullary thymic epithelial cells. Nat. Immunol. 2007, 8, 351–358. [Google Scholar] [CrossRef]
- Anderson, M.S.; Venanzi, E.S.; Klein, L.; Chen, Z.; Berzins, S.P.; Turley, S.J.; Von Boehmer, H.; Bronson, R.; Dierich, A.; Benoist, C.; et al. Projection of an immunological self shadow within the thymus by the aire protein. Science 2002, 298, 1395–1401. [Google Scholar] [CrossRef] [Green Version]
- Ngiow, S.F.; Young, A. Re-education of the tumor microenvironment with targeted therapies and immunotherapies. Front. Immunol. 2020, 11, 1633. [Google Scholar] [CrossRef]
- Wei, S.C.; Duffy, C.R.; Allison, J.P. Fundamental mechanisms of immune checkpoint blockade therapy. Cancer Discov. 2018, 8, 1069–1086. [Google Scholar] [CrossRef] [Green Version]
- Chamoto, K.; Hatae, R.; Honjo, T. Current issues and perspectives in PD-1 blockade cancer immunotherapy. Int. J. Clin. Oncol. 2020, 25, 790–800. [Google Scholar] [CrossRef] [Green Version]
- Hakozaki, T.; Richard, C.; Elkrief, A.; Hosomi, Y.; Benlaïfaoui, M.; Mimpen, I.; Terrisse, S.; DeRosa, L.; Zitvogel, L.; Routy, B.; et al. The gut microbiome associates with immune checkpoint inhibition outcomes in patients with advanced non-small cell lung cancer. Cancer Immunol. Res. 2020, 8, 1243–1250. [Google Scholar] [CrossRef] [PubMed]
- Salgia, N.J.; Bergerot, P.G.; Maia, M.C.; Dizman, N.; Hsu, J.; Gillece, J.D.; Folkerts, M.; Reining, L.; Trent, J.; Highlander, S.K.; et al. Stool microbiome profiling of patients with metastatic renal cell carcinoma receiving anti-PD-1 immune checkpoint inhibitors. Eur. Urol. 2020, 78, 498–502. [Google Scholar] [CrossRef]
- Coutzac, C.; Jouniaux, J.-M.; Paci, A.; Schmidt, J.; Mallardo, D.; Seck, A.; Asvatourian, V.; Cassard, L.; Saulnier, P.; Lacroix, L.; et al. Systemic short chain fatty acids limit antitumor effect of CTLA-4 blockade in hosts with cancer. Nat. Commun. 2020, 11, 2168. [Google Scholar] [CrossRef] [PubMed]
- Wikoff, W.R.; Anfora, A.T.; Liu, J.; Schultz, P.G.; Lesley, S.A.; Peters, E.C.; Siuzdak, G. Metabolomics analysis reveals large effects of gut microflora on mammalian blood metabolites. Proc. Natl. Acad. Sci. USA 2009, 106, 3698–3703. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hashimoto, T.; Perlot, T.; Rehman, A.; Trichereau, J.; Ishiguro, H.; Paolino, M.; Sigl, V.; Hanada, T.; Hanada, R.; Lipinski, S.; et al. ACE2 links amino acid malnutrition to microbial ecology and intestinal inflammation. Nature 2012, 487, 477–481. [Google Scholar] [CrossRef]
- Gao, J.; Xu, K.; Liu, H.; Liu, G.; Bai, M.; Peng, C.; Li, T.; Yin, Y. Impact of the gut microbiota on intestinal immunity mediated by tryptophan metabolism. Front. Cell. Infect. Microbiol. 2018, 8, 13. [Google Scholar] [CrossRef] [Green Version]
- Smith, T. A modification of the method for determining the production of indol by bacteria. J. Exp. Med. 1897, 2, 543–547. [Google Scholar] [CrossRef]
- Eme, L.; Gentekaki, E.; Curtis, B.; Archibald, J.M.; Roger, A.J. Lateral gene transfer in the adaptation of the anaerobic parasite Blastocystis to the gut. Curr. Biol. 2017, 27, 807–820. [Google Scholar] [CrossRef] [Green Version]
- Denoeud, F.; Roussel, M.; Noel, B.; Wawrzyniak, I.; Da Silva, C.; Diogon, M.; Viscogliosi, E.; Brochier-Armanet, C.; Couloux, A.; Poulain, J.; et al. Genome sequence of the stramenopile Blastocystis, a human anaerobic parasite. Genome Biol. 2011, 12, R29. [Google Scholar] [CrossRef]
- Wawrzyniak, I.; Courtine, D.; Osman, M.; Hubans-Pierlot, C.; Cian, A.; Nourrisson, C.; Chabé, M.; Poirier, P.; Bart, A.; Polonais, V.; et al. Draft genome sequence of the intestinal parasite Blastocystis subtype 4-isolate WR1. Genom. Data 2015, 4, 22–23. [Google Scholar] [CrossRef]
- Beghini, F.; Pasolli, E.; Truong, T.D.; Putignani, L.; Cacciò, S.M.; Segata, N. Large-scale comparative metagenomics of Blastocystis, a common member of the human gut microbiome. ISME J. 2017, 11, 2848–2863. [Google Scholar] [CrossRef] [PubMed]
- Yason, J.A.; Liang, Y.R.; Png, C.W.; Zhang, Y.; Tan, K.S.W. Interactions between a pathogenic Blastocystis subtype and gut microbiota: In vitro and in vivo studies. Microbiome 2019, 7, 30. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Venkatesh, M.; Mukherjee, S.; Wang, H.; Li, H.; Sun, K.; Benechet, A.P.; Qiu, Z.; Maher, L.; Redinbo, M.R.; Phillips, R.S.; et al. Symbiotic bacterial metabolites regulate gastrointestinal barrier function via the xenobiotic sensor PXR and toll-like receptor 4. Immunity 2014, 41, 296–310. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bessede, A.; Gargaro, M.; Pallotta, M.T.; Matino, D.; Servillo, G.; Brunacci, C.; Bicciato, S.; Mazza, E.M.C.; Macchiarulo, A.; Vacca, C.; et al. Aryl hydrocarbon receptor control of a disease tolerance defence pathway. Nature 2014, 511, 184–190. [Google Scholar] [CrossRef] [Green Version]
- Wheeler, J.L.H.; Martin, K.C.; Resseguie, E.; Lawrence, B.P. Differential consequences of two distinct AhR ligands on innate and adaptive immune responses to influenza a virus. Toxicol. Sci. 2013, 137, 324–334. [Google Scholar] [CrossRef] [Green Version]
- Ehrlich, A.K.; Pennington, J.M.; Bisson, W.H.; Kolluri, S.K.; Kerkvliet, N.I. TCDD, FICZ, and other high affinity AhR ligands dose-dependently determine the fate of CD4+ T cell differentiation. Toxicol. Sci. 2018, 161, 310–320. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gutiérrez-Vázquez, C.; Quintana, F.J. Regulation of the immune response by the aryl hydrocarbon receptor. Immunity 2018, 48, 19–33. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tian, L.X.; Tang, X.; Zhu, J.Y.; Luo, L.; Ma, X.Y.; Cheng, S.W.; Zhang, W.; Tang, W.Q.; Ma, W.; Yang, X.; et al. Cytochrome P450 1A1 enhances inflammatory responses and impedes phagocytosis of bacteria in macrophages during sepsis. Cell Commun. Signal. 2020, 18, 70. [Google Scholar] [CrossRef] [PubMed]
- Bettelli, E.; Carrier, Y.; Gao, W.; Korn, T.; Strom, T.B.; Oukka, M.; Weiner, H.L.; Kuchroo, V.K. Reciprocal developmental pathways for the generation of pathogenic effector TH17 and regulatory T cells. Nature 2006, 441, 235–238. [Google Scholar] [CrossRef]
- Dodd, D.; Spitzer, M.H.; Van Treuren, W.; Merrill, B.D.; Hryckowian, A.J.; Higginbottom, S.K.; Le, A.; Cowan, T.M.; Nolan, G.P.; Fischbach, M.A.; et al. A gut bacterial pathway metabolizes aromatic amino acids into nine circulating metabolites. Nature 2017, 551, 648–652. [Google Scholar] [CrossRef]
- Dubrac, S.; Elentner, A.; Ebner, S.; Horejs-Hoeck, J.; Schmuth, M. Modulation of T lymphocyte function by the pregnane X receptor. J. Immunol. 2010, 184, 2949–2957. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Veldhoen, M.; Hirota, K.; Christensen, J.; O’Garra, A.; Stockinger, B. Natural agonists for aryl hydrocarbon receptor in culture medium are essential for optimal differentiation of Th17 T cells. J. Exp. Med. 2009, 206, 43–49. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Smirnova, A.; Wincent, E.; Bergander, L.V.; Alsberg, T.; Bergman, J.; Rannug, A.; Rannug, U. Evidence for new light-independent pathways for generation of the endogenous aryl hydrocarbon receptor agonist FICZ. Chem. Res. Toxicol. 2015, 29, 75–86. [Google Scholar] [CrossRef] [PubMed]
- Veldhoen, M.; Hirota, K.; Westendorf, A.M.; Buer, J.; Dumoutier, L.; Renauld, J.-C.; Stockinger, B. The aryl hydrocarbon receptor links TH17-cell-mediated autoimmunity to environmental toxins. Nature 2008, 453, 106–109. [Google Scholar] [CrossRef] [PubMed]
- Quintana, F.J.; Basso, A.S.; Iglesias, A.H.; Korn, T.; Farez, M.F.; Bettelli, E.; Caccamo, M.; Oukka, M.; Weiner, H.L. Control of Treg and TH17 cell differentiation by the aryl hydrocarbon receptor. Nature 2008, 453, 65–71. [Google Scholar] [CrossRef]
- Singh, N.P.; Singh, U.P.; Singh, B.; Price, R.L.; Nagarkatti, M.; Nagarkatti, P.S. Activation of Aryl hydrocarbon receptor (AhR) leads to reciprocal epigenetic regulation of Foxp3 and IL-17 expression and amelioration of experimental colitis. PLoS ONE 2011, 6, e23522. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wei, Y.-D.; Bergander, L.; Rannug, U.; Rannug, A. Regulation of CYP1A1 transcription via the metabolism of the tryptophan-derived 6-formylindolo[3,2-b]carbazole. Arch. Biochem. Biophys. 2000, 383, 99–107. [Google Scholar] [CrossRef]
- Schiering, C.; Wincent, E.; Metidji, A.; Iseppon, A.; Li, Y.; Potocnik, A.J.; Omenetti, S.; Henderson, C.J.; Wolf, C.R.; Nebert, D.W.; et al. Feedback control of AHR signalling regulates intestinal immunity. Nature 2017, 542, 242–245. [Google Scholar] [CrossRef] [Green Version]
- Kimura, A.; Naka, T.; Nohara, K.; Fujii-Kuriyama, Y.; Kishimoto, T. Aryl hydrocarbon receptor regulates Stat1 activation and participates in the development of Th17 cells. Proc. Natl. Acad. Sci. USA 2008, 105, 9721–9726. [Google Scholar] [CrossRef] [Green Version]
- Mascanfroni, I.D.; Takenaka, M.C.; Yeste, A.; Patel, B.; Wu, Y.; Kenison, J.E.; Siddiqui, S.; Basso, A.S.; Otterbein, L.E.; Pardoll, E.M.; et al. Metabolic control of type 1 regulatory T cell differentiation by AHR and HIF1-α. Nat. Med. 2015, 21, 638–646. [Google Scholar] [CrossRef]
- Joller, N.; Lozano, E.; Burkett, P.R.; Patel, B.; Xiao, S.; Zhu, C.; Xia, J.; Tan, T.G.; Sefik, E.; Yajnik, V.; et al. Treg cells expressing the coinhibitory molecule TIGIT selectively inhibit proinflammatory Th1 and Th17 cell responses. Immunity 2014, 40, 569–581. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jurado-Manzano, B.B.; Zavala-Reyes, D.; Turrubiartes-Martínez, E.A.; Portales-Pérez, D.P.; González-Amaro, R.; Layseca-Espinosa, E. FICZ generates human tDCs that induce CD4+ CD25high Foxp3+ Treg-like cell differentiation. Immunol. Lett. 2017, 190, 84–92. [Google Scholar] [CrossRef] [PubMed]
- Yeste, A.; Takenaka, M.C.; Mascanfroni, I.D.; Nadeau, M.; Kenison, J.E.; Patel, B.; Tukpah, A.-M.; Babon, J.A.B.; DeNicola, M.; Kent, S.C.; et al. Tolerogenic nanoparticles inhibit T cell-mediated autoimmunity through SOCS2. Sci. Signal. 2016, 9, ra61. [Google Scholar] [CrossRef] [Green Version]
- Li, Y.; Innocentin, S.; Withers, D.R.; Roberts, N.A.; Gallagher, A.R.; Grigorieva, E.F.; Wilhelm, C.; Veldhoen, M. Exogenous stimuli maintain intraepithelial lymphocytes via aryl hydrocarbon receptor activation. Cell 2011, 147, 629–640. [Google Scholar] [CrossRef] [Green Version]
- Cervantes-Barragan, L.; Colonna, M. AHR signaling in the development and function of intestinal immune cells and beyond. Semin. Immunopathol. 2018, 40, 371–377. [Google Scholar] [CrossRef] [PubMed]
- Goettel, J.A.; Gandhi, R.; Kenison, J.E.; Yeste, A.; Murugaiyan, G.; Sambanthamoorthy, S.; Griffith, A.E.; Patel, B.; Shouval, D.S.; Weiner, H.L.; et al. AHR activation is protective against colitis driven by T cells in humanized mice. Cell Rep. 2016, 17, 1318–1329. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, J.; Ueno, A.; Iacucci, M.; Gasia, M.F.; Jijon, H.B.; Panaccione, R.; Kaplan, G.G.; Beck, P.L.; Luider, J.; Barkema, H.W.; et al. Crossover subsets of CD4+ T lymphocytes in the intestinal lamina propria of patients with Crohn’s disease and ulcerative colitis. Dig. Dis. Sci. 2017, 62, 2357–2368. [Google Scholar] [CrossRef]
- Műzes, G.; Molnár, B.; Tulassay, Z.; Sipos, F. Changes of the cytokine profile in inflammatory bowel diseases. World J. Gastroenterol. 2012, 18, 5848–5861. [Google Scholar] [CrossRef] [Green Version]
- Boland, A.B.S.; He, Z.; Tsai, M.S.; Olvera, J.G.; Kyla, D.; Duong, H.G.; Kim, E.S.; Limary, A.E.; Milner, J.J.; Yu, B.; et al. Heterogeneity and clonal relationships of adaptive immune cells in ulcerative colitis revealed by single-cell RNA and antigen-receptor sequencing analyses. Sci. Immunol. 2020, 5, eabb4432. [Google Scholar] [CrossRef]
- Roosenboom, B.; Wahab, P.J.; Smids, C.; Groenen, M.J.M.; Van Koolwijk, E.; Van Lochem, E.G.; Horjus-Talabur Horje, C.S. Intestinal CD103+CD4+ and CD103+CD8+ T-cell subsets in the gut of inflammatory bowel disease patients at diagnosis and during follow-up. Inflamm. Bowel Dis. 2019, 25, 1497–1509. [Google Scholar] [CrossRef]
- Monteleone, I.; Rizzo, A.; Sarra, M.; Sica, G.; Sileri, P.; Biancone, L.; Macdonald, T.T.; Pallone, F.; Monteleone, G. Aryl hydrocarbon receptor-induced signals up-regulate IL-22 production and inhibit inflammation in the gastrointestinal tract. Gastroenterol. 2011, 141, 237–248. [Google Scholar] [CrossRef] [PubMed]
- Gevers, D.; Kugathasan, S.; Denson, L.A.; Vázquez-Baeza, Y.; Van Treuren, W.; Ren, B.; Schwager, E.; Knights, D.; Song, S.J.; Yassour, M.; et al. The treatment-naive microbiome in new-onset Crohn’s disease. Cell Host Microbe 2014, 15, 382–392. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sokol, H.; Leducq, V.; Aschard, H.; Pham, H.-P.; Jegou, S.; Landman, C.; Cohen, D.; Liguori, G.; Bourrier, A.; Nion-Larmurier, I.; et al. Fungal microbiota dysbiosis in IBD. Gut 2017, 66, 1039–1048. [Google Scholar] [CrossRef] [Green Version]
- Wlodarska, M.; Luo, C.; Kolde, R.; D’Hennezel, E.; Annand, J.W.; Heim, C.E.; Krastel, P.; Schmitt, E.K.; Omar, A.S.; Creasey, E.A.; et al. Indoleacrylic acid produced by commensal Peptostreptococcus species suppresses inflammation. Cell Host Microbe 2017, 22, 25–37. [Google Scholar] [CrossRef] [Green Version]
- Franzosa, E.A.; Sirota-Madi, A.; Avila-Pacheco, J.; Fornelos, N.; Haiser, H.J.; Reinker, S.; Vatanen, T.; Hall, A.B.; Mallick, H.; McIver, L.J.; et al. Gut microbiome structure and metabolic activity in inflammatory bowel disease. Nat. Microbiol. 2019, 4, 898. [Google Scholar] [CrossRef]
- Peled, J.U.; Devlin, S.M.; Staffas, A.; Lumish, M.; Khanin, R.; Littmann, E.R.; Ling, L.; Kosuri, S.; Maloy, M.; Slingerland, J.B.; et al. Intestinal microbiota and relapse after hematopoietic-cell transplantation. J. Clin. Oncol. 2017, 35, 1650–1659. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Michonneau, D.; Latis, E.; Curis, E.; Dubouchet, L.; Ramamoorthy, S.; Ingram, B.; De Latour, R.P.; Robin, M.; De Fontbrune, F.S.; Chevret, S.; et al. Metabolomics analysis of human acute graft-versus-host disease reveals changes in host and microbiota-derived metabolites. Nat. Commun. 2019, 10, 5695. [Google Scholar] [CrossRef] [Green Version]
- Swimm, A.; Giver, C.R.; DeFilipp, Z.; Rangaraju, S.; Sharma, A.; Antonova, A.U.; Sonowal, R.; Capaldo, C.; Powell, D.; Qayed, M.; et al. Indoles derived from intestinal microbiota act via type I interferon signaling to limit graft-versus-host disease. Blood 2018, 132, 2506–2519. [Google Scholar] [CrossRef] [Green Version]
- Dant, T.A.; Lin, K.L.; Bruce, D.W.; Montgomery, S.A.; Kolupaev, O.V.; Bommiasamy, H.; Bixby, L.M.; Woosley, J.T.; McKinnon, K.P.; Gonzalez, F.J.; et al. T-cell expression of AhR inhibits the maintenance of pTreg cells in the gastrointestinal tract in acute GVHD. Blood 2017, 130, 348–359. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ridlon, J.M.; Harris, S.C.; Bhowmik, S.; Kang, D.-J.; Hylemon, P.B. Consequences of bile salt biotransformations by intestinal bacteria. Gut Microbes 2016, 7, 22–39. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, J.; Cao, Z.; Smith, A.D.; Carlson, P.E.C., Jr.; Coryell, M.; Chen, H.; Beger, R.D. Bile acid profile and its changes in response to cefoperazone treatment in MR1 deficient mice. Metabolites 2020, 10, 127. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hang, S.; Paik, D.; Yao, L.; Kim, E.; Trinath, J.; Lu, J.; Ha, S.; Nelson, B.N.; Kelly, S.P.; Wu, L.; et al. Bile acid metabolites control TH17 and Treg cell differentiation. Nat. Cell Biol. 2019, 576, 143–148. [Google Scholar] [CrossRef]
- Campbell, C.; McKenney, P.T.; Konstantinovsky, D.; Isaeva, O.I.; Schizas, M.; Verter, J.; Mai, C.; Jin, W.-B.; Guo, C.-J.; Violante, S.; et al. Bacterial metabolism of bile acids promotes generation of peripheral regulatory T cells. Nature 2020, 581, 475–479. [Google Scholar] [CrossRef]
- Lloyd-Price, J.; Arze, C.; Ananthakrishnan, A.N.; Schirmer, M.; Avila-Pacheco, J.; Poon, T.W.; Andrews, E.; Ajami, N.J.; Bonham, K.S.; Brislawn, C.J.; et al. Multi-omics of the gut microbial ecosystem in inflammatory bowel diseases. Nature 2019, 569, 655–662. [Google Scholar] [CrossRef] [PubMed]
- Gherardin, N.A.; Keller, A.N.; Woolley, R.E.; Le Nours, J.; Ritchie, D.S.; Neeson, P.J.; Birkinshaw, R.W.; Eckle, S.B.; Waddington, J.N.; Liu, L.; et al. Diversity of T cells restricted by the MHC class I-related molecule MR1 facilitates differential antigen recognition. Immunity 2016, 44, 32–45. [Google Scholar] [CrossRef] [Green Version]
- Reantragoon, R.; Corbett, A.J.; Sakala, I.G.; Gherardin, N.A.; Furness, J.B.; Chen, Z.; Eckle, S.B.; Uldrich, A.P.; Birkinshaw, R.W.; Patel, O.; et al. Antigen-loaded MR1 tetramers define T cell receptor heterogeneity in mucosal-associated invariant T cells. J. Exp. Med. 2013, 210, 2305–2320. [Google Scholar] [CrossRef] [PubMed]
- McWilliam, H.E.; Villadangos, J.A. MR1: A multi-faceted metabolite sensor for T cell activation. Curr. Opin. Immunol. 2020, 64, 124–129. [Google Scholar] [CrossRef]
- Legoux, F.; Bellet, D.; Daviaud, C.; El Morr, Y.; Darbois, A.; Niort, K.; Procopio, E.; Salou, M.; Gilet, J.; Ryffel, B.; et al. Microbial metabolites control the thymic development of mucosal-associated invariant T cells. Science 2019, 366, 494–499. [Google Scholar] [CrossRef]
- Dumas, A.; Corral, D.; Colom, A.; Levillain, F.; Peixoto, A.; Hudrisier, D.; Poquet, Y.; Neyrolles, O. The host microbiota contributes to early protection against lung colonization by mycobacterium tuberculosis. Front. Immunol. 2018, 9, 2656. [Google Scholar] [CrossRef]
- Le Bourhis, L.; Martin, E.; Péguillet, I.; Guihot, A.; Froux, N.; Coré, M.; Lévy, E.; Dusseaux, M.; Meyssonnier, V.; Premel, V.; et al. Antimicrobial activity of mucosal-associated invariant T cells. Nat. Immunol. 2010, 11, 701–708. [Google Scholar] [CrossRef] [Green Version]
- Seach, N.; Guerri, L.; Le Bourhis, L.; Mburu, Y.; Cui, Y.; Bessoles, S.; Soudais, C.; Lantz, O. Double positive thymocytes select mucosal-associated invariant T cells. J. Immunol. 2013, 191, 6002–6009. [Google Scholar] [CrossRef] [Green Version]



{kind=link}
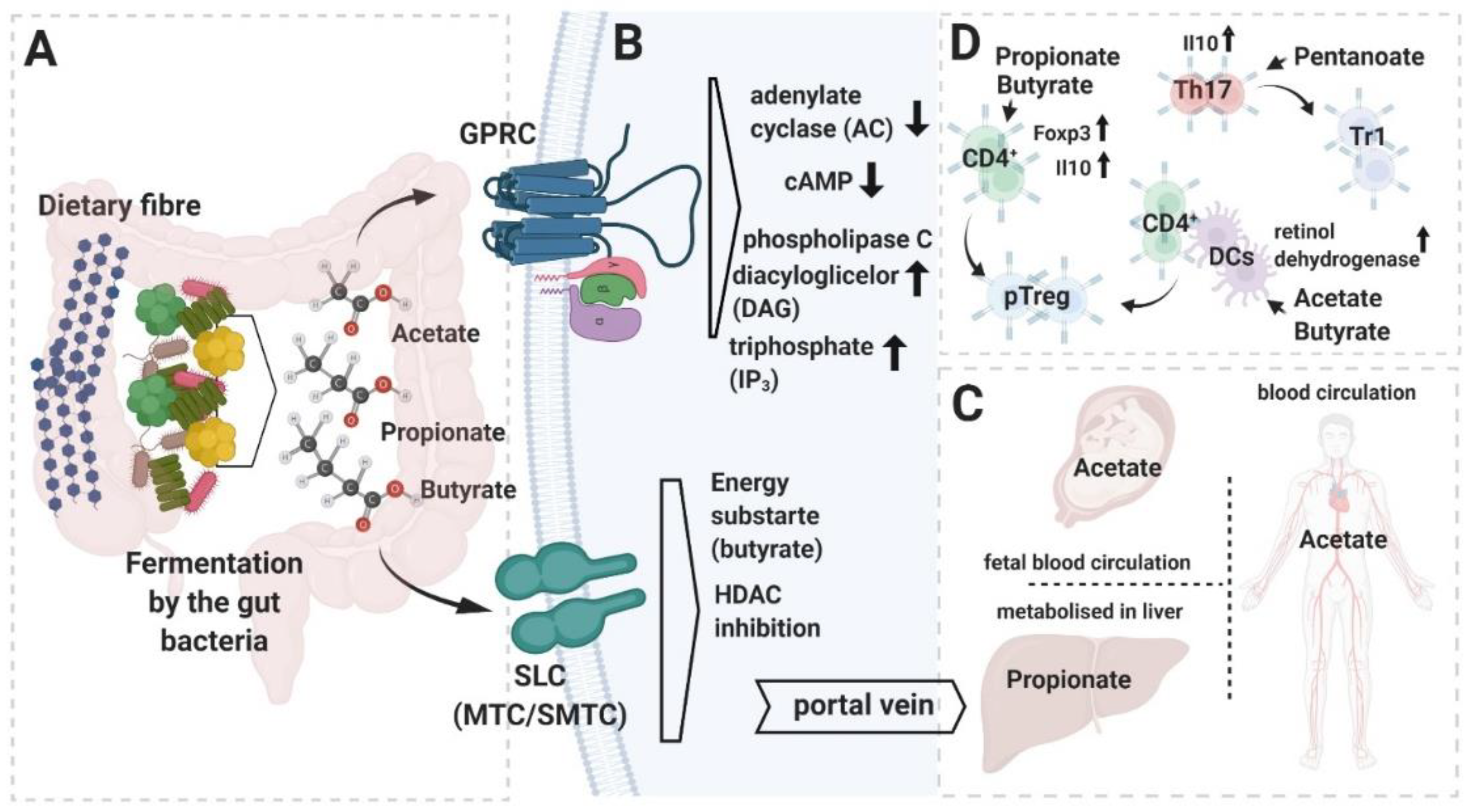{kind=link}
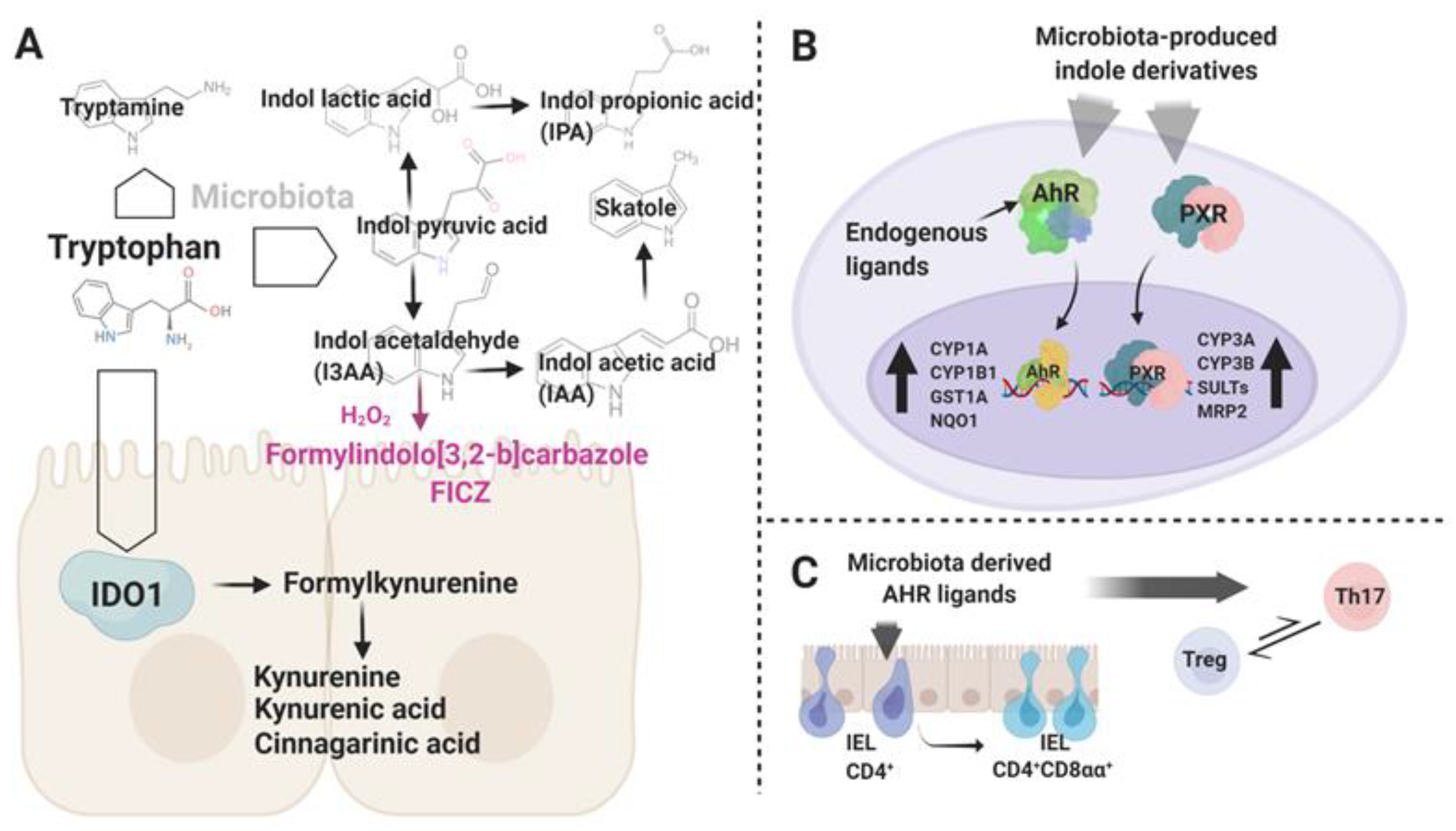{kind=link}
| SCFA | Species | Ref |
|---|---|---|
| Butyrate | Roseburia intestinalis | [26] |
| Eubacterium rectale | ||
| Eubacterium hallii | ||
| Ruminococcus obeum | ||
| Ruminococcus gnavus | ||
| Propionate | Clostridium ramosum | [27] |
| Clostridium bifermentans | ||
| Bacteroides fragilis | ||
| Acetate | Clostridium ramosum | [26,27] |
| Clostridium bifermentans | ||
| Bacteroides fragilis | ||
| Ruminococcus obeum | ||
| Ruminococcus gnavus | ||
| Valerate/Pentanoate | Clostridium luticellarii | [28] |
| Candidatus methanogranum |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wojciech, L.; Tan, K.S.W.; Gascoigne, N.R.J. Taming the Sentinels: Microbiome-Derived Metabolites and Polarization of T Cells. Int. J. Mol. Sci. 2020, 21, 7740. https://doi.org/10.3390/ijms21207740
Wojciech L, Tan KSW, Gascoigne NRJ. Taming the Sentinels: Microbiome-Derived Metabolites and Polarization of T Cells. International Journal of Molecular Sciences. 2020; 21(20):7740. https://doi.org/10.3390/ijms21207740
Chicago/Turabian StyleWojciech, Lukasz, Kevin S. W. Tan, and Nicholas R. J. Gascoigne. 2020. "Taming the Sentinels: Microbiome-Derived Metabolites and Polarization of T Cells" International Journal of Molecular Sciences 21, no. 20: 7740. https://doi.org/10.3390/ijms21207740
APA StyleWojciech, L., Tan, K. S. W., & Gascoigne, N. R. J. (2020). Taming the Sentinels: Microbiome-Derived Metabolites and Polarization of T Cells. International Journal of Molecular Sciences, 21(20), 7740. https://doi.org/10.3390/ijms21207740